
Abstract
Parkinson’s disease (PD) is a neurodegenerative disease that is characterized by specific pathological features such as the death of dopaminergic neurons and formation of Lewy bodies. Another marker of stress that is found in patients and PD animal models is an increase in inflammation. Different cells are found to be allocated to this process, such as resident brain cells, specifically microglia and astrocytes, and peripheral infiltrated cells such as T cells and monocytes. Those inflammatory cells may secrete different stress factors such as IL-1β and reactive oxygen species that might exacerbate neuronal death. However, there is also evidence that those cells may also secrete neuroprotective factors that may support stressed cells. This review will aim to address the role of those inflammatory cells in the progression of PD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
Inflammation is a natural process within tissues in which immune cells, such as macrophages and dendritic cells in the periphery and microglia in the brain, mount a response against external infection or injury to the tissue [1]. This response is characterized by vasodilation (expansion of blood vessels), increased capillary permeability, and migration of phagocytes into the tissue [2]. Such an inflammatory response can be either acute , i.e., short and powerful around the time of insult, or chronic, which lasts over a long period of time. As neurons appear to be more vulnerable to inflammatory responses compared to other tissues, chronic inflammation in the brain could be harmful to the tissue [3].
Inflammation might play a double-edged sword in neurodegenerative diseases . It might be linked to neuronal stress and death signals through cytokines such as interleukin 1 beta (IL-1β) [4], tumor necrosis factor alpha (TNFα) [5], and interferon gamma (IFNγ) [6], increased secretion of reactive oxygen species (ROS) [7], and by activation of the complement system [8]. However, inflammation may also be linked to increased secretion of antiapoptotic cytokines such as IL-10 [9] and IL-4 or neurotrophic factors such as brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT3), or insulin-like growth factor 1 (IGF-1) [10–12]. It has been suggested that within the central nervous system, astrocytes might play a role as sensors and modulate the inflammation in the microenvironment [13]. In many neurodegenerative diseases, evidence for such inflammatory processes is seen both at the site of the degeneration, as well as in the peripheral system [14–16].
4.1.1 Inflammation and PD
Parkinson’s disease (PD) is characterized by progressive degeneration of neuromelanin-containing dopaminergic neurons throughout the substantia nigra pars compacta (SNc) within the basal nuclei [17]. This degeneration is accompanied by the formation of intracellular inclusion bodies, termed Lewy bodies , within neurons [18]. These inclusion bodies are comprised of several proteins, the most studied of which are ubiquitin and α-synuclein [19].
It has been reported that the inflammatory processes may play a role in pathogenesis of the disease. In PD patients’ brains , there is evidence of chronic inflammation, seen in the elevated levels of proinflammatory cytokines such as IL-1β and IL-6 in the cerebrospinal fluid (CSF) and TNFα in the substantia nigra of patients [20]. This reaction is attributed, at least in part, to the activation of microglia and infiltrated monocyte cells, marked by [11C](R)-PK11195 reactivity in the midbrain of patients [21] and an increase in EBM11 (CD68)-positive cells in the substantia nigra, in postmortem [22]. In addition to proinflammatory cytokines , activated microglia and monocytes also exhibit a higher expression of inducible nitric-oxide synthase (iNOS) and cyclooxygenases 1 and 2 (COX1 and COX2) which can exert cytotoxic effects through oxidative stress [23]. Since these findings were observed in patients at different disease stages and disease durations, it is possible that the increase in the presence of microglia is linked to chronic inflammation, rather than an ad hoc activation.
In contrast to microglia , the role of astrocytes in chronic inflammation in PD is not clear. In some PD patients’ brains, there is no morphological evidence for reactive astrocytes in the substantia nigra, compared to control samples [24]. In comparison, others have found astrocytic activation in the substantia nigra accompanied with increased expression of intercellular adhesion molecule-1 (ICAM-1) [25].
The complement system also appears to play a role in the pathogenesis of PD, as early-stage complement proteins such as iC3b, as well as late-stage complement proteins such as C9, surround Lewy bodies in PD patients’ brains [26]. Of note, iC3b was also observed around melanized neurons in the SN, i.e., dopaminergic neurons still containing neuromelanin. This suggests that complement activation could play a role in triggering the death of dopaminergic neurons in the SN [26].
