
Abstract
Imbalance in excitatory/inhibitory signal in the brain has been proposed as one of the main pathological features in autism spectrum disorders, although the underlying cellular and molecular mechanism is unclear yet. Because excitatory/inhibitory imbalance can be induced by aberration in glutamatergic/GABAergic neuronal differentiation, we investigated the mechanism of dysregulated neuronal differentiation between excitatory and inhibitory neurons in the embryonic and postnatal brain of prenatally valproic acid-exposed rat offspring, which is often used as an animal model of autism spectrum disorders. Transcription factor Pax6, implicated in glutamatergic neuronal differentiation, was transiently increased in embryonic cortex by valproate exposure, which resulted in the increased expression of glutamatergic proteins in postnatal brain of offspring. Chromatin immunoprecipitation showed increased acetylated histone binding on Pax6 promoter region, which may underlie the transcriptional up-regulation of Pax6. Other histone deacetylase (HDAC) inhibitors including TSA and SB but not valpromide, which is devoid of HDAC inhibitor activity, induced Pax6 up-regulation. Silencing Pax6 expression in cultured rat primary neural progenitor cells demonstrated that up-regulation of Pax6 plays an essential role in valproate-induced glutamatergic differentiation. Blocking glutamatergic transmission with MK-801 or memantine treatment, and to a lesser extent with MPEP treatment, reversed the impaired social behaviors and seizure susceptibility of prenatally valproate-exposed offspring. Together, environmental factors may contribute to the imbalance in excitatory/inhibitory neuronal activity in autistic brain by altering expression of transcription factors governing glutamatergic/GABAergic differentiation during fetal neural development, in conjunction with the genetic preload.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autism spectrum disorder (ASD) is a prototypic pervasive developmental disorder, which results from abnormal process of brain development. ASD is a clinically heterogeneous group of disorders diagnosed before 3 years of age and characterized by three behavioral symptoms: social deficits, impaired language and communication, and stereotyped and repetitive behaviors [1, 2]. The brain of ASD patients shows specific anatomical and functional features including increased prevalence of macrocephaly [3, 4], reduced GABAergic signaling system [5], increased glutamatergic signaling system [6], aberrant minicolumnar organization [7], and excessive synapse formation in the cortex [8]. Although there is no unifying theory explaining all the ASD cases, imbalanced signal between excitation and inhibition in the autistic brain has been recently proposed as one of the main endophenotypes of ASD [9, 10]. Decreased expression of GABAA receptor subunit [11, 12], glutamic acid decarboxylase [13], and disrupted inhibitory architecture [14] was reported in autistic brains. In spite of ample anatomical or functional evidence that supports glutamatergic hyper-functionality in autistic brains, the relevant mechanistic studies in the ASD brain is relatively sparse.
Exposure to valproic acid (VPA) in and around the first trimester of pregnancy causes damage to the fetal brain [15] and induces cognitive impairment, similar to symptoms of ASD [16]. Accordingly, VPA exposure was firstly used to induce an animal model of autism [17], which used as alternative to thalidomide. And many researchers established the animal model of ASD using VPA during pregnancy. Similar to human autistic symptoms, prenatally VPA-exposed rats showed defects in social behaviors [18] and abnormalities in GABAergic neurotransmission and enhanced long-term potentiation in the cortex [19]. Recently, we reported that prenatal VPA exposure-induced macrocephalic phenotype in the rat offspring by inducing excessive neuronal differentiation [20]. These observations prompted us to investigate whether these animals show increased glutamatergic differentiation during early developmental periods along with the mechanism governing the processes.
As a molecular link to explain VPA-mediated hyper-glutamaterigic differentiation, we focused on the role of Pax6 during cortical development. Pax6, a member of the paired box gene family, encodes a transcriptional factor that assigns glutamatergic identity among cortical progenitors [21]. Expression of Pax6 is elevated in neuronal precursor cells at pre-differentiation state [22, 23], which may contribute to the down-regulation of Pax6 by negative auto-feedback mechanism at later stage of development. Subsequently, Tbr2, NeuroD1 and Tbr1 are sequentially expressed during neurogenesis of pyramidal neurons [24], poising Pax6 as an important and foremost transcription factor governing glutamatergic neuronal differentiation as well as stemness of neural stem cells. In a recent study, we also reported that gestational exposure to an environmental factor such as ethanol increases expression of Pax6, Ngn2, and NeuroD1 that induced glutamatergic neuronal differentiation in rodent brains [25].
In this study, we investigated the imbalance between excitatory and inhibitory neuronal differentiation in prenatally VPA-exposed rat brain, focusing on regulation of Pax6. Using siRNA knock-down of Pax6 in neural progenitor cells (NPCs), we demonstrated that transient up-regulation of Pax6 is essential for VPA-induced glutamatergic neuronal differentiation. Finally, we used NMDA receptor antagonists MK-801 and memantine [26, 27] and mGluR5 antagonist MPEP [28–30], which has been investigated in some forms of animal model of ASD, as therapeutic strategies to treat autism-like behaviors of prenatally VPA-exposed rat offspring.
Materials and Methods
Materials
Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12), fetal bovine serum (FBS), penicillin/streptomycin, and 0.25 % trypsin–EDTA were purchased from GibcoBRL (Grand Island, NY). Agarose, PIPES, potassium hydroxide, Tween® 20, 2-mercaptoethanol, trypsin, and VPA were purchased from Sigma (St. Louis, MO). ECLTM Western blotting detection reagents were obtained from Amersham Life Science (Arlington Heights, IL). SuperScriptTM II Reverse Transcriptase, Trizol® reagent, Pax6 siRNA was from Invitrogen (Carlsbad, CA). DNase Ι was purchased from Roche (Mannheim, Germany). Protein G agarose was obtained from Millipore Corporation (Billerica, MA). Taq polymerase and dNTP were form Takara (Shiga, Japan). Protease inhibitor cocktail was from Calbiochem (La Jolla, CA).
Antibodies were purchased from the following companies: anti-β-actin from Sigma (St. Louis, MO), α-CaMKII, Mash1, NeuroD1, PSD-95, reelin, synaptophysin, Tbr2, and vGluT1 antibody from Abcam (Cambrigeshire, England). Nestin, Pax6, Ngn2, GAD, and GFAP antibody were from Millipore (Billerica, MA). Histone H3, acetyl-histone H3, GSK-3β, phospho-GSK-3β, and phospho-histone H3 antibody were from Cell Signaling (Boston, MA). Tuj-1 antibody was from Covance (Princeton, NJ).
Animals
Timed-pregnant Sprague–Dawley rats were purchased from DaeHan BioLink (Daejeon, Korea). Animals were maintained on a 12:12-h circadian cycle with lights on at 0600 hours, at a constant temperature (22 ± 2 °C) and humidity (55 ± 5 %). Animal treatment and maintenance were carried out in accordance with the Principle of Laboratory Animal Care (NIH publication no. 85-23, revised 1985) and were approved by the Animal Care and Use Committee of Konkuk University, Korea (KU12115). All efforts were made to minimize the number of animals as well as their suffering. Behavioral experiments were performed between 1000 and 1600 hours in dedicated test room. The number of animals used in each experiment was provided in supplementary information (Table S1).
Subcutaneous Injection of VPA to Pregnant Rat
The sodium salt of VPA (Sigma, St. Louis, MO) was dissolved in 0.9 % saline for a concentration of 100 mg/ml, pH 7.3. The dosage was adjusted according to the body weight of the pregnant rat on the day of injection. Pregnant rats were received a single subcutaneous injection of 400 mg/kg VPA on gestational day 12 [20, 31]. Control rats were treated with saline.
Culture of Rat Primary Neural Progenitor Cells
The preparation of cortical progenitors from embryonic day 14 (E14) rat embryos was based on a method previously reported [32, 33] with minor modifications. NPC culture was prepared from E14 embryos of Sprague–Dawley rats. Cortices were dissociated into single cells by mechanical trituration, and the cells were incubated with DMEM/F12 supplemented with B27 and 20 ng/ml epidermal growth factor (EGF) in a 5 % CO2 incubator. The culture medium was changed every 2 days, and the cells grew into floating neurospheres. The primary neurosphere was dissociated into single cells with trypsin–EDTA (Invitrogen, Carlsbad, CA, USA), and the cells were re-grown into neurospheres in EGF-containing media. This procedure was repeated, and neurosphere colonies were again dissociated into single cells (1 × 106 cells per a single well of 6-well plate) and plated on poly-l-ornithine-coated plates with DMEM/F12 media supplemented with B27.
Transient Transfection
Transient transfection to NPCs was carried out using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions with minor modifications. Briefly, Pax6 siRNA (Invitrogen, Pax6-RSS352201) and Lipofectamine were mixed in serum- and antibiotics-free OPTI-MEM media. Twenty picomoles of siRNA was transfected on 1 × 106 cells on 6-well plate. The siRNA-lipofectamine complex was added to the 60–70 % confluent NPCs 3 h after NPC subculture. After 3 h-incubation, the medium was changed and further incubated for 24 h at 37 °C to rescue cells. Control siRNA with similar GC contents was purchased from Invitrogen and was used as a control.
