
Abstract
The amyloid-beta peptide (Aβ) cascade hypothesis posits that Aβ accumulation is the fundamental initiator of Alzheimer's disease (AD), and mounting evidence suggests that impaired Aβ clearance rather than its overproduction is the major pathogenic event for AD. Recent genetic studies have identified cluster of differentiation 33 (CD33) as a strong genetic locus linked to AD. As a type I transmembrane protein, CD33 belongs to the sialic acid-binding immunoglobulin-like lectins, mediating the cell–cell interaction and inhibiting normal functions of immune cells. In the brain, CD33 is mainly expressed on microglial cells. The level of CD33 was found to be increased in the AD brain, which positively correlated with amyloid plaque burden and disease severity. More importantly, CD33 led to the impairment of microglia-mediated clearance of Aβ, which resulted in the formation of amyloid plaques in the brain. In this article, we review the recent epidemiological findings of CD33 that related with AD and discuss the levels and pathogenic roles of CD33 in this disease. Based on the contributing effects of CD33 in AD pathogenesis, targeting CD33 may provide new opportunities for AD therapeutic strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer's disease (AD) is a complex and multifactorial neurodegenerative disease, which is characterized by the formation of extracellular amyloid plaques containing the amyloid-beta peptide (Aβ) within the parenchyma of the brain [1]. Emerging evidence indicates that impaired Aβ clearance rather than its overproduction is the central pathogenic event in AD, as the clearance rates for Aβ are declined in AD patients, while its production rates stay unchanged compared with those in healthy controls [2]. Recent genome-wide association studies (GWASs) have identified cluster of differentiation 33 (CD33) as a strong genetic locus associated with late-onset AD (LOAD) in white populations [3–5], and these findings have been successfully replicated in other ethnic groups [6–11] (Table 1). CD33 is a type I transmembrane protein belonging to the sialic acid-binding immunoglobulin-like lectins (Siglecs) family, which is thought to mediate the cell–cell interaction and to inhibit normal functions of immune cells [12]. In the brain, CD33 is mainly expressed on microglial cells, and compelling evidence implicating that CD33 facilitates Aβ-related pathology in AD by impairing microglia-mediated Aβ clearance [13–16]. In this article, we review the recent epidemiological findings of CD33 that related with LOAD. The evidence about the levels and pathogenic functions of CD33 in AD has also been discussed. Moreover, we present the advances and challenges in targeting CD33 for AD therapeutic strategies.
Structure, Localization, and Physiological Function of CD33
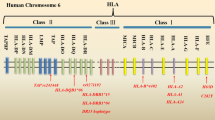
CD33 gene maps on chromosome 19q13.33 in humans, encoding a 67-kD protein CD33 (Fig. 1), and CD33 protein consists of an extracellular N-terminal V-set immunoglobulin domain responsible for sialic acid recognition, followed by a C2-type immunoglobulin repeat [12, 17]. Intracellularly, human CD33 has two conserved cytoplasmic tyrosine-based motifs: a membrane-proximal immunoreceptor tyrosine-based inhibitory motif (ITIM) and a membrane-distal ITIM-like motif. However, the ITIM is lacking in mouse CD33 (Fig. 2a) [18]. Both ITIM and ITIM-like motif are involved in inhibitory signal transduction via the recruitment of SHP [Src homology 2 (SH2) domain-containing protein tyrosine phosphatase]-1 and SHP-2 tyrosine phosphatases as well as other SH2 domain-containing effector proteins [19, 20]. In mammals, CD33 has been found to be expressed on hematopoietic and phagocytic cells, including hematopoietic progenitors, myelomonocytic precursors, macrophages, monocytes, dendritic cells, and microglial cells [21, 22]. With respect to its functions, CD33 is known to participate in adhesion processes of human primary immune cells. CD33 is able to bind to high-affinity sialoglycans on target cells, mediating cell–cell interaction [23]. Similarly, CD33 can interact with sialylated pathogens and viruses, which are decorated on their surface with host-derived or self-synthesized sialoglycans. Binding of this pathogen to CD33 via sialylated ligands could facilitate their endocytosis and clearance by host phagocytes or, as opposed, could mediate their internalization to spread infection [24]. Interestingly, the endocytic function of CD33 has been exploited for cell-directed therapies in acute myeloid leukemia (AML) [25]. Another important role of CD33 is its ability to inhibit immune cell functions [26]. CD33 appeared to inhibit the human monocyte production of pro-inflammatory cytokines, such as interleukin (IL)-1β, tumor necrosis factor alpha (TNF-α), and IL-8 via phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase-dependent pathway [27]. Conversely, reduction of CD33 resulted in the increased secretion of TNF-α in human monocyte [28]. CD33 has also been reported to regulate cell growth and survival, through the inhibition of proliferation and induction of apoptosis. Activation of CD33 on CD34+ myeloid progenitors as well as CD33 on monocyte-derived dendritic cells by a specific antibody markedly inhibited their growth [29]. In addition, engagement of CD33 expressed on AML cells led to their apoptosis [30].