Immune cells in the periphery also show abnormalities in PD patients: PD patients exhibit lower levels of circulating CD4+ T-helper 1 lymphocytes and lower levels of B cells, concomitant with increased levels of natural killer (NK) cells [27]. Similarly, PD patients show lower levels of induced secretion of IL-2 [28]. In line with these findings, PD patients’ monocyte-derived macrophages also showed impairment in inducible expression of CD200R, the ligand for the T-cell-expressed CD200 protein [29]. As the CD200-CD200R signaling is thought to exert an inhibitory effect on myeloid-lineage cells, such as macrophages and microglia [30], impairment in this signaling could lead to excessive activation of proinflammatory functions among these cells. Finally, PD patients also exhibit increased levels of infiltrating CD4+ and CD8+ T cells within the substantia nigra, suggesting that peripheral lymphocytes play a role in degenerative processes in PD [31].
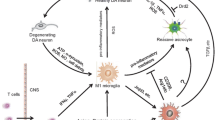
Together, these findings point to an abnormality in lymphocyte cells and their interaction with macrophages in the peripheral system of PD patients, which has possible implications on processes within the central nervous system (see Fig. 4.1).

The role of inflammation in triggering and exacerbating dopaminergic neurons death in PD. Inflammation can cause neuronal death through two potential mechanisms: (1) As a result from stress signals of apoptotic dopaminergic neurons. Toxin-based models can induce mitochondrial dysfunction and inhibition of dopamine synthesis in dopaminergic neurons, leading to secretion of stress signals that induce neurotoxic inflammatory responses. (2) As an initiation stage by which inflammatory activation of brain-resident cells and infiltrating cells may trigger dopaminergic neuronal stress. Inflammation-based models (lipopolysaccharide, or LPS) induce inflammatory activation of brain-resident cells and infiltrating cells, which in turn induce toxicity in dopaminergic neurons by Inflammatory signals such as nitric oxide (NO), ROS, IFNγ, TNFα, IL-1β, and complement proteins. Abbreviations: 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
4.2 Parkinson Genes and Inflammation
Several genes have been linked to familial forms of PD, which can present either autosomal-dominant or autosomal-recessive inheritance, and often cause early onset of the disease [32]. Furthermore, recent studies regarding PD biomarkers in patients reveal an association between PD cases and variation in the human leukocyte antigen (HLA) gene , which is linked to regulation of immune functions, suggesting that variations in immune functions affect the risk of developing PD [33].
It has been reported that some of the genes which cause familial forms of PD can also directly modulate a proinflammatory response or their expressed proteins might trigger inflammation.
α-Synuclein : Alpha-synuclein is a protein predominantly expressed in the central nervous system and found in presynaptic terminals of neurons [34], as well as in astrocytes, microglia, and oligodendrocytes [35]. Mutations in the α-synuclein gene are linked to dominant inheritance of PD [36], although duplication mutations of the gene are also implicated in the disease [32].
Several α-synuclein mouse models for PD exist, with either overexpression of wild-type α-synuclein or expression of mutant forms of α-synuclein, through different promoters [37]. While such models exhibit only some dopaminergic degeneration in older ages, they exhibit chronic microglial activation in the SN, accompanied by increased expression of inflammatory markers such as ICAM-1, IL-1β, IL-6, TNF-α, and iNOS [38, 39].
Interestingly, α-synuclein has been shown to be a negative regulator of cellular degradation processes such as autophagy in T cells [40]. As autophagy has been shown to degrade aggregated α-synuclein in these cells, this could represent a positive-feedback mechanism, in which α-synuclein accumulation prevents its own degradation, furthering the accumulation and cellular burden [40]. Likewise, α-synuclein has been shown to play a role in mediating B-cell-dependent immune responses, as α-synuclein −/− animals have lower levels of B cells, and reduced production of IgG antibodies in response to immune challenges [41].
PINK-1: Phosphotensin-induced kinase 1 (PINK1) is a serine/threonine kinase which is located both in the cytosol and in the mitochondrial membrane [42, 43] and is important for various mitochondrial functions [44]. As PD-related mutations are thought to act through a loss of function mechanism [44], most of the research on PINK1 pathology is carried through knockout or knockdown models.