Western Blot Analysis
Cells were washed twice with phosphate-buffered saline (PBS) and lysed with 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (120 mM Tris–HCl (pH 6.8), 20 % glycerol, 4 % SDS, 28.8 mM 2-mercaptoethanol, 0.01 % bromophenol blue). Brain tissues were homogenized using RIPA buffer (150 mM sodium chloride, 1 % Triton X-100, 0.5 % sodium deoxycholate, 0.1 % SDS, 50 mM Tris, pH 8.0), and the resulting lysates were diluted with 2× SDS-PAGE sample buffer. Same amounts of protein (50 μg) determined by BCA protein assay was separated by 10 % SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with 1 % polyvinylalcohol in PBS containing 0.2 % Tween 20 for 1 h. The membranes were incubated with primary antibody overnight at 4 °C and then with peroxidase-conjugated secondary antibody (Santa Cruz, CA) for 2 h at room temperature. Dilution factor and catalog number of each primary antibodies were described in supplementary information (Table S2). Specific bands were detected using the ECL system (Amersham, Buckinghamshire, UK) and exposed to Bio-Rad electrophoresis image analyzer (Bio-Rad, Hemel Hempstead, UK). β-Actin was used as loading control, and Western blot band intensity was normalized with β-actin immunoreactivity.
RT-PCR
Total RNA was isolated from NPCs or cortical tissues using Trizol reagent (Invitrogen, Carlsbad, CA), and 1 μg of total RNA was converted to cDNA using Superscript II reverse transcriptase (Invitrogen), according to the manufacturer’s instructions. Specific DNA bands were amplified by PCR. The amplified DNA products were resolved by 1.0 % agarose gel electrophoresis and visualized by staining with ethidium bromide and exposed to Bio-Rad electrophoresis image analyzer (Bio-Rad). All primers were purchased from Invitrogen. The primers used in this analysis are for Pax6, Ngn2, Mash1, NeuroD1, GAPDH, PSD-95, and GAD1.
Pax6, 5′ AAGCAAAATAGCCCAGTATAAACG (forward)
5′ TAATGGGTCCTCTCAAACTCTTTC (reverse)
Ngn2, 5′ AAGAGGTAGGAGAGCTACTTGTGC (forward)
5′ CAGTGTTTTCAGAATGATGAGAGG (reverse)
Mash1, 5′ GTTTCTCCCCTCTTCTTTTTCTTC (forward)
5′ CACCTTGCTCATCTTCTTGTTG (reverse)
NeuroD1, 5′ TTAATGCCATCTTTCACGATTAGA (forward)
5′ ATTCGTGGTACACATTTTCCTTTT (reverse)
GAPDH, 5′ TCCCTCAAGATTGTCAGCAA (forward)
5′ AGATCCACAACGGATACATT (reverse)
PSD-95, 5′ TATGTAACGAAGATCATCGAAGGA (forward)
5′ GAGAATACGAGGTTGTGATGTCTG (reverse)
GAD1, 5′ TTCTTACTGGAGGTGGTTGACATA (forward)
5′ TGATCTCTCGCATCTTCTTAAGTG (reverse)
Isolation of Post-synaptic Density Fraction
Isolation of post-synaptic density (PSD) fraction was performed according to the previous report [34] with several modifications. Briefly, brain tissues of Sprague–Dawley rat were homogenized in ice-cold sucrose/HEPES (ISH) buffer. Lysates were centrifuged, and the supernatants were sedimented to isolate crude synaptosomal fraction. After centrifugation, pellet (crude synaptosomal fraction) was re-suspended in 2 ml of ISH buffer. The suspension was added to 2 ml of 40 %, 2 ml of 35 %, 2 ml of 30 %, and 2 ml of 25 % sucrose gradient and was centrifuged with SW 41 Ti swinging bucket rotor (Beckman Instruments, Palo Alto, CA) at 25,000 rpm for 2 h in Beckman Optima XL-100K preparative ultracentrifuge. Purified synaptosomal fraction was enriched in the border between 35 and 40 % sucrose solution. Isolated fraction was re-suspended in 0.5 % Triton X-100 and was incubated for 15 min. The suspension was added to 2 ml of 65 %, 2 ml of 55 %, 2 ml of 45 %, and 2 ml of 35 % sucrose gradient and was centrifuged at 40,000 rpm for 2 h. PSD fraction was enriched in the border between 55 and 65 % sucrose solution. Isolated PSD fraction was re-suspended in 0.5 % Triton X-100 and was centrifuged at 40,000 rpm for 20 min. Pellets were used for Western blot experiment.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed according to the reported method [35] with minor modifications. Briefly, 43 μl of 37 % formaldehyde was added to 1.6 ml of overlaying medium of NPC culture, and incubated for 15 min at room temperature. After incubation, 225 μl of 1 M glycine was added and incubated for 5 min. Cells was scraped and collected by centrifugation (2,000 g for 5 min at 4 °C), then washed twice with cold PBS. Collected cells were lysed with IP buffer (150 mM sodium chloride, 50 mM Tris–HCl pH 7.5, 5 mM EDTA, 0.5 % IGEPAL CA-630, 1.0 % Triton X-100) on ice. Pellet was resuspended with IP buffer by pipetting and washed by centrifugation (12,000 g for 1 min at 4 °C). To shear the chromatin, 1 mL of the washed and resuspended pellet was sonicated on ice. After centrifugation (12,000 g for 10 min at 4 °C), supernatants were used for immunoprecipitation. Primary antibody (1 μg) was added to 1 ml of supernatant, and the samples were incubated for 12 hrs at 4 °C on a rotating platform. IgG was used as a control antibody. After incubation, mixture of 20 μl of IP buffer and 20 μl of Protein G Agarose (Millipore) was added to the sample, and incubated for 45 min at 4 °C on a rotating platform. After incubation, samples were washed five times by centrifugation (2,000 g for 3 min at 4 °C), and the supernatants were removed. 100 μl of 10 % Chelex 100 was added to the washed beads for DNA isolation, and the samples were boiled for 10 min at 90 °C. After centrifugation (12,000 g for 1 min at 4 °C), 80 μl of supernatant was transferred to new tube, and 120 μl of DDW was added to beads. After centrifugation (12,000 g for 1 min at 4 °C), 120 μl of supernatant was collected and added to the previous supernatant. Isolated DNA was used for PCR reaction. The primers used in this analysis are:
Pax6, 5′ AGGACCTCGTAGAGATGATGAAAC (forward)
5′ AAAAGAGTTGCTCGTGAGAGTTTT (reverse)
NeuroD1, 5′ GGTCTCTCTGAAAGGACAGTCAAT (forward)
5′ ATAGGGACAACTGACTCCATGAAT (reverse)
Mash1, 5′ TTTTCCAAGTTCTCAAGAGACTCC (forward)
5′ GGTTTTAAAAGAGGAAAGGGAAAA (reverse)
Immunocytochemistry
Cultured rat primary NPCs on cover glass (Fisher Scientific, PA) were washed and fixed with 4 % paraformaldehyde at 4 °C for 2 h. The cells were treated with 0.3 % Triton X-100 for 15 min at room temperature and blocked for 30 min with blocking buffer (1 % BSA, 5 % FBS in PBS) at room temperature. The cells were incubated overnight at 4 °C with primary antibodies and washed with washing buffer (0.1 % BSA, 0.5 % FBS in PBS). Secondary antibodies conjugated with TMRE (anti-mouse) or FITC (anti-rabbit) were diluted in blocking buffer and incubated for 2 h at room temperature. After washed three times with washing buffer, the cover glass were mounted in Vectashield (Vector Laboratories, Burlingame, CA) and viewed with a confocal microscope (TCS-SP, Leica, Heidelberg, Germany). Where appropriate, specific antibody-labeled cells were counted and quantified by a researcher blind to the experiments. For nuclear proteins including Pax6, AcH3, Ngn2, and NeuroD1, labeled cells with DAPI counter-staining were counted and quantified. For synaptic proteins including PSD-95 and synaptophysin, labeled puncta with Tuj-1 staining were analyzed using Image J (National Institutes of Health) with a colocalization plug-in downloaded from the program’s website (http://rsb.info.nih.gov/ij/plugins/colocalization.html). Following thresholding, points of colocalization were defined as regions greater than 4 pixels in size where the intensity ratio of the two channels was greater than 50 % [36].