Schematic of CD33 gene. The CD33 gene structure spans 51,728,335–51,743,274 bp on chromosome 19q13.33 (hg19) and encodes seven exons (represented by orange boxes). Note that the single-nucleotide polymorphisms in CD33 that have a significant association with AD risk are highlighted on this figure

Structure of CD33 and its relation with Aβ pathology in AD. a CD33 consists of an extracellular N-terminal V-set immunoglobulin domain responsible for sialic acid recognition, followed by a C2-type immunoglobulin repeat. Intracellularly, human CD33 has two conserved cytoplasmic tyrosine-based motifs: a membrane-proximal ITIM and a membrane-distal ITIM-like motif. However, the ITIM is lacking in mouse CD33. b Binding with sialic acid is necessary for CD33 to exert its inhibitory effects on microglia-mediated Aβ uptake [37]. In the AD brain, amyloid plaques are often aggregated with several proteins and lipids which are highly sialylated [38–40]. Hence, the binding of sialylated glycoproteins and glycolipids on amyloid plaques to CD33 expressed on microglial cells may account for the immune avoidance for amyloid plaques in AD, which avoided the uptake and clearance by microglial cells
Genetics of CD33 Gene in AD
In last 5 years, three independent GWASs have identified CD33 as a strong genetic locus linked to LOAD (Table 1) [3–5]. The main single-nucleotide polymorphisms (SNPs) associated with LOAD are rs3865444 and rs3826656, which are located 373 and 1,722 bp upstream of CD33 coding region, respectively.
In regard to rs3865444, the minor allele has been identified as a protective allele in two GWASs in white populations [4, 5], and this result has been successfully replicated in cohorts from Europe, North America, and Korea [6, 7]. By contrast, the T allele of rs3865444 appears to be a risky rather than a protective allele in AD in Han Chinese population, as demonstrated by studies from Deng et al. and our group [8, 9]. In fact, the minor allele frequency of rs3865444 in Han Chinese cohort was 17 % [9], which was significant lower than that of the white populations (30 %) [4]. Besides, some epidemiological researchers suggested that the rs3865444 polymorphism may be in linkage disequilibrium (LD) with a functional polymorphism that causes stronger influences on AD risk, and the degrees of LD among these variants usually differ across populations [31], which may be responsible for the contradictory findings on the association between rs3865444 and AD. Another possible explanation is that the association between rs3865444 and AD was influenced by environmental and lifestyle factors in different populations, which led to the phenomenon that rs3865444 conferred a risk for AD in some populations while provided protective effects against AD in other populations [32]. Aside from rs3865444, Bertram et al. found that the minor allele of rs3826656 conferred a risk for AD in a large family-based GWAS containing 1,376 samples from 410 European families [3]. The association between rs3826656 and AD was unable to replicate in a more recent GWAS by Naj and colleagues [4]. However, it should be noted that only stage 1 analysis (containing 8,309 AD patients and 7,366 cognitively normal controls) was conducted in the study of Naj, not the entire sample, so this finding was not yet conclusive. Nevertheless, in contrast to the findings in Caucasians, Yuan et al. reported that the minor allele of rs3826656 was associated with a reduced risk of AD in Han Chinese population with apolipoprotein E (APOE) ε4 alleles [10]. It is worth noting that rs3826656 is only 1,348 bp from rs3865444, and they are in a complete linkage with each other in both Caucasian and Han Chinese populations (for Caucasian population, D′ = 1.0, r 2 = 0.11; for Han Chinese population, D′ = 1, r 2 = 0.47). The relatively low r 2 may be accountable by the vastly different minor allele frequencies of these two SNPs [for Caucasian population, rs3826656 = 19.5 % and rs3865444 = 31.9 %; for Han Chinese population, rs3826656 = 29.9 % and rs3865444 = 17.6 %; frequencies taken from the HapMap database (release no. 28, August 2010, http://www.hapmap.org)].