PINK1 −/− mice do not exhibit major abnormalities compared to WT mice, except for some mitochondrial impairments [37]. In contrast, organotypic cortical slices of PINK1 −/− mice show higher expression of proinflammatory genes such as TNF-α, IL-6, and IL-1β [45], and these mice produce larger amounts of IL-1β, IL-12, and IL-10 in response to a peripheral injection of lipopolysaccharides (LPS) [46].
DJ-1: DJ-1 is almost ubiquitously expressed in human tissues [47] and is located both in the cells’ cytosols and around mitochondria [48]. DJ-1 acts as an oxidative-stress response protein which protects neurons from various oxidative-stress conditions [49]. Several mutations observed in human PD patients have been found to generate unstable proteins, generating an “effective knockout” or knockdown of DJ-1 [50], suggesting that DJ-1-related pathology is due to DJ-1 loss of function.
DJ-1 −/− mice do not show prominent Parkinsonian symptoms or marked neurodegeneration [37]. In vitro models of DJ-1 deficiency, however, reveal impairments in immune responses to various challenges: primary astrocytes derived from DJ-1 −/− mice exhibited stronger proinflammatory and neurotoxic effects in response to an LPS challenge, compared to astrocytes from WT animal [51], and the same phenomena are observed in microglia [52]. Of note, glial cells cultured from DJ-1 KO mice exhibit increased phosphorylation of the inflammatory signaling molecule signal transducers and activators of transcription 1 (STAT1) , resulting in enhanced inflammatory responses following INF-γ stimulation [53]. These findings suggest that DJ-1 exerts anti-inflammatory effects and that loss of DJ-1 function can lead to exacerbation of inflammatory processes [52].
4.3 Inflammation and Toxin Animal Model of PD
Dopaminergic death in PD has been found to be accompanied by an increase in inflammatory markers. However, it is hard to define whether one is a trigger for the other. Indeed, experiments in animal models of PD suggest two potential roles of inflammation in PD: (1) inflammation exacerbates neuronal death following stress signals from neurons, and (2) inflammation triggers dopaminergic neuronal stress and death (see Fig. 4.1).
4.3.1 Toxin-Mediated Animal Model
Parkinson’s disease research employs several toxins which cause dopaminergic degeneration and induce PD-like symptoms in animal models, enabling diverse in vitro and in vivo experimental models.
6-OHDA animal model : One of the most studied toxins is 6-hydroxydopamine (6-OHDA) , a hydroxylated derivative of dopamine. 6-OHDA enters catecholaminergic neurons via natural reuptake mechanisms [54], where it exerts its toxic effects by two main processes: the first involves the generation of reactive oxygen species through activation of NADPH oxidase [55], and the second involves impairment in mitochondrial activity through inhibition of mitochondrial complex I and complex IV [56].
In addition to its direct effects on dopaminergic neurons, research in recent years has shown that the deleterious effects of 6-OHDA involve significant inflammatory activation, both in vitro and in vivo.
Administration of 6-OHDA to neuronal cells induces rapid translocation of the inflammatory nuclear factor-kB (NF-kB) to the nucleus, where it binds to the DNA [57]. Moreover, striatal injection of 6-OHDA causes astrogliosis, marked by increased numbers of astrocytes and increased expression of GFAP within the astrocytes [58].
Microglia cells show a robust reaction following striatal 6-OHDA injection, around dopaminergic neurons, and these cells also have been shown to participate in phagocytosis of dopaminergic neurons [59]. Microglia-induced degeneration of dopaminergic neurons is attenuated in a mice harboring knock-in of DNAX adaptor protein 12 (DAP-12) [59]. Conversely, enhancement of microglia activity, by blocking the inhibitory CD200-CD200R signaling in microglia, results in increased neurodegeneration after 6-OHDA administration, accompanied by increased secretion of the proinflammatory cytokines IL-6 and TNFα [60].
Intrastriatal injection of 6-OHDA also has been shown to induce disruption of the blood–brain barrier (BBB), leading to SN blood leakage, which co-localizes with degeneration of dopaminergic neurons [61]. Moreover, increased levels of MHC-II-reactive microglia were also observed in the same areas [61]. These findings suggest that 6-OHDA administration induces a secondary reaction by glial cells that contributes to the progressive neurodegeneration.