Immunohistochemistry
Brains of embryos and postnatal pups were fixed with PBS containing 4 % paraformaldehyde and sectioned with a cryostat (CM 3050, Leica Instruments). For embryonic experiments, each of single E14 embryo was randomly selected from four control and VPA-exposed pregnant mother rats, respectively (n = 4). For postnatal experiments, each of single P28 rat offspring was randomly selected from four control and VPA-exposed litters, respectively (n = 4). Selected rats were sacrificed and perfused with ice-cold 4 % PFA in PBS (pH 7.4) for 20 min. For each animals, at least five sections were immunostained for examination. Sections (12 μm thickness) were treated with 0.1 % Triton X-100 for 30 min at room temperature and blocked for 60 min with blocking buffer (1 % BSA) at room temperature. Then sections were incubated overnight at 4 °C with primary antibodies and washed with PBS plus 0.2 % Tween 20 (PBST). Secondary antibodies were diluted in PBST and incubated for 2 h at room temperature. After rigorous washing with PBST, the sections were mounted in Vectashield (Vector laboratories, Burlingame, CA) and examined using epifluorescence microscope equipped with appropriate filters. Images were captured with digital acquisition systems (Nikon DS-U1 or DEI-750, Optronics, Goletta, GA). For quantification, labeled cells were counted and quantified by a researcher blind to experiments.
Open-Field Locomotor Activity
The observation apparatus consisted for five plastic boxes (42 × 42 cm) with a field bordered by 42 cm high sidewalls. Rats were moved into test boxes 5 min before recording for habituation. The total distance moved and the duration of movement were monitored for 10 min [37, 38] using CCD camera-assisted motion tracking apparatus and software (EthoVision 3.1, Noldus information Technology, the Netherlands).
Social Interaction Test
The social interaction was adapted from Crawley [39] and performed with several modifications using 4-week-old rats as we reported previously [31]. The test took place in an environment unknown to the rat being tested in the form of a cage with three communicating compartments. At the beginning of sociability test, test rat was placed in the empty central compartment. In the left compartment (stranger 1 side), a conspecific stranger rat unexposed to rats being tested was placed under small wire cage. In the right compartment (empty side), only a wire cage was placed. After 5 min habituation period, sociability test was performed for 10 min by placing test rats in the central compartment. Moving on social preference test, another stranger rat (stranger 2) was added in the right compartment inside wire cage. The test rat was again placed in the empty central compartment, and social preference test was conducted for 10 min directly after the termination of the sociability test. Sociability index and social preference index was calculated as described [31]. The trace of rat movements during experiments was automatically recorded using Ethovision software. The stranger rats were selected from control rats of same gender as test rats, and rats used as a stranger 1 or stranger 2 were not applied in social interaction tests.
Measurement of Electroshock Seizure Threshold
Electroshock seizure threshold was measured with minor modifications with 4 weeks old rats, as we reported previously [38]. Briefly, seizure was evoked by a constant current stimulator through ear clip for 1 s in 2-mA interval individually depending on whether the previous animal exhibits seizure or not. The seizure was defined as overt hind limb extension. To determine the electroshock seizure threshold, convulsive current 50 (CC50), which elicits convulsion in 50 % of animals, was determined by a “staircase” procedure [40], which was designed to minimize the number of animals required for the CC50 determination, and calculated by the Litchfield–Wilcoxon II method [41].
Statistical Analysis
Data were expressed as mean ± standard error of mean (SEM) and analyzed for statistical significance using one-way analysis of variance (ANOVA) followed by Newman–Keuls test as a post-hoc test. Two-way ANOVA was used to identify the effect of VPA exposure or Pax6 siRNA transfection, or interaction between the two factors. In the behavioral analysis, two-way ANOVA was used to identify the effect of VPA exposure or drug treatment, or interaction between the two factors. If significant effects were found in any one of the factors, post-hoc comparisons were conducted using Bonferroni’s post-tests. Differences were considered statistically significant when the P value was less than 0.05 (p < 0.05). All statistical analyses were conducted using PASW Statistics (18.0; SPSS Inc, Chicago, IL, USA).
Results
VPA Exposure Leads to Sequential Expression of Transcription Factors Pax6, Ngn2, and NeuroD1
Pregnant rats were injected with VPA at embryonic day 12 (E12) as we reported previously [20, 31], which did not produce gross health problem such as weight loss in mother rats. Expression of Pax6 protein in frontal cortex was transiently increased at E14 and decreased below control level at E18 by prenatal VPA exposure in vivo (Fig. 1a). At E16 and E18, protein expression of Ngn2 and Tbr2 was elevated as compared with control. Subsequently, NeuroD1 expression was also increased in postnatal days in the cortex of VPA-exposed offspring. Mash1, a transcription factor which leads to GABAergic neuronal differentiation, was not significantly changed by VPA. The expression patterns of mRNAs were similar with those of protein expression in vivo (Fig. 1a). Immunohistochemical images also showed that VPA increased intensity of Pax6 immunostaining in the ventricular and subventricular zone of E14 brain (Fig. 1c). Sequential increased expression of Pax6, Ngn2, and NeuroD1 was also observed in cultured NPCs, treated with 0.5 mM VPA in vitro. Treatment of VPA in differentiating NPCs increased Pax6 expression as compared with control at DIV 1 and 3 followed by Ngn2 and NeuroD1 expression (Fig. 1b). In the immunocytochemical studies, in vitro VPA exposure induced Pax6 expression on DIV 1 (Fig. 1d) and also increased the expression of Ngn2 and NeuroD1 thereafter (Fig. 1g).

VPA exposure induced sequential expression of glutamatergic transcription factors. a Prenatal VPA exposure regulated expression of transcription factors including Pax6, Ngn2, and NeuroD1, which showed sequential temporal peak expression profiles along the course of development. Double band was observed for Ngn2 and NeuroD1. b VPA exposure in vitro induced sequential expression of Pax6, Ngn2, and NeuroD1 in cultured rat primary NPCs. Quantitative graphs were expressed as percentage increment of VPA-exposed groups compared with each control groups. c Immunohistochemical localization of Pax6 and nestin in vivo. Scale bar represents 100 μm. d Co-localization of Pax6 and AcH3 in NPCs. Cultured NPCs were treated with 0.5 mM VPA for 1 day. Scale bar represents 100 μm. e ChIP analysis was conducted as described in materials and methods. VPA enhanced the acetylation of histone H3 bound to Pax6 gene and detached HDAC1 protein from Pax6 gene. VPA also induced HDAC1 protein dissociation from genes encoding NeuroD1 and Mash1. Interaction between Pax6 protein and genes encoding NeuroD1 and Mash1 was enhanced by VPA exposure. f, g In vitro VPA exposure induces expression of nestin (1 day), Tuj-1, Ngn2, and NeuroD1 (7 day) from cultured NPCs. Scale bar represents 100 μm. All data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control (n = 4)
Role of HDAC Inhibition on VPA-Induced Pax6 Expression
Using VPA, a histone deacetylase inhibitor (HDACi), we examined whether the interaction between HDAC1 protein and Pax6 gene is changed in cultured NPCs (Fig. 1e). Protein binding to Pax6 promoter region was assessed by chromatin immunoprecipitation. In VPA-treated groups, dissociation of bound HDAC1 as well as the increased acetylation of histone H3 in the Pax6 gene promoter region was observed (Fig. 1e). In cultured NPCs, cells with increased Pax6 expression by VPA was co-localized with increased acetylated histone H3 (Fig. 1d). These results suggested that VPA enhanced the transcription of Pax6 gene by driving histone acetylation. Other HDAC inhibitors such as trichostatin A (TSA) or sodium butyrate (SB) but not valpromide, a structural analog of VPA devoid of HDACi activity, similarly increased Pax6 expression in cultured NPCs (Fig. 2). Because VPA also possesses GSK-3β inhibitory effect, we also tested GSK-3β inhibitors LiCl and TDZD, which slightly decreased the expression level of Pax6 in our experimental conditions (Fig. 2).