More recently, a novel SNP rs114282264 that is located in the intron region of CD33 has been identified to be significantly associated with AD in African Americans [11], and this association requires to be confirmed in the other ethnic groups in the future.
CD33 Levels in AD
In the brain, the levels of CD33 protein were increased in AD patients, as revealed by the increase in the count of CD33-immunoreactive microglial cells [13]. Regarding the transcription level, CD33 mRNA was revealed to be dramatically increased in AD, suggesting a potential upregulation of CD33 transcription in microglial cells [13]. These findings were consistent with a recent study from Karch et al., which demonstrated a significant correlation between mRNA levels of CD33 and Iba-1 (a biomarker of microglial cells) in brain tissue from the parietal lobe of AD cases. In addition, when normalized to Iba-1 expression, the CD33 mRNA expression was revealed to correlate with disease status and Clinical Dementia Rating scores [14].
As mentioned above, most of the genetic studies indicated a protective role of minor (T) allele of the CD33 SNP rs3865444 in AD [4–7]. The mechanism by which T allele protects against AD has been uncovered recently, as T allele was associated with reduced CD33 protein levels on microglial cells both in the AD and control subject. Coincidentally, a recent study from Bradshaw et al. demonstrated that the risk allele (C) of rs3865444 was associated with elevated CD33 levels, as a sevenfold increase in CD33 expression on monocytes was observed in young cognitively normal individuals carrying the CC genotype versus those carrying the TT genotype. Meanwhile, this finding has been successfully replicated in old person either with or without AD [15]. It is worth noting that T allele is not associated with significant alterations in CD33 mRNA levels in those carriers [13]. One reasonable explanation is that the rs3865444 SNP is in linkage disequilibrium with functional variant(s) located in the coding region, which influences mRNA translation rather than its stability.
Taken together, these findings suggested a possible association of CD33 levels with the etiology and pathogenesis of AD.
CD33, Phagocytes, and Aβ Pathology
Amyloid hypothesis have emphasized the crucial role of Aβ pathology in the pathogenesis of AD, which is thought to drive a pathologic cascade including hyperphosphorylation of tau protein and neuroinflammation, ultimately leading to cognitive impairment [1]. Accumulating evidence has indicated a strong association of CD33 with Aβ pathology in AD progression. In a recent study from Griciuc et al., the T allele of SNP rs3865444, which led to the reduction of CD33 levels in the brain, was found to be linked with decreased amyloid plaque burdens in the brain cortex of AD patients [13]. In agreement with this finding, Bradshaw and colleagues showed that the C allele of SNP rs3865444, which caused the elevation in CD33 levels, was associated with a greater burden of fibrillar amyloid in older asymptomatic individuals and with neuritic amyloid plaques in the brains of older individuals at autopsy [15]. More direct evidence on the association between CD33 and Aβ pathology has been gathered from animal models of AD, as amyloid precursor protein/presenilin 1 (APP/PS1) transgenic mice lacking CD33 exhibited significant lower Aβ levels as well as reduced amyloid plaque burden in the brain [13]. These observations clearly indicated a pathogenic role of CD33 in facilitation of Aβ pathology. More importantly, deletion of CD33 in APP/PS1 transgenic mice did not alter the APP processing or the levels of pro-inflammatory cytokines in the brain, implying that CD33 contributed to Aβ pathology by interfering Aβ clearance rather than promoting its generation [13].