MTPT animal model : MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine ) is another frequently used substance in PD research. It is rapidly converted into MPP+ (1-methyl-4-phenylpyridinium) by the MAO-B enzyme within astrocytes [62], and in this ionized form, it is readily taken by dopaminergic neurons’ dopamine transporter (DAT) [63]. Once inside the cells, MPP+ inhibits the generation of dopamine by nitration of tyrosine hydroxylase, directly contributing to dopamine depletion in the brain [64]. Moreover, MPP+ inhibits the activity of the mitochondrial complex I and causes a depletion of ATP levels and the generation of ROS [65].
Aside from its direct effects on dopaminergic neurons, MPTP exerts marked inflammatory responses in the brain: shortly after the exposure to MPTP, there is activation of both microglia and astrocytes in the SN, accompanied with infiltration of CD4+ and CD8+ T cells [66]. Moreover, several days after the administration of the toxin, increased expression of MHC-II and ICAM-1 on microglia cells is observed in the mouse brain [66], and increased expression of astrocytic ICAM-1 and microglia leukocyte function antigen 1 (LFA-1) is observed in the monkey brain [25].
Mice deficient of iNOS show a similar glial response to MPTP, compared to WT mice, but the neurodegeneration is almost completely abolished in the iNOS −/− mice. Interestingly, dopamine levels are still decreased in this phenotype [67]. These findings suggest an active role for inflammatory responses in the MPTP model that occur simultaneously with the direct effects of this toxin on dopaminergic neurons.
Rotenone animal model : Rotenone is an inhibitor of the mitochondrial complex I, and it exerts toxic effects through disruption of cell respiration and ATP synthesis, as well as enhanced production of ROS by the mitochondria [68].
Rotenone induces dopaminergic neurodegeneration in the SN after chronic administration, accompanied by microglial activation [69]. Interestingly, however, in vitro experiments on primary microglia cultures show that microglia do not exhibit inflammatory responses to rotenone [70], suggesting that the activation of microglia cells is not a direct effect of rotenone, but rather a secondary effect, perhaps by signals from damaged neurons.
Paraquat animal model : Paraquat is an inhibitor of mitochondrial complex I, which causes a reduction in cell respiration and increased free radical formation [71].
When administered to lab animals, prolonged exposure to paraquat induces dopaminergic degeneration, simultaneously with oxidative damage [72]. The oxidative damage appears to be a causative neurotoxic factor, as transgenic mice which are more resistant to oxidative damage show no susceptibility to this toxin [72]. Finally, similarly to rotenone, paraquat does not elicit inflammatory reactions in microglia cells [70].
4.3.2 Inflammatory Mediated Animal Model
As inflammation appears to play a role in the pathology of PD, some experimental models utilize inflammatory agents to initiate dopaminergic neuronal pathological processes. These models commonly involve the use of lipopolysaccharide (LPS) , a macromolecule found on the outer membrane of gram-negative bacteria, which elicits inflammatory responses in mammalian cells through Toll-like receptor 4 (TLR4) signaling [73].
A single intraperitoneal injection of LPS can elicit rapid proinflammatory responses in microglia at the substantia nigra and cause a significant reduction in the numbers of dopaminergic neurons 7–10 months after the injection [74]. Furthermore, a single injection of LPS into the substantia nigra can cause selective dopaminergic degeneration and dopamine depletion, starting 4 days after the injection and persisting for 12 months. This effect is preceded by microgliosis, but not astrocyte proliferation in the injection site, starting only 2 days after the injection [75]. These findings suggest that the neurodegeneration is not a primary result of the inflammatory toxin, but rather a secondary process to microglia activation and neurotoxic inflammatory responses.
Of note, several reports have suggested that inflammation and α-synuclein aggregation create a positive-feedback loop, whereas oxidative stress causes α-synuclein aggregation, and α-synuclein aggregation causes inflammation and oxidative stress [20]. Since α-synuclein deposition in PD patients appears to begin in the vagus nerve and in the anterior olfactory nucleus [76], it is possible that inflammatory processes in the gastric epithelium and olfactory epithelium, which are exposed to the external environment and external pathogens, are a key factor in the pathogenesis of PD [20]. Together, various lines of evidence link inflammatory processes to neurotoxic responses from microglia, aggregation of α-synuclein, and the possible spreading of α-synuclein between neighboring cells.