HDACi inhibitor activity of VPA is involved in up-regulation of Pax6 expression in NPCs. Each chemicals were treated on cultured NPCs on day 1 at the following concentrations: VPA (0.5 mM), VPM (valpromide; 0.5 mM), TSA (trichostatin A; 0.2 μM), SB (sodium butyrate; 0.1 mM), LiCl (Lithium Chloride; 0.2 mM), and TDZD-8 (5 μM). TSA and SB were used as HDAC inhibitors. LiCl and TDZD-8 were used as GSK-3β inhibitors. VPM was used as a structural analog of VPA, which is devoid of HDACi activity. All data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control. (n = 3)
VPA Exposure Induces Glutamatergic Neuronal Differentiation from NPCs
Next, we investigated the differentiation of glutamatergic and GABAergic neuron in the cortex of prenatally VPA-exposed rats. Glutamatergic neuronal proteins including PSD-95, α-CaMKII, and vGluT1 were increased in VPA-exposed group during all the postnatal days examined (Fig. 3a), consistent with our previous report showing increased glutamatergic synaptic proteins in the cortex of 4-week-old rats prenatally exposed to VPA [42]. However, GAD, which is a marker protein of GABAergic neuron, was reduced by VPA exposure. Reelin, released from GABAergic neuron, was also decreased in VPA-exposed group. The expression of mRNAs encoding PSD-95 and GAD showed similar tendency (Fig. 3a). Treatment with VPA in cultured NPCs in vitro also increased expression of PSD-95 and α-CaMKII whereas decreased the expression of GAD (Fig. 3b). Excessive glutamatergic neuronal differentiation by VPA was also demonstrated using immunohistochemistry (Fig. 3c) and immunocytochemistry (Fig. 3d). In prefrontal cortical region of VPA-exposed rat offspring, expression of PSD-95 and vGluT1 was increased, whereas expression of GAD was decreased as compared with saline-treated rat offspring (Fig. 3c). Immunochemical staining showed increased synaptic localization of PSD-95 and synaptophysin in neurons differentiated from NPCs exposed to VPA in vitro (Fig. 3d). To determine whether the increased glutamatergic proteins localized at functional synapse, we biochemically isolated PSD fraction from cortical lysates obtained at postnatal day 28 and performed Western blot. We found that the level of PSD-95 and α-CaMKII in the PSD fraction was significantly increased by VPA exposure (Fig. 3e). In addition, synaptophysin, which exists in pre-synaptic area, was enriched in the synaptosomal fraction by VPA exposure. These results suggest that VPA-induces functional localization of PSD-95 and α-CaMKII in postsynaptic density.

VPA exposure increased glutamatergic neuronal differentiation. Prenatal VPA exposure increases glutamatergic neuronal maturation both in vivo (a) and in vitro (b) as evidenced by the expression of marker proteins. Quantitative graphs were expressed as percentage increment of VPA-exposed groups compared with each control groups. c Prenatal VPA exposure increases glutamatergic neuron (stained for PSD95 or vGluT), whereas decreases GABAergic neuron (stained for GAD) in 4-week-old rat brains. Scale bar represents 200 μm. d VPA exposure in vitro enhances synaptic localization of PSD-95 and synaptophysin. Cultured cells were differentiated from NPCs for 12 days and were subjected to immunocytochemistry. Scale bar represents 500 μm. All data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control. (n = 4). e Subcellular localization of pre- and post-synaptic marker proteins. The up-regulated PSD-95 and α-CaMKII were predominantly localized in post-synaptic density fraction by prenatal VPA exposure, and synaptophysin was localized in synaptosomal fraction. Postnatal brains (4 weeks old) were derived from each three litters (n = 3). All data are expressed as mean ± SEM. *p < 0.05, **p < 0.01 vs. control
siRNA Knock-down of Pax6 Leads to Reduced Glutamatergic Neuronal Differentiation
To examine whether Pax6 plays an essential role in VPA-induced glutamatergic neuronal differentiation, we performed transient Pax6 siRNA transfection on NPCs. siRNA containing similar GC contents was used as a control (Invitrogen). Two-way ANOVA was used to identify the effect of VPA or Pax6 siRNA (Supplementary Table. S3). Consistent with our previous reports [20], nestin expression was increased by VPA, which is blocked by Pax6 siRNA (Fig. 4a). These results are consistent with the role of Pax6 in NPCs proliferation as well as neuronal subtype differentiation. Under Pax6 knock-down, protein expression of Ngn2 and NeuroD1 was decreased (Fig. 4a). Knock-down of Pax6 expression inhibited VPA-induced increased expression of glutamatergic neuronal markers including PSD-95, α-CaMKII, and vGluT1 in differentiated NPCs, whereas it induced expression of GAD, which is reduced by VPA (Fig. 4b). In contrast to the changes in neuronal differentiation, glial differentiation was affected neither by VPA [20] nor Pax6 siRNA, determined by GFAP protein levels in Western blot.

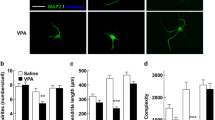
siRNA knock-down of Pax6 expression in NPCs decreases glutamatergic neuronal differentiation. Knock-down of Pax6 expression using siRNA transfection in NPCs was described in methods. a On day 1 after transfection, expression of Pax6 and Nestin was decreased. On day 7, expression of Ngn2 and NeuroD1 was decreased. b On culture day 12, expression of neuronal and synaptic markers was examined. by Western blot. All data are expressed as mean ± SEM (n = 3). **p < 0.01, ***p < 0.001 vs. Control siRNA; ### p < 0.001 vs. VPA; α p < 0.01 vs. Pax6 siRNA control, as revealed by post-hoc Bonferroni’s comparisons following two-way ANOVA. c Immunocytochemical staining of Pax6 and Nestin in VPA (0.5 mM) exposed NPCs after Pax6 knock-down. Scale bar represents 100 μm. d Effects of Pax6 knock-down on proliferation of NPCs. NPCs were treated with 0.5 mM VPA during 1 day. The number of phospho-histone H3 positive NPCs was increased by VPA, which is prevented by Pax6 siRNA transfection. Immunocytochemical localization of PSD-95 (e) or synaptophysin (f) on NPCs differentiated for 12 days in vitro. White arrows indicate localized PSD-95 puncta in Tuj-1-stained neuronal axon. Scale bar represents 20 μm (E; PSD-95) or 500 μm (F; synaptophysin). All data are expressed as mean ± SEM (n = 4). *p < 0.05, ***p < 0.001 vs. control siRNA; ### p < 0.001 vs. VPA, as revealed by post-hoc Bonferroni’s comparisons following two-way ANOVA
In the immunocytochemical experiments, VPA-induced Pax6 expression was inhibited by knock-down of Pax6 (Fig. 4c). Consistent with the decreased nestin expression by Pax6 siRNA transfection, diminished proliferation of NPCs was also observed under the same conditions. pH3-positive NPCs were increased by VPA, which was blocked by Pax6 siRNA transfection (Fig. 4d). VPA-induced glutamatergic neuronal differentiation was also prevented by knock-down of Pax6 that is evidenced by decreased synaptic puncta of PSD-95 (Fig. 4e) and synaptophysin (Fig. 4f).
In this study, the transient increase in Pax6 expression reprograms NPCs toward hyper-glutamatergic differentiation so that NPCs isolated and cultured from E14 brain of fetus previously injected with VPA to their dam at E12 (ex vivo experiments) showed increased transcription factors and glutamatergic markers expression similar to in vivo and in vitro situation (Fig. 5). These results suggest that VPA exposure in critical period, in this case, E12 in rats [31], renders the NPCs into a state opt to dysregulated neuronal differentiation.

Prenatal VPA exposure increases glutamatergic neuronal differentiation ex vivo. NPCs were isolated from VPA-exposed embryonic cortices and differentiated in vitro (ex vivo experiments). a Sequential expression of transcription factors initiated from Pax6 was increased by VPA exposure ex vivo. Subsequently, expression of Ngn2 and NeuroD1 was increased. b Glutamatergic neuronal differentiation was determined by Western blot and RT-PCR. Glutamatergic neuronal proteins including PSD-95, α-CaMKII, and vGluT1 were increased by VPA exposure ex vivo. GABAergic protein GAD was decreased. Pre-synaptic marker protein synaptophysin was also increased by VPA. Quantitative graphs were expressed as percentage increment of VPA-exposed groups compared with each control groups. Ex vivo culture NPCs were derived from ten embryos randomly selected from each litters, and four pregnant mother rats was used for each experimental groups. All data are expressed as mean ± SEM (n = 4). *p < 0.05, ** p < 0.01, ***p < 0.001 vs. control
NMDA Receptor Antagonist MK-801 Reverses ASD-Like Behavioral Symptoms Induced by VPA
Excessive glutamatergic neuronal differentiation in medial prefrontal cortex by prenatal VPA along with up-regulation of NR2A and NR2B expression by VPA [19] may increase glutamatergic neurotransmission. We also reported the up-regulation of PSD-95 and α-CAMKII, which compose NMDA receptor cluster by VPA [42]. In this study, we treated MK-801, an NMDA receptor antagonist, to the prenatally VPA-exposed rat offspring at 4 weeks of age and investigated ASD-related behavioral parameters. MK-801 (0.3 mg/kg, dissolved in saline as 0.03 mg/ml) was intraperitoneally injected 30 min before the tests. Vehicle (saline) was injected as a control. Two-way ANOVA was used to identify the effect of VPA or MK-801 (Table S4).