Microglial cells are considered to be the main phagocytes in the brain, which plays a nuanced and complex role in the progression of AD [33]. Despite of its detrimental role in exacerbation of neuroinflammation, microglial cells is also known to the uptake and degradation of Aβ, which prevents the formation of amyloid plaque in the AD brain [34]. In view of the fact that CD33 is mainly expressed on the surface of phagocytes including microglial cells, some researches inferred that CD33 may facilitate amyloid pathology via impairing the microglia-mediated clearance of Aβ. Considering the fact that microglial cells are replenished partly by the immigration of circulating monocytes under pathological conditions including AD [35, 36], Bradshaw and colleagues [15] employed circulating monocytes to study the role of CD33 in Aβ clearance. They found that a higher expression of CD33 on the surface of circulating monocytes directly inhibited their abilities to phagocytose Aβ. In addition, they also observed that CD33 levels positively correlated with the mean proportion of terminally activated microglial cells in the inferior temporal lobe, which is an early target of Aβ pathology in the brain. This observation can be explained by the hypothesis that a higher CD33 levels inhibited the functions of microglial cells in Aβ uptake, which subsequently led to the accumulation of less functional microglial cells in plaque-associated brain regions.
The observations from the study of Bradshaw [15] have been proven by Griciuc and colleagues [13] using microglial cells. In mouse primary microglial cells lacking CD33, increased uptake of Aβ was observed to be relative to wide-type (WT) cells. Intriguingly, there is no difference in Aβ degradation rate between CD33-deficient and WT cells. By contrast, BV2 microglial cells overexpressing WT CD33 protein exhibited a significant impairment in their capacity to uptake Aβ, but the ability to degrade Aβ remained unaffected. These findings were further confirmed by transfecting microglial cells with an ubiquitylation-defective CD33 mutant (CD33K7R), which led to enhanced cell surface expression of CD33. As expected, microglial cells expressing CD33K7R exhibited a dramatic reduction in capacity of Aβ uptake when compared with WT cells [13]. Taken together, the findings from Bradshaw et al. [15] and Griciuc et al. [13] indicated that CD33 impaired Aβ clearance via directly inhibiting its uptake by microglial cells. As discussed above, CD33 could exert their physiological functions by interacting with sialic acids [12]. To explore the requirement of sialic acid binding in the CD33-mediated inhibition on microglial uptake of Aβ, Griciuc et al. [13] transfected BV2 microglial cells with a CD33 mutant that lead to the deficiency in the sialic acid-binding V-type immunoglobulin-like domain (CD33ΔV-Ig protein). Cells expressing the CD33ΔV-Ig protein demonstrated no impairment in their capacity to uptake Aβ, indicating that sialic acid binding is required for the inhibition of CD33 on microglia-mediated uptake of Aβ [13].
Moreover, the above observations could be linked to a recent proposed hypothesis that amyloid plaque is able to evade the clearance of microglial cells with the help of CD33-related Siglecs, which subsequently leads to neurodegeneration [37]. In the AD brain, amyloid plaques are often aggregated with several proteins and lipids which are highly sialylated, such as clusterin, APOE proteins, and gangliosides [38–40]. As discussed above, sialic acid binding is necessary for the inhibition of CD33 and its related Siglecs on Aβ uptake. Hence, the binding of sialylated glycoproteins and glycolipids on amyloid plaques to CD33 expressed on microglial cells may account for the immune avoidance for amyloid plaques in AD, which avoided the uptake and clearance by microglial cells [37] (Fig. 2b).