4.4 Anti-inflammatory Treatment and PD
The link between inflammation and PD pathology suggests a potential for an anti-inflammatory approach for treating PD. Indeed, this approach is supported by results in a PD animal model, where attenuation of inflammatory processes in MPTP experimental models appears to mitigate the deleterious effects of this toxin: Inhibition of the signaling of peptide angiotensin II, an inducer of inflammatory responses, reduces microglia activation and mitigates MPTP-induced neurotoxicity [77]. Similarly, administration of the anti-inflammatory drug dexamethasone reduces MPTP-induced upregulation of MHC-II and ICAM-1, reduces microglia reactivity, and significantly abolishes T-cell infiltration into the SN; these processes coincide with reduced neurotoxicity [66]. These findings suggest an active role for inflammatory responses in the MPTP model, which occurs simultaneously with direct effects of this toxin on dopaminergic neurons and significantly contributes to the neurotoxic effects of this substance.
Epidemiological studies among regular users of aspirin and nonsteroidal anti-inflammatory drugs (NSAIDs) suggest a reduction in the risk for developing PD [78]. Nevertheless, a clinical trial with aspirin and other NAISDs, except ibuprofen [79], did not show significant effects in reducing the risk for PD [80]. Of note, the positive results with ibuprofen might be mediated through its effect on other genes such as APOE4 [81] and not through its effect on inflammation. Interestingly, some therapeutic approaches in animal models suggest induction of immune responses specifically against α-synuclein may also result in neuroprotection [82].
4.5 Conclusion
Inflammation plays a major role in processes in which the body preserves homeostasis and protects itself from various insults. Those insults may be linked to abnormalities that prompt the development of neurodegenerative diseases such as PD. Further understanding the role of each inflammatory biomarker in the etiology and the progression of PD may elucidate the role of inflammation in the disease and may lead to the rational development of immunomodulation approaches in PD.
References
Dantzer, R., O’Connor, J.C., Freund, G.G., Johnson, R.W., Kelley, K.W.: From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56 (2008). doi:10.1038/nrn2297
Kindt, T.J., Goldsby, R.A., Osborne, B.A., Kuby, J.: Kuby immunology. W.H. Freeman, New York (2007)
Gao, H.M., Liu, B., Zhang, W., Hong, J.S.: Novel anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol. Sci. 24, 395–401 (2003). doi:10.1016/S0165-6147(03)00176-7
Tanaka, S., et al.: Activation of microglia induces symptoms of Parkinson’s disease in wild-type, but not in IL-1 knockout mice. J. Neuroinflammation 10, 143 (2013). doi:10.1186/1742-2094-10-143
McCoy, M.K., Ruhn, K.A., Blesch, A., Tansey, M.G.: TNF: a key neuroinflammatory mediator of neurotoxicity and neurodegeneration in models of Parkinson’s disease. Adv. Exp. Med. Biol. 691, 539–540 (2011). doi:10.1007/978-1-4419-6612-4_56
Barcia, C., et al.: IFN-gamma signaling, with the synergistic contribution of TNF-alpha, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 3, e379 (2012). doi:10.1038/cddis.2012.123
Miller, R.L., James-Kracke, M., Sun, G.Y., Sun, A.Y.: Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem. Res. 34, 55–65 (2009). doi:10.1007/s11064-008-9656-2
McGeer, P.L., McGeer, E.G.: Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat. Disord. 10(Suppl 1), S3–S7 (2004). doi:10.1016/j.parkreldis.2004.01.005
Kwilasz, A.J., Grace, P.M., Serbedzija, P., Maier, S.F., Watkins, L.R.: The therapeutic potential of interleukin-10 in neuroimmune diseases. Neuropharmacology (2014). doi:10.1016/j.neuropharm.2014.10.020