VPA-exposed rats showed increased locomotor activity as reported previously [42], which was restored by MK-801 treatment. Both moved distance and moved duration in the open-field were increased by VPA exposure and decreased by MK-801 (Fig. 6a). VPA-exposed rats at 4 weeks of age exhibited impaired social interactions [31], and MK-801 treatment restored social impairments (Fig. 6b). In the sociability test, stay duration in a side chamber with a conspecific rat (stranger 1) was decreased by VPA exposure and restored by MK-801. Duration in central compartment was increased by VPA exposure and decreased by MK-801. Duration in empty wire cage side was not changed both by VPA exposure and MK-801. Sociability index (SI) was defined as the ratio between duration in stranger 1 side and duration in empty side [31]. SI was significantly decreased by VPA exposure and restored by MK-801. In the social preference test, stay duration in familiar side (stranger 1 side) was increased by VPA exposure and decreased by MK-801. Duration in central compartment was increased by VPA exposure and decreased by MK-801. Duration in novel side (stranger 2 side) was decreased by VPA exposure and restored by MK-801. Social preference index (SPI) was defined as the ratio between duration in novel side and duration in familiar side [31]. SPI was decreased by VPA exposure and restored by MK-801. We next examined the effects of MK-801 treatment on the electroshock seizure threshold (Fig. 6c). Electroshock seizure thresholds of the VPA-exposed group were significantly lower than those of the control group, suggesting increased excitability of the prenatally VPA-exposed animals ([42], p < 0.001), and treatment of MK-801 restored sensitivity to electroshock (VPA and MK-801-treated group vs. VPA-exposed group, p < 0.001). Memantine, an NMDA receptor antagonist, which was intraperitoneally injected 30 min before the tests as 30 mg/kg, also rescued autism-like behaviors of prenatally VPA-exposed rat offspring (Fig. 7). Memantine also restored hyperactivity in open-field test (Fig. 7a), impaired social interactions (Fig. 7b, Table S5), and sensitivity to electroshock (Fig. 7c) of VPA-exposed rat offspring. In addition, MPEP (30 mg/kg), an mGluR5 antagonist, showed a similar restorative effect on autism-like behaviors which is consistent with previous report [30], although MPEP did not rescue a defect in sociability (Fig. 8b, Table S6).

MK-801 rescues impaired social interactions and sensitivity to electroshock seizure induced by prenatal VPA exposure. Rat offspring were pre-treated with MK-801 30 min before each behavioral analysis as described. Vehicle-treated group was used as a control. a Open-field locomotor activity test was performed as described in materials and methods. b Sociability and social preference test were performed as described in materials and methods. All data are expressed as mean ± SEM (n = 8). *p < 0.05, ** p < 0.01, ***p < 0.001 vs. saline-treated group; # p < 0.05, ## p < 0.01, ### p < 0.001 vs. VPA-treated group; β p < 0.05, β′ p < 0.01 vs. saline and MK-801-treated group, as revealed by post-hoc Bonferroni’s comparisons following two-way ANOVA. c Measurement of electroshock seizure threshold was performed as described in “Materials and Methods.” CC50 of VPA-exposed rats was significantly lower than that of control rats (p < 0.001), which was prevented by MK-801 treatment (p < 0.001). For each experimental group, 16 rat offspring from 8 litters were used for the determination of CC50. Table showed actual value of CC50 with upper and lower confidence limits

Memantine rescues impaired social interactions and sensitivity to electroshock seizure induced by prenatal VPA exposure. Rat offspring were pre-treated with memantine 30 min before each behavioral analysis as described. Memantine (30 mg/kg, dissolved in saline as 3 mg/ml) was intraperitoneally injected 30 min before the tests. Vehicle (saline) was injected as a control. Two-way ANOVA was used to identify the effect of VPA or memantine (Table S5). a Open-field locomotor activity test was performed as described in materials and methods. b Sociability and social preference test were performed as described in “Materials and Methods.” All data are expressed as mean ± SEM (n = 8). *p < 0.05, **p < 0.01, ***p < 0.001 vs. saline-treated group; ## p < 0.01, ### p < 0.001 vs. VPA-treated group; β p < 0.05, β′ p < 0.01, β″ p < 0.001 vs. saline and memantine-treated group; α p < 0.05, α′ p < 0.01, α″ p < 0.001 for saline-treated group vs. saline and memantine-treated group, as revealed by post-hoc Bonferroni’s comparisons following two-way ANOVA. c Measurement of electroshock seizure threshold was performed as described in “Materials and Methods.” CC50 of VPA-exposed rats was significantly lower than that of control rats (p < 0.001), which was prevented by memantine treatment (p < 0.001). For each experimental groups, 16 rat offspring from 8 litters were used for the determination of CC50. Table showed actual value of CC50 with upper and lower confidence limits

MPEP rescues impaired social interactions and sensitivity to electroshock seizure induced by prenatal VPA exposure. Rat offspring were pre-treated with MPEP 30 min before each behavioral analysis as described. MPEP (30 mg/kg, dissolved in saline as 3 mg/ml) was intraperitoneally injected 30 min before the tests. Vehicle (saline) was injected as a control. Two-way ANOVA was used to identify the effect of VPA or MPEP (Table S6). a Open-field locomotor activity test was performed as described in “Materials and Methods.” b Sociability and social preference test were performed as described in “Materials and Methods.” All data are expressed as mean ± SEM (n = 8). *p < 0.05, **p < 0.01, ***p < 0.001 vs. saline-treated group; ### p < 0.001 vs. VPA-treated group; β p < 0.05, β′ p < 0.01, β″ p < 0.001 vs. saline and MPEP-treated group; α p < 0.05, α″ p < 0.001 for saline-treated group vs. saline and MPEP-treated group, as revealed by post-hoc Bonferroni’s comparisons following two-way ANOVA. c Measurement of electroshock seizure threshold was performed as described in materials and methods. CC50 of VPA-exposed rats was significantly lower than that of control rats (p < 0.001), which was prevented by MPEP treatment (p < 0.001). For each experimental group, 16 rat offspring from 8 litters were used for the determination of CC50. Table showed actual value of CC50 with upper and lower confidence limits
Discussion
Excitatory/inhibitory Imbalance in ASD
One of the key pathological features of the human autistic brain and animal models of ASD is the imbalance between excitatory/inhibitory neurotransmission in the brain [9, 43]. Inhibitory architecture disruption [44] and even insufficiency of GABAergic proteins [12, 13] were reported in the autistic patients.
Prenatal VPA exposure model has been widely used for the study of structural and functional alterations in the autistic brain [18, 45, 46]. Although contradictory results exist in terms of the exact nature of aberrant neuronal differentiation induced by VPA, hyperactive glutamatergic activity and microcircuit connectivity may render the brain highly plastic, more autonomous, and difficult to control once activated [19, 46], which may be consistent with the reduced seizure threshold and increased hyperactivity in VPA animal model of ASD [31]. Similarly, one-third of human ASD patients showed increased seizure susceptibility [47].
Here, we provided epigenetic and cellular mechanisms that transient up-regulation of Pax6 induced by VPA exposure at mid-gestation stage is critical for enhanced glutamatergic differentiation. Perturbed social interaction in VPA-exposed offspring was normalized with MK-801 or memantine that blocks glutamatergic neurotransmission, suggesting the possible neurochemical and neuropharmacological regulation of autism-like behaviors. Our study suggests that environmental (epigenetic) factors implicated in ASD may have profound effects on differentiation of specific subsets of neurons (for example, glutamatergic neurons or GABAergic neurons). The altered functional and mechanistic balance of the different subtypes of neurons by epigenetic regulation may determine the manifestation of autistic symptoms in conjunction with the contribution from the genetic predisposition.
Roles of Pax6 in the Developmental Process of Embryonic Brain
Pax6 is a paired box family member and plays an essential role in the brain development. It prevents precocious neuronal differentiation and a depletion of NPCs [48]. Pax6 also instructs glutamatergic neuronal identity among basal progenitor cells/NPCs by inducing Tbr2 and Ngn2 [49–51]. Subsequently, Pax6 becomes down-regulated by negative feedback and NeuroD1 and Tbr1 are induced instead, which leads to glutamatergic neuronal differentiation [23, 52]. Thus, among transcriptional cascade with a sequence of Pax6 → Ngn2 → Tbr2 → NeuroD1 → Tbr1 [24], Pax6 is the major factor that initiates and triggers glutamaterigic neuronal differentiation as well as maintains a pool of NPCs. In this study, VPA-exposed embryonic brains or NPCs triggered transient up-regulation of Pax6, which coincides with dissociation of HDAC1 protein and increased association of acetylated histone at Pax6 promoter. Other HDAC inhibitors also mimicked the effect of VPA [53, 54], indicating that the transient up-regulation of Pax6 is mediated by HDACi activity.