CD33 as a Potential Therapeutic Target for AD
The Aβ cascade hypothesis posits that accumulation of Aβ is the fundamental initiator of AD [1, 41]. Meanwhile, it is widely believed that impaired Aβ clearance is a major pathogenic event for LOAD [2, 42]. As mentioned earlier, microglial cells have been found to play critical roles in Aβ clearance in the brain, and the recent study from Griciuc et al. [13] demonstrated that CD33 contributed to the pathogenesis of AD by impairing microglia-mediated clearance of Aβ. Meanwhile, deletion of CD33 both in vitro and in vivo led to the increased Aβ clearance and reduced Aβ levels. Remarkably, CD33 deletion in vivo was viable, as CD33 knockout mice were fertile and exhibited no anatomical defect, implying that targeting CD33 may be a safety option in AD treatment. It should be noted that therapies targeting CD33 have already been developed in AML, as approximately 90 % of AML patients have myeloblasts expressing CD33 [43]. Naked humanized anti-CD33 and calicheamicin-conjugated humanized murine anti-CD33 antibodies have been developed and tested in several phase III clinical trials of AML [25, 44]. Meanwhile, development of a CD33 antibody that is able to cross the blood–brain barrier (BBB) is technically feasible, as a chimeric antibody in which the CD33 antibody is fused to a monoclonal antibody against the human insulin receptor was found to facilitate the receptor-mediated passage of the chimera across the BBB [45]. Alternatively, development of small compounds, such as sialic acid-based antagonists that specifically target CD33 and inhibit its function should also be considered as a therapeutic strategy for AD.
Conclusions and Future Perspectives
CD33 is a type I transmembrane protein expressed on brain microglial cells, exerting its inhibitory effects on microglia-mediated Aβ uptake. To date, three SNPs in CD33 have been identified to be significantly associated with LOAD risk, and these findings ought to be further confirmed in the other ethnic groups. In the future, more in-depth genetic screenings should be performed to uncover functional variants in CD33 that related to LOAD. In addition to the inhibition on Aβ clearance, the other biological mechanisms by which CD33 contributed to the pathogenesis of AD should also be investigated, such as its role in neuroinflammation and neuronal apoptosis. Asides from the CD33 in the brain, the plasma level of CD33 in AD patient and its correlation with this disease should be evaluated by future studies, as a better understanding of CD33 action both in the brain and in the periphery will hopefully lead to the development of novel therapeutics for the prevention and treatment of AD.
References
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297(5580):353–356. doi:10.1126/science.1072994
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 330(6012):1774. doi:10.1126/science.1197623
Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I, Jiang H, Laird N, Moscarillo T, Ohlsen KL, Elliott K, Wang X, Hu-Lince D, Ryder M, Murphy A, Wagner SL, Blacker D, Becker KD, Tanzi RE (2008) Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet 83(5):623–632. doi:10.1016/j.ajhg.2008.10.008
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 43(5):436–441. doi:10.1038/ng.801
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O'Donovan M, Amouyel P, Williams J (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 43(5):429–435. doi:10.1038/ng.803
Carrasquillo MM, Belbin O, Hunter TA, Ma L, Bisceglio GD, Zou F, Crook JE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, Passmore P, Morgan K, Younkin SG (2011) Replication of EPHA1 and CD33 associations with late-onset Alzheimer's disease: a multi-centre case–control study. Mol Neurodegener 6(1):54. doi:10.1186/1750-1326-6-54
Chung SJ, Lee JH, Kim SY, You S, Kim MJ, Lee JY, Koh J (2012) Association of GWAS top hits with late-onset Alzheimer disease in Korean population. Alzheimer Dis Assoc Disord 27(3):250–257. doi:10.1097/WAD.0b013e31826d7281