Nagatsu, T., Mogi, M., Ichinose, H., Togari, A.: Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural Transm. Suppl. 60, 277–290 (2000)
Lee, Y.H., Song, G.G.: BDNF 196 G/A and 270 C/T polymorphisms and susceptibility to Parkinson’s disease: a meta-analysis. J. Mot. Behav. 46, 59–66 (2014). doi:10.1080/00222895.2013.862199
Huang, B., et al.: Maternal exposure to bisphenol A may increase the risks of Parkinson’s disease through down-regulation of fetal IGF-1 expression. Med. Hypotheses 82, 245–249 (2014). doi:10.1016/j.mehy.2013.10.023
Segev-Amzaleg, N., Trudler, D., Frenkel, D.: Preconditioning to mild oxidative stress mediates astroglial neuroprotection in an IL-10-dependent manner. Brain Behav. Immun. 30, 176–185 (2013). doi:10.1016/j.bbi.2012.12.016
Akiyama, H., et al.: Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421 (2000)
McGeer, P.L., McGeer, E.G.: Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 26, 459–470 (2002). doi:10.1002/mus.10191
Moller, T.: Neuroinflammation in Huntington’s disease. J. Neural Transm. 117, 1001–1008 (2010). doi:10.1007/s00702-010-0430-7
Damier, P., Hirsch, E.C., Agid, Y., Graybiel, A.M.: The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122(Pt 8), 1437–1448 (1999)
Gibb, W.R., Lees, A.J.: The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 51, 745–752 (1988)
Spillantini, M.G., et al.: Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997). doi:10.1038/42166
Lema Tome, C.M., et al.: Inflammation and alpha-synuclein’s prion-like behavior in Parkinson’s disease—is there a link? Mol. Neurobiol. 47, 561–574 (2013). doi:10.1007/s12035-012-8267-8
Ouchi, Y., et al.: Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 57, 168–175 (2005). doi:10.1002/ana.20338
Banati, R.B., Daniel, S.E., Blunt, S.B.: Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov. Disord. 13, 221–227 (1998). doi:10.1002/mds.870130205
Knott, C., Stern, G., Wilkin, G.P.: Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 16, 724–739 (2000). doi:10.1006/mcne.2000.0914
Mirza, B., Hadberg, H., Thomsen, P., Moos, T.: The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 95, 425–432 (2000)
Miklossy, J., et al.: Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 197, 275–283 (2006). doi:10.1016/j.expneurol.2005.10.034
Loeffler, D.A., Camp, D.M., Conant, S.B.: Complement activation in the Parkinson’s disease substantia nigra: an immunocytochemical study. J. Neuroinflammation 3, 29 (2006). doi:10.1186/1742-2094-3-29
Niwa, F., Kuriyama, N., Nakagawa, M., Imanishi, J.: Effects of peripheral lymphocyte subpopulations and the clinical correlation with Parkinson’s disease. Geriatr. Gerontol. Int. 12, 102–107 (2012). doi:10.1111/j.1447-0594.2011.00740.x
Kluter, H., Vieregge, P., Stolze, H., Kirchner, H.: Defective production of interleukin-2 in patients with idiopathic Parkinson’s disease. J. Neurol. Sci. 133, 134–139 (1995)
Luo, X.G., et al.: Altered regulation of CD200 receptor in monocyte-derived macrophages from individuals with Parkinson’s disease. Neurochem. Res. 35, 540–547 (2010). doi:10.1007/s11064-009-0094-6
Hoek, R.M., et al.: Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290, 1768–1771 (2000)
Brochard, V., et al.: Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Invest. 119, 182–192 (2009). doi:10.1172/JCI36470
Wirdefeldt, K., Adami, H.O., Cole, P., Trichopoulos, D., Mandel, J.: Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur. J. Epidemiol. 26(Suppl 1), S1–S58 (2011). doi:10.1007/s10654-011-9581-6
Hamza, T.H., et al.: Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 42, 781–785 (2010). doi:10.1038/ng.642
Burre, J., et al.: Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 (2010). doi:10.1126/science.1195227
Golovko, M.Y., et al.: The role of alpha-synuclein in brain lipid metabolism: a downstream impact on brain inflammatory response. Mol. Cell. Biochem. 326, 55–66 (2009). doi:10.1007/s11010-008-0008-y