The protein level of Pax6 is critical for the balance between the maintenance of stem cells and neuronal differentiation [55]. Thus, the final outcome of neural differentiation could vary depending on the level and duration of Pax6 expression during embryonic brain development. For instance, when Pax6 expression is down-regulated, neurogenesis could be enhanced at early period, but later became inhibited with reduced cortical thickness and smaller number of neurons [55]. Similarly, pax77 TG mice, which over-express Pax6, showed an increased cell cycle length among apical progenitors, which facilitates neurogenesis [48]. Upon VPA exposure, Pax6 was transiently increased in E14 brain, but later further down-regulated below the control level at E18. Interestingly, VPA injection only at E12 but not in other embryonic stages induced autism-like behaviors in rat offspring [31]. Therefore, the transient up-regulation of Pax6 by a brief exposure to VPA at mid-gestation period appears to be an ideal time window to efficiently increase NPCs. It is of interest that over-expression of Pax6 also induced increased layer 5 pyramidal neurons at E12.5 mice brain [55], consistent with the increased glutamatergic differentiation observed in this study. Together, we propose that level of Pax6 is critical in the regulation of neuronal differentiation and subtype specification in VPA animal model of ASD.
Effect of VPA on Pax6 Expression
In this study, we observed the induction of Pax6 from differentiating NPCs by VPA exposure both in vivo and in vitro. Few studies investigated the effects of VPA on Pax6 expression in differentiating NPCs. In Xenopus laevis, high concentration of VPA (5 and 10 mM) induced lethality, somite abnormalities, and eye malformations and decreased expression of Pax6 in eye [56]. In chicken embryo studies, 5 ∼ 20 mg/ml (34.7 ∼ 138.7 mM) of VPA was used [57], and application of 15 mg/ml of VPA produced gross abnormalities, multiple eye abnormalities with decreased Pax6 expression. In our experimental conditions, prenatally VPA (400 mg/kg)-exposed rat offspring did not show abnormalities including birth rate, body weight [31], and rota-rod function [42]. We also used 1 mM concentration of VPA in vitro, which did not produce immediate cellular toxicity in cultured NPCs. These results suggested that high doses of VPA may produce decreased Pax6 expression during early embryonic development, which might be related to the teratogenic activity of VPA, and the final outcome of in utero exposure of VPA might be differentially modulated by the dosage of VPA exposure as well as critical time windows of exposure [31].
Recently, Balmer and his colleagues reported the effect of VPA on human-derived embryonic stem cell line (hESC) [58]. Balmer reported that VPA (0.25 ∼ 1.0 mM) increased mRNA expression of OCT4 and NANOG, which are used as stem cell markers, whereas VPA decreased mRNA expression of PAX6, which is a marker for neuroepithelial precursor (NEP) cells. These results suggested that VPA delayed normal differentiation from hESC into NEP, and these altered differentiation may underlie abnormal neurodevelopment such as neural tube defect [58], which again supports the idea that the effects of prenatal exposure to VPA on neural differentiation including the regulation of Pax6 expression might be modulated differentially in different development periods.
Relationship Between ASD and Pax6
Pax6 is a pivotal gene in the WAGR (Wilms’ tumor, aniridia, genitourinary and mental retardation) syndrome, caused by deletions of the 11p14-p12 chromosome region including Pax6 and WT1 gene. Recently, deletion of Pax6 gene has been implicated in ASD [59, 60]. Pax6 heterozygous mutant rats (rSey2/+ rats) showed impaired prepulse inhibition [61] and aggressive social behaviors [62] as a part of ASD phenotypes. Seemingly contradictory to our observation that transient up-regulation of Pax6 increased glutamatergic neuronal differentiation, it is notable that many of critical genes relevant to psychiatric disorders such as schizophrenia and ASD often yields a similar behavioral phenotype in gain or loss of function studies. Due to synaptic imbalance, “too much” or “too little” synaptic activity may lead to the same dysfunctional output [63]. Zoghbi and Bear recently proposed that optimal synaptic function occurs within a limited dynamic range, and the pathophysiology at both ends of this range can cause autistic behavior [63]. Therefore, the results obtained from genetic backgrounds (human and Pax6 TG mice) and epigenetic backgrounds (current study) suggest that perturbed Pax6 signal during embryonic development may explain abnormal synaptic development and behavioral characteristics in ASD. Next question would be what is the nature of the genetic and epigenetic interactions in regard to the role of Pax6 in neuronal subtype differentiation and synaptic maturation.
Therapeutic Effects of Glutamate Receptor Antagonists on ASD Animal Models
In this study, blocking glutamatergic neurotransmission with MK-801 or memantine reversed the perturbed social behaviors in VPA-exposed rat offspring at week 4 (Figs. 6b and 7b). In prenatally poly(I:C) exposure model of ASD [64, 65], MK-801 treatment restored aberrant locomotor activity [66]. Moreover, recent studies reported memantine as a possible adjunctive therapy either alone [26] or with risperidone in ASD children [27]. On structural level, memantine restored dendritic synapse formation of cultured cerebellar granule cells from Fmr1 KO mice [67]. Considering MPEP produced weaker behavioral reversal compared with MK801, NMDA pathway may play most prominent role in the regulation of ASD-like behaviors in VPA ASD model, although the contribution of AMPA signaling pathway should be investigated further in the future.
Although we described the therapeutic effects of glutamate receptor antagonists on behavioral phenotypes of VPA animal model of ASD, early exposure to NMDA antagonist itself may produce detrimental developmental effects such as neuro-degeneration and schizophrenia-like symptoms [68, 69], which mandate the careful investigation of the optimal therapeutic time windows and dosage in developing subjects. In addition, systemic administration of MK-801 has been frequently used to make a hypo-functional state of NMDA system, which is believed to underlie schizophrenia-spectrum disorders including psychosis [70, 71]. Modeling schizophrenia, MK-801 induced deficits in hippocampal LTP and synaptic plasticity [72] and decreased neurite outgrowth [73], which were opposite to VPA animal model of ASD, which showed hyper-plasticity [46] and increased synaptic connections [42, 46]. These results may suggest that at least some symptoms of schizophrenia-spectrum disorder and ASD lie on the opposite end of a neurobehavioral spectrum and use of NMDA antagonists may shift the behavioral phenotypes on the continuum depending on the several factors, which may include dosage, timing of administration, treatment period and the systemic milieu where NMDA antagonists are administered, etc. Therefore, care should be given to select the optimal condition of MK-801 treatment to control ASD phenotypes.
In the prenatally VPA-exposed rat models, Rinaldi et al. reported the increase of CaMKII, NR2A and NR2B in the somatosensory cortex at week 2 [19] as well as hyper-plasticity in the medial prefrontal cortex at week 2 [46]. We also found the increased expression of glutamate receptor subtypes including NMDA receptors (NR1 and NR2B), AMPA receptors (GluR1 and GluR2), and mGluR5 (mGluR5, Homer1, Homer1a and Shank1-3) at week 4 (manuscript in preparation). Considering previous reports and recent results together, glutamate receptor antagonists may provide therapeutic effect through modulation of glutamatergic nervous system in prenatally VPA-exposed rat offspring.
In this study, we only used an acute administration (intraperitoneal injection 30 min before the tests) of NMDA receptor antagonists and an mGluR5 antagonist. Chronic treatment with low dose of drug is an important approach to rescue autism-like behavioral symptoms, which may alleviate the possible side effects of drug treatment. Further studies might be required to determine an optimal condition for chronic administrations such as starting time point to treat, drug dosage, and administration route.
Altogether, these results suggested that VPA induces overt expression of Pax6 during brain developmental process, which induces glutamatergic neuronal differentiation from NPCs. The dysregulated neuronal differentiation was mediated by sequential expression of transcription factors such as Pax6, Ngn2, and NeuroD1. Pax6 expression plays essential roles in this process as evidenced by blockade of VPA-induced dysregulation of glutamatergic neuronal expression when Pax6 is down-regulated. Finally and most importantly, the blockade of the overt glutamatergic transmission in VPA group using MK801 or memantine restored the altered social behaviors and seizure susceptibility suggesting the essential role of glutamatergic transmission in autism-like behaviors of prenatally VPA-exposed rat model of ASD.
Until now, there is no cure for core symptoms of ASD, such as impairments in social interaction and communication, although many studies suggest beneficial effects of risperidone on repetitive behavior of ASD patients. Extensive investigation is focused on several pharmacological targets such as mGluR5 antagonist MPEP [28–30] and CTEP [74], GABAB agonist arbaclofen [75], ERK inhibitor lovastatin [76], mToR inhibitor rapamycin [77], D2 receptor antagonist risperidone [78], and endocannabinoid receptor antagonists [79] (for a review, see [80]) However, most of these therapeutic strategies only restored non-core symptoms such as anxiety, seizure, and hyperactivity, which requires extensive efforts in this field. Defining the pathophysiological mechanisms of subsets of ASD such as presented here may provide rational approach to develop the therapeutic options against ASD. Obviously, investigating the general applicability of glutamate antagonists in other models of ASD, at least showing increased excitatory/inhibitory balance, would be one of the first directions to further develop a plausible therapeutic option using these groups of molecules.