Deng YL, Liu LH, Wang Y, Tang HD, Ren RJ, Xu W, Ma JF, Wang LL, Zhuang JP, Wang G, Chen SD (2012) The prevalence of CD33 and MS4A6A variant in Chinese Han population with Alzheimer's disease. Hum Genet 131(7):1245–1249. doi:10.1007/s00439-012-1154-6
Tan L, Yu JT, Zhang W, Wu ZC, Zhang Q, Liu QY, Wang W, Wang HF, Ma XY, Cui WZ (2013) Association of GWAS-linked loci with late-onset Alzheimer's disease in a northern Han Chinese population. Alzheimer's Dement J Alzheimer's Assoc 9(5):546–553. doi:10.1016/j.jalz.2012.08.007
Yuan Q, Chu C, Jia J (2012) Association studies of 19 candidate SNPs with sporadic Alzheimer's disease in the North Chinese Han population. Neurol Sci 33(5):1021–1028. doi:10.1007/s10072-011-0881-0
Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, Valladares O, Lin CF, Larson EB, Graff-Radford NR, Evans D, De Jager PL, Crane PK, Buxbaum JD, Murrell JR, Raj T, Ertekin-Taner N, Logue M, Baldwin CT, Green RC, Barnes LL, Cantwell LB, Fallin MD, Go RC, Griffith P, Obisesan TO, Manly JJ, Lunetta KL, Kamboh MI, Lopez OL, Bennett DA, Hendrie H, Hall KS, Goate AM, Byrd GS, Kukull WA, Foroud TM, Haines JL, Farrer LA, Pericak-Vance MA, Schellenberg GD, Mayeux R (2013) Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 309(14):1483–1492. doi:10.1001/jama.2013.2973
Crocker PR, Paulson JC, Varki A (2007) Siglecs and their roles in the immune system. Nat Rev Immunol 7(4):255–266. doi:10.1038/nri2056
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE (2013) Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78(4):631–643. doi:10.1016/j.neuron.2013.04.014
Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM (2012) Expression of novel Alzheimer's disease risk genes in control and Alzheimer's disease brains. PloS One 7(11):e50976. doi:10.1371/journal.pone.0050976
Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, Morris MC, Evans DA, Johnson K, Sperling RA, Schneider JA, Bennett DA, De Jager PL (2013) CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci 16(7):848–850. doi:10.1038/nn.3435
Malpass K (2013) Alzheimer disease: functional dissection of CD33 locus implicates innate immune response in Alzheimer disease pathology. Nat Rev Neurol 9(7):360. doi:10.1038/nrneurol.2013.119
Varki A, Angata T (2006) Siglecs—the major subfamily of I-type lectins. Glycobiology 16(1):1R–27R. doi:10.1093/glycob/cwj008
von Gunten S, Bochner BS (2008) Basic and clinical immunology of Siglecs. Ann N Y Acad Sci 1143:61–82. doi:10.1196/annals.1443.011
Ravetch JV, Lanier LL (2000) Immune inhibitory receptors. Science 290(5489):84–89
Walter RB, Raden BW, Zeng R, Hausermann P, Bernstein ID, Cooper JA (2008) ITIM-dependent endocytosis of CD33-related Siglecs: role of intracellular domain, tyrosine phosphorylation, and the tyrosine phosphatases, Shp1 and Shp2. J Leukoc Biol 83(1):200–211. doi:10.1189/jlb.0607388
Andrews RG, Torok-Storb B, Bernstein ID (1983) Myeloid-associated differentiation antigens on stem cells and their progeny identified by monoclonal antibodies. Blood 62(1):124–132
Griffin JD, Linch D, Sabbath K, Larcom P, Schlossman SF (1984) A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res 8(4):521–534
Jandus C, Simon HU, von Gunten S (2011) Targeting Siglecs—a novel pharmacological strategy for immuno- and glycotherapy. Biochem Pharmacol 82(4):323–332. doi:10.1016/j.bcp.2011.05.018
Vimr E, Lichtensteiger C (2002) To sialylate, or not to sialylate: that is the question. Trends Microbiol 10(6):254–257
Ricart AD (2011) Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res 17(20):6417–6427. doi:10.1158/1078-0432.CCR-11-0486
Crocker PR, Redelinghuys P (2008) Siglecs as positive and negative regulators of the immune system. Biochem Soc Trans 36(Pt 6):1467–1471. doi:10.1042/BST0361467