Polymeropoulos, M.H., et al.: Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997)
Dawson, T.M., Ko, H.S., Dawson, V.L.: Genetic animal models of Parkinson’s disease. Neuron 66, 646–661 (2010). doi:10.1016/j.neuron.2010.04.034
Theodore, S., Cao, S., McLean, P.J., Standaert, D.G.: Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 67, 1149–1158 (2008). doi:10.1097/NEN.0b013e31818e5e99
Su, X., Federoff, H.J., Maguire-Zeiss, K.A.: Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox. Res. 16, 238–254 (2009). doi:10.1007/s12640-009-9053-x
Colasanti, T., et al.: Role of alpha-synuclein in autophagy modulation of primary human T lymphocytes. Cell Death Dis. 5, e1265 (2014). doi:10.1038/cddis.2014.211
Xiao, W., Shameli, A., Harding, C.V., Meyerson, H.J., Maitta, R.W.: Late stages of hematopoiesis and B cell lymphopoiesis are regulated by alpha-synuclein, a key player in Parkinson’s disease. Immunobiology 219, 836–844 (2014). doi:10.1016/j.imbio.2014.07.014
Springer, W., Kahle, P.J.: Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 7, 266–278 (2011)
Chu, C.T.: A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum. Mol. Genet. 19, R28–R37 (2010). doi:10.1093/hmg/ddq143
Deas, E., Plun-Favreau, H., Wood, N.W.: PINK1 function in health and disease. EMBO Mol. Med. 1, 152–165 (2009). doi:10.1002/emmm.200900024
Kim, J., et al.: PINK1 deficiency enhances inflammatory cytokine release from acutely prepared brain slices. Exp. Neurobiol. 22, 38–44 (2013). doi:10.5607/en.2013.22.1.38
Akundi, R.S., et al.: Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One 6, e16038 (2011). doi:10.1371/journal.pone.0016038
Nagakubo, D., et al.: DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 231, 509–513 (1997). doi:10.1006/bbrc.1997.6132
Zhang, L., et al.: Mitochondrial localization of the Parkinson’s disease related protein DJ-1: implications for pathogenesis. Hum. Mol. Genet. 14, 2063–2073 (2005). doi:10.1093/hmg/ddi211
Taira, T., et al.: DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5, 213–218 (2004). doi:10.1038/sj.embor.7400074
Blackinton, J., et al.: Effects of DJ-1 mutations and polymorphisms on protein stability and subcellular localization. Brain Res. Mol. Brain Res. 134, 76–83 (2005). doi:10.1016/j.molbrainres.2004.09.004
Waak, J., et al.: Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 23, 2478–2489 (2009). doi:10.1096/fj.08-125153
Trudler, D., Weinreb, O., Mandel, S.A., Youdim, M.B., Frenkel, D.: DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J. Neurochem. 129, 434–447 (2014). doi:10.1111/jnc.12633
Kim, J.H., et al.: DJ-1 facilitates the interaction between STAT1 and its phosphatase, SHP-1, in brain microglia and astrocytes: a novel anti-inflammatory function of DJ-1. Neurobiol. Dis. 60, 1–10 (2013). doi:10.1016/j.nbd.2013.08.007
Luthman, J., Fredriksson, A., Sundstrom, E., Jonsson, G., Archer, T.: Selective lesion of central dopamine or noradrenaline neuron systems in the neonatal rat: motor behavior and monoamine alterations at adult stage. Behav. Brain Res. 33, 267–277 (1989)
Rodriguez-Pallares, J., et al.: Mechanism of 6-hydroxydopamine neurotoxicity: the role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 103, 145–156 (2007). doi:10.1111/j.1471-4159.2007.04699.x
Glinka, Y.Y., Youdim, M.B.: Inhibition of mitochondrial complexes I and IV by 6-hydroxydopamine. Eur. J. Pharmacol. 292, 329–332 (1995)
Levites, Y., Youdim, M.B., Maor, G., Mandel, S.: Attenuation of 6-hydroxydopamine (6-OHDA)-induced nuclear factor-kappaB (NF-kappaB) activation and cell death by tea extracts in neuronal cultures. Biochem. Pharmacol. 63, 21–29 (2002)
Gomide, V.C., Silveira, G.A., Chadi, G.: Transient and widespread astroglial activation in the brain after a striatal 6-OHDA-induced partial lesion of the nigrostriatal system. Int. J. Neurosci. 115, 99–117 (2005)