References
World Health Organization. (2004) International statistical classification of diseases and related health problems. 10th revision, 2nd edition. World Health Organization, Geneva
Lewis G (1996) DSM-IV. Diagnostic and statistical manual of mental disorders, 4th edn. AmerPsychiatAssoc Psychol Med 26(3):651–652
Woodhouse W, Bailey A, Rutter M, Bolton P, Baird G, Le Couteur A (1996) Head circumference in autism and other pervasive developmental disorders. J Child Psychol Psychiatry 37(6):665–671
Fidler DJ, Bailey JN, Smalley SL (2000) Macrocephaly in autism and other pervasive developmental disorders. Dev Med Child Neurol 42(11):737–740
Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML (2001) Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord 31(6):537–543
Bejjani A, O’Neill J, Kim JA, Frew AJ, Yee VW, Ly R, Kitchen C, Salamon N, McCracken JT, Toga AW, Alger JR, Levitt JG (2012) Elevated glutamatergic compounds in pregenual anterior cingulate in pediatric autism spectrum disorder demonstrated by 1H MRS and 1H MRSI. PLoS One 7(7):e38786. doi:10.1371/journal.pone.0038786
Casanova MF, Buxhoeveden DP, Switala AE, Roy E (2002) Neuronal density and architecture (Gray Level Index) in the brains of autistic patients. J Child Neurol 17(7):515–521
Hutsler JJ, Zhang H (2010) Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res 1309:83–94. doi:10.1016/j.brainres.2009.09.120
Rubenstein JL, Merzenich MM (2003) Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2(5):255–267
Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB (2005) Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A 102(35):12560–12565. doi:10.1073/pnas.0506071102
Samaco RC, Hogart A, LaSalle JM (2005) Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet 14(4):483–492. doi:10.1093/hmg/ddi045
Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD (2009) GABA(A) receptor downregulation in brains of subjects with autism. J Autism Dev Disord 39(2):223–230. doi:10.1007/s10803-008-0646-7
Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR (2002) Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry 52(8):805–810
Casanova MF, Buxhoeveden D, Gomez J (2003) Disruption in the inhibitory architecture of the cell minicolumn: implications for autisim. Neuroscientist 9(6):496–507. doi:10.1177/1073858403253552
Arnon J, Shechtman S, Ornoy A (2000) The use of psychiatric drugs in pregnancy and lactation. Isr J Psychiatry Relat Sci 37(3):205–222
Moore SJ, Turnpenny P, Quinn A, Glover S, Lloyd DJ, Montgomery T, Dean JC (2000) A clinical study of 57 children with fetal anticonvulsant syndromes. J Med Genet 37(7):489–497
Rodier PM, Ingram JL, Tisdale B, Nelson S, Romano J (1996) Embryological origin for autism: developmental anomalies of the cranial nerve motor nuclei. J Comp Neurol 370 (2):247–261. doi:10.1002/(SICI)1096-9861(19960624)370:2<247::AID-CNE8>3.0.CO;2–2
Schneider T, Przewlocki R (2005) Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology 30(1):80–89. doi:10.1038/sj.npp.1300518.1300518
Rinaldi T, Kulangara K, Antoniello K, Markram H (2007) Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc Natl Acad Sci U S A 104(33):13501–13506. doi:10.1073/pnas.0704391104
Go HS, Kim KC, Choi CS, Jeon SJ, Kwon KJ, Han SH, Lee J, Cheong JH, Ryu JH, Kim CH, Ko KH, Shin CY (2012) Prenatal exposure to valproic acid increases the neural progenitor cell pool and induces macrocephaly in rat brain via a mechanism involving the GSK-3beta/beta-catenin pathway. Neuropharmacology 63(6):1028–1041. doi:10.1016/j.neuropharm.2012.07.028
Kroll TT, O’Leary DD (2005) Ventralized dorsal telencephalic progenitors in Pax6 mutant mice generate GABA interneurons of a lateral ganglionic eminence fate. Proc Natl Acad Sci U S A 102(20):7374–7379. doi:10.1073/pnas.0500819102
Schuurmans C, Armant O, Nieto M, Stenman JM, Britz O, Klenin N, Brown C, Langevin LM, Seibt J, Tang H, Cunningham JM, Dyck R, Walsh C, Campbell K, Polleux F, Guillemot F (2004) Sequential phases of cortical specification involve neurogenin-dependent and -independent pathways. EMBO J 23(14):2892–2902. doi:10.1038/sj.emboj.76002787600278
Bel-Vialar S, Medevielle F, Pituello F (2007) The on/off of Pax6 controls the tempo of neuronal differentiation in the developing spinal cord. Dev Biol 305(2):659–673. doi:10.1016/j.ydbio.2007.02.012
Hevner RF, Hodge RD, Daza RA, Englund C (2006) Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res 55(3):223–233. doi:10.1016/j.neures.2006.03.004
Kim KC, Go HS, Bak HR, Choi CS, Choi I, Kim P, Han SH, Han SM, Shin CY, Ko KH (2010) Prenatal exposure of ethanol induces increased glutamatergic neuronal differentiation of neural progenitor cells. J Biomed Sci 17:85. doi:10.1186/1423-0127-17-85
Chez MG, Burton Q, Dowling T, Chang M, Khanna P, Kramer C (2007) Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J Child Neurol 22(5):574–579. doi:10.1177/0883073807302611
Ghaleiha A, Asadabadi M, Mohammadi MR, Shahei M, Tabrizi M, Hajiaghaee R, Hassanzadeh E, Akhondzadeh S (2012) Memantine as adjunctive treatment to risperidone in children with autistic disorder: a randomized, double-blind, placebo-controlled trial. Int J Neuropsychopharmacol:1–7. doi: 10.1017/S1461145712000880
Westmark CJ, Westmark PR, O’Riordan KJ, Ray BC, Hervey CM, Salamat MS, Abozeid SH, Stein KM, Stodola LA, Tranfaglia M, Burger C, Berry-Kravis EM, Malter JS (2011) Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS One 6(10):e26549. doi:10.1371/journal.pone.0026549
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP (2005) Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49(7):1053–1066. doi:10.1016/j.neuropharm.2005.06.004
Mehta MV, Gandal MJ, Siegel SJ (2011) mGluR5-antagonist mediated reversal of elevated stereotyped, repetitive behaviors in the VPA model of autism. PLoS One 6(10):e26077. doi:10.1371/journal.pone.0026077
Kim KC, Kim P, Go HS, Choi CS, Yang SI, Cheong JH, Shin CY, Ko KH (2011) The critical period of valproate exposure to induce autistic symptoms in Sprague–Dawley rats. Toxicol Lett 201(2):137–142. doi:10.1016/j.toxlet.2010.12.018
Benoit BO, Savarese T, Joly M, Engstrom CM, Pang L, Reilly J, Recht LD, Ross AH, Quesenberry PJ (2001) Neurotrophin channeling of neural progenitor cell differentiation. J Neurobiol 46(4):265–280. doi:10.1002/1097-4695(200103)46:4<265::AID-NEU1007>3.0.CO;2-B
Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying QL, Cattaneo E, Smith A (2005) Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 3(9):e283. doi:10.1371/journal.pbio.0030283
Carlin RK, Grab DJ, Cohen RS, Siekevitz P (1980) Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol 86(3):831–845
Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1(1):179–185. doi:10.1038/nprot.2006.27
O’Connor TP, Cockburn K, Wang W, Tapia L, Currie E, Bamji SX (2009) Semaphorin 5B mediates synapse elimination in hippocampal neurons. Neural Dev 4:18. doi:10.1186/1749-8104-4-18
Noldus LP, Spink AJ, Tegelenbosch RA (2001) EthoVision: a versatile video tracking system for automation of behavioral experiments. Behavior research methods, instruments, & computers. J Psychon Soc 33(3):398–414, Inc
Park HG, Yoon SY, Choi JY, Lee GS, Choi JH, Shin CY, Son KH, Lee YS, Kim WK, Ryu JH, Ko KH, Cheong JH (2007) Anticonvulsant effect of wogonin isolated from Scutellaria baicalensis. Eur J Pharmacol 574(2–3):112–119. doi:10.1016/j.ejphar.2007.07.011
Crawley JN (2004) Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment Retard Dev Disabil Res Rev 10(4):248–258. doi:10.1002/mrdd.20039
Browning RA, Wang C, Lanker ML, Jobe PC (1990) Electroshock- and pentylenetetrazol-induced seizures in genetically epilepsy-prone rats (GEPRs): differences in threshold and pattern. Epilepsy Res 6(1):1–11
Litchfield JT Jr, Wilcoxon F (1949) A simplified method of evaluating dose-effect experiments. J Pharmacol Exp Ther 96(2):99–113
Kim KC, Kim P, Go HS, Choi CS, Park JH, Kim HJ, Jeon SJ, Dela Pena IC, Han SH, Cheong JH, Ryu JH, Shin CY (2013) Male-specific alteration in excitatory postsynaptic development and social interaction in prenatal valproic acid exposure model of autism spectrum disorder. J Neurochem. doi:10.1111/jnc.12147
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K (2011) Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477(7363):171–178. doi:10.1038/nature10360