Lajaunias F, Dayer JM, Chizzolini C (2005) Constitutive repressor activity of CD33 on human monocytes requires sialic acid recognition and phosphoinositide 3-kinase-mediated intracellular signaling. Eur J Immunol 35(1):243–251. doi:10.1002/eji.200425273
Gonzalez Y, Herrera MT, Soldevila G, Garcia-Garcia L, Fabián G, Pérez-Armendariz EM, Bobadilla K, Guzmán-Beltran S, Sada E, Torres M (2012) High glucose concentrations induce TNF-alpha production through the down-regulation of CD33 in primary human monocytes. BMC Immunol 13:19. doi:10.1186/1471-2172-13-19
Vitale C, Romagnani C, Falco M, Ponte M, Vitale M, Moretta A, Bacigalupo A, Moretta L, Mingari MC (1999) Engagement of p75/AIRM1 or CD33 inhibits the proliferation of normal or leukemic myeloid cells. Proc Natl Acad Sci U S A 96(26):15091–15096
Balaian L, Zhong RK, Ball ED (2003) The inhibitory effect of anti-CD33 monoclonal antibodies on AML cell growth correlates with Syk and/or ZAP-70 expression. Exp Hematol 31(5):363–371
Lin PI, Vance JM, Pericak-Vance MA, Martin ER (2007) No gene is an island: the flip-flop phenomenon. Am J Hum Genet 80(3):531–538. doi:10.1086/512133
Allen M, Cox C, Belbin O, Ma L, Bisceglio GD, Wilcox SL, Howell CC, Hunter TA, Culley O, Walker LP, Carrasquillo MM, Dickson DW, Petersen RC, Graff-Radford NR, Younkin SG, Ertekin-Taner N (2012) Association and heterogeneity at the GAPDH locus in Alzheimer's disease. Neurobiol Aging 33(1):203.e25–203.e33. doi:10.1016/j.neurobiolaging.2010.08.002
Cameron B, Landreth GE (2010) Inflammation, microglia, and Alzheimer's disease. Neurobiol Dis 37(3):503–509. doi:10.1016/j.nbd.2009.10.006
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K (2006) Role of toll-like receptor signalling in Abeta uptake and clearance. Brain: J Neurol 129(Pt 11):3006–3019. doi:10.1093/brain/awl249
Simard AR, Rivest S (2004) Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J 18(9):998–1000. doi:10.1096/fj.04-1517fje
Hickman SE, El Khoury J (2013) The neuroimmune system in Alzheimer's disease: the glass is half full. J Alzheimer's Dis 33(Suppl 1):S295–S302. doi:10.3233/JAD-2012-129027
Salminen A, Kaarniranta K (2009) Siglec receptors and hiding plaques in Alzheimer's disease. J Mol Med (Berl) 87(7):697–701. doi:10.1007/s00109-009-0472-1
Yu JT, Tan L (2012) The role of clusterin in Alzheimer's disease: pathways, pathogenesis, and therapy. Mol Neurobiol 45(2):314–326. doi:10.1007/s12035-012-8237-1
Bu G (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10(5):333–344. doi:10.1038/nrn2620
Ariga T, McDonald MP, Yu RK (2008) Role of ganglioside metabolism in the pathogenesis of Alzheimer's disease—a review. J Lipid Res 49(6):1157–1175. doi:10.1194/jlr.R800007-JLR200
Selkoe DJ (2000) Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci 924:17–25
Wang YJ, Zhou HD, Zhou XF (2006) Clearance of amyloid-beta in Alzheimer's disease: progress, problems and perspectives. Drug Discov Today 11(19–20):931–938. doi:10.1016/j.drudis.2006.08.004
Legrand O, Perrot JY, Baudard M, Cordier A, Lautier R, Simonin G, Zittoun R, Casadevall N, Marie JP (2000) The immunophenotype of 177 adults with acute myeloid leukemia: proposal of a prognostic score. Blood 96(3):870–877
Sievers EL (2001) Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukaemia in first relapse. Expert Opin Biol Ther 1(5):893–901. doi:10.1517/14712598.1.5.893
Paulson JC, Macauley MS, Kawasaki N (2012) Siglecs as sensors of self in innate and adaptive immune responses. Ann N Y Acad Sci 1253:37–48. doi:10.1111/j.1749-6632.2011.06362.x
Acknowledgments
This work was supported in part by grants from the National Natural Science Foundation of China (81000544 and 81171209), the Shandong Provincial Natural Science Foundation, China (ZR2010HQ004 and ZR2011HZ001), and the Shandong Provincial Outstanding Medical Academic Professional Program.
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Jiang, T., Yu, JT., Hu, N. et al. CD33 in Alzheimer's Disease. Mol Neurobiol 49, 529–535 (2014). https://doi.org/10.1007/s12035-013-8536-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8536-1