Casanova MF, Buxhoeveden DP, Brown C (2002) Clinical and macroscopic correlates of minicolumnar pathology in autism. J Child Neurol 17(9):692–695
Dufour-Rainfray D, Vourc’h P, Le Guisquet AM, Garreau L, Ternant D, Bodard S, Jaumain E, Gulhan Z, Belzung C, Andres CR, Chalon S, Guilloteau D (2010) Behavior and serotonergic disorders in rats exposed prenatally to valproate: a model for autism. Neurosci Lett 470(1):55–59. doi:10.1016/j.neulet.2009.12.054
Rinaldi T, Perrodin C, Markram H (2008) Hyper-connectivity and hyper-plasticity in the medial prefrontal cortex in the valproic acid animal model of autism. Front Neural Circ 2:4. doi:10.3389/neuro.04.004.2008
Spence SJ, Schneider MT (2009) The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr Res 65(6):599–606. doi:10.1203/01.pdr.0000352115.41382.65
Georgala PA, Manuel M, Price DJ (2011) The generation of superficial cortical layers is regulated by levels of the transcription factor Pax6. Cereb Cortex 21(1):81–94. doi:10.1093/cercor/bhq061
Quinn JC, Molinek M, Martynoga BS, Zaki PA, Faedo A, Bulfone A, Hevner RF, West JD, Price DJ (2007) Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev Biol 302(1):50–65. doi:10.1016/j.ydbio.2006.08.035
Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P (2001) Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105(1):43–55
Scardigli R, Baumer N, Gruss P, Guillemot F, Le Roux I (2003) Direct and concentration-dependent regulation of the proneural gene Neurogenin2 by Pax6. Development 130(14):3269–3281
Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25(1):247–251. doi:10.1523/JNEUROSCI.2899-04.2005
Kim SN, Kim NH, Lee W, Seo DW, Kim YK (2009) Histone deacetylase inhibitor induction of P-glycoprotein transcription requires both histone deacetylase 1 dissociation and recruitment of CAAT/enhancer binding protein beta and pCAF to the promoter region. Mol Cancer Res 7(5):735–744. doi:10.1158/1541-7786.MCR-08-0296
Smith KT, Martin-Brown SA, Florens L, Washburn MP, Workman JL (2010) Deacetylase inhibitors dissociate the histone-targeting ING2 subunit from the Sin3 complex. Chem Biol 17(1):65–74. doi:10.1016/j.chembiol.2009.12.010
Sansom SN, Griffiths DS, Faedo A, Kleinjan DJ, Ruan Y, Smith J, van Heyningen V, Rubenstein JL, Livesey FJ (2009) The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet 5(6):e1000511. doi:10.1371/journal.pgen.1000511
Pennati R, Groppelli S, de Bernardi F, Sotgia C (2001) Action of valproic acid on Xenopus laevis development: teratogenic effects on eyes. Teratog Carcinog Mutagen 21(2):121–133
Whitsel AI, Johnson CB, Forehand CJ (2002) An in ovo chicken model to study the systemic and localized teratogenic effects of valproic acid. Teratology 66(4):153–163. doi:10.1002/tera.10093
Balmer NV, Weng MK, Zimmer B, Ivanova VN, Chambers SM, Nikolaeva E, Jagtap S, Sachinidis A, Hescheler J, Waldmann T, Leist M (2012) Epigenetic changes and disturbed neural development in a human embryonic stem cell-based model relating to the fetal valproate syndrome. Hum Mol Genet 21(18):4104–4114. doi:10.1093/hmg/dds239
Davis LK, Meyer KJ, Rudd DS, Librant AL, Epping EA, Sheffield VC, Wassink TH (2008) Pax6 3′ deletion results in aniridia, autism and mental retardation. Hum Genet 123(4):371–378. doi:10.1007/s00439-008-0484-x
Maekawa M, Iwayama Y, Nakamura K, Sato M, Toyota T, Ohnishi T, Yamada K, Miyachi T, Tsujii M, Hattori E, Maekawa N, Osumi N, Mori N, Yoshikawa T (2009) A novel missense mutation (Leu46Val) of PAX6 found in an autistic patient. Neurosci Lett 462(3):267–271. doi:10.1016/j.neulet.2009.07.021
Maekawa M, Takashima N, Matsumata M, Ikegami S, Kontani M, Hara Y, Kawashima H, Owada Y, Kiso Y, Yoshikawa T, Inokuchi K, Osumi N (2009) Arachidonic acid drives postnatal neurogenesis and elicits a beneficial effect on prepulse inhibition, a biological trait of psychiatric illnesses. PLoS One 4(4):e5085. doi:10.1371/journal.pone.0005085
Umeda T, Takashima N, Nakagawa R, Maekawa M, Ikegami S, Yoshikawa T, Kobayashi K, Okanoya K, Inokuchi K, Osumi N (2010) Evaluation of Pax6 mutant rat as a model for autism. PLoS One 5(12):e15500. doi:10.1371/journal.pone.0015500
Zoghbi HY, Bear MF (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4 (3). doi: 10.1101/cshperspect.a009886.a009886
Patterson PH (2009) Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res 204(2):313–321. doi:10.1016/j.bbr.2008.12.016
Meyer U, Feldon J, Dammann O (2011) Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res 69(5 Pt 2):26R–33R. doi:10.1203/PDR.0b013e318212c196
Howland JG, Cazakoff BN, Zhang Y (2012) Altered object-in-place recognition memory, prepulse inhibition, and locomotor activity in the offspring of rats exposed to a viral mimetic during pregnancy. Neuroscience 201:184–198. doi:10.1016/j.neuroscience.2011.11.011.S0306-4522(11)01278-4
Wei H, Dobkin C, Sheikh AM, Malik M, Brown WT, Li X (2012) The therapeutic effect of memantine through the stimulation of synapse formation and dendritic spine maturation in autism and fragile X syndrome. PLoS One 7(5):e36981. doi:10.1371/journal.pone.0036981
Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283(5398):70–74
du Bois TM, Huang XF (2007) Early brain development disruption from NMDA receptor hypofunction: relevance to schizophrenia. Brain Res Rev 53(2):260–270. doi:10.1016/j.brainresrev.2006.09.001
Coyle JT, Tsai G, Goff D (2003) Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann N Y Acad Sci 1003:318–327
Vrajova M, Stastny F, Horacek J, Lochman J, Sery O, Pekova S, Klaschka J, Hoschl C (2010) Expression of the hippocampal NMDA receptor GluN1 subunit and its splicing isoforms in schizophrenia: postmortem study. Neurochem Res 35(7):994–1002. doi:10.1007/s11064-010-0145-z
Wiescholleck V, Manahan-Vaughan D (2013) Persistent deficits in hippocampal synaptic plasticity accompany losses of hippocampus-dependent memory in a rodent model of psychosis. Frontiers in integrative neuroscience 7:12. doi: 10.3389/fnint.2013.00012
Ringler SL, Aye J, Byrne E, Anderson M, Turner CP (2008) Effects of disrupting calcium homeostasis on neuronal maturation: early inhibition and later recovery. Cell Mol Neurobiol 28(3):389–409. doi:10.1007/s10571-007-9255-9
Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L (2012) Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74(1):49–56. doi:10.1016/j.neuron.2012.03.009
Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR, Brynczka C, Rush R, Thomas A, Paylor R, Warren ST, Vanderklish PW, Kind PC, Carpenter RL, Bear MF, Healy AM (2012) Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABA(B) receptors with arbaclofen. Sci Transl Med 4(152):152ra128. doi:10.1126/scitranslmed.3004218
Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF (2013) Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77(2):243–250. doi:10.1016/j.neuron.2012.01.034
Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ (2008) Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med 14(8):843–848. doi:10.1038/nm1788
Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH (2011) Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147(1):235–246. doi:10.1016/j.cell.2011.08.040
Busquets-Garcia A, Gomis-Gonzalez M, Guegan T, Agustin-Pavon C, Pastor A, Mato S, Perez-Samartin A, Matute C, de la Torre R, Dierssen M, Maldonado R, Ozaita A (2013) Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 19(5):603–607. doi:10.1038/nm.3127
Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T (2013) Progress toward treatments for synaptic defects in autism. Nat Med 19(6):685–694. doi:10.1038/nm.3193
Acknowledgments
This work was supported by Mid-career Researcher Program (2011–0014258) and the framework of international cooperation program (2012K2A1A2032549) through the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST).
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 54 kb)
Rights and permissions
About this article
Cite this article
Kim, K.C., Lee, DK., Go, H.S. et al. Pax6-Dependent Cortical Glutamatergic Neuronal Differentiation Regulates Autism-Like Behavior in Prenatally Valproic Acid-Exposed Rat Offspring. Mol Neurobiol 49, 512–528 (2014). https://doi.org/10.1007/s12035-013-8535-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8535-2