Virgone-Carlotta, A., et al.: Mapping and kinetics of microglia/neuron cell-to-cell contacts in the 6-OHDA murine model of Parkinson’s disease. Glia 61, 1645–1658 (2013). doi:10.1002/glia.22546
Zhang, S., et al.: CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. J. Neuroinflammation 8, 154 (2011). doi:10.1186/1742-2094-8-154
Carvey, P.M., et al.: 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 22, 1158–1168 (2005). doi:10.1111/j.1460-9568.2005.04281.x
Brooks, W.J., Jarvis, M.F., Wagner, G.C.: Astrocytes as a primary locus for the conversion MPTP into MPP+. J. Neural Transm. 76, 1–12 (1989)
Kitayama, S., Wang, J.B., Uhl, G.R.: Dopamine transporter mutants selectively enhance MPP+ transport. Synapse 15, 58–62 (1993). doi:10.1002/syn.890150107
Ara, J., et al.: Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc. Natl. Acad. Sci. U. S. A. 95, 7659–7663 (1998)
Przedborski, S., Tieu, K., Perier, C., Vila, M.: MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J. Bioenerg. Biomembr. 36, 375–379 (2004). doi:10.1023/B:JOBB.0000041771.66775.d5
Kurkowska-Jastrzebska, I., Wronska, A., Kohutnicka, M., Czlonkowski, A., Czlonkowska, A.: The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp. Neurol. 156, 50–61 (1999). doi:10.1006/exnr.1998.6993
Dehmer, T., Lindenau, J., Haid, S., Dichgans, J., Schulz, J.B.: Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J. Neurochem. 74, 2213–2216 (2000)
Li, N., et al.: Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 278, 8516–8525 (2003). doi:10.1074/jbc.M210432200
Zhou, F., et al.: Iptakalim alleviates rotenone-induced degeneration of dopaminergic neurons through inhibiting microglia-mediated neuroinflammation. Neuropsychopharmacology 32, 2570–2580 (2007). doi:10.1038/sj.npp.1301381
Klintworth, H., Garden, G., Xia, Z.: Rotenone and paraquat do not directly activate microglia or induce inflammatory cytokine release. Neurosci. Lett. 462, 1–5 (2009). doi:10.1016/j.neulet.2009.06.065
Tawara, T., et al.: Effects of paraquat on mitochondrial electron transport system and catecholamine contents in rat brain. Arch. Toxicol. 70, 585–589 (1996)
McCormack, A.L., et al.: Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J. Neurochem. 93, 1030–1037 (2005). doi:10.1111/j.1471-4159.2005.03088.x
Triantafilou, M., Triantafilou, K.: The dynamics of LPS recognition: complex orchestration of multiple receptors. J. Endotoxin Res. 11, 5–11 (2005). doi:10.1179/096805105225006641
Qin, L., et al.: Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462 (2007). doi:10.1002/glia.20467
Herrera, A.J., Castano, A., Venero, J.L., Cano, J., Machado, A.: The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol. Dis. 7, 429–447 (2000). doi:10.1006/nbdi.2000.0289
Braak, H., et al.: Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003)
Joglar, B., et al.: The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J. Neurochem. 109, 656–669 (2009). doi:10.1111/j.1471-4159.2009.05999.x
Wahner, A.D., Bronstein, J.M., Bordelon, Y.M., Ritz, B.: Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 69, 1836–1842 (2007). doi:10.1212/01.wnl.0000279519.99344.ad
Casper, D., Yaparpalvi, U., Rempel, N., Werner, P.: Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci. Lett. 289, 201–204 (2000)
Chen, H., et al.: Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol. 58, 963–967 (2005). doi:10.1002/ana.20682
Sastre, M., Gentleman, S.M.: NSAIDs: how they work and their prospects as therapeutics in Alzheimer’s disease. Front. Aging Neurosci. 2, 20 (2010). doi:10.3389/fnagi.2010.00020
Masliah, E., et al.: Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46, 857–868 (2005). doi:10.1016/j.neuron.2005.05.010
Acknowledgments
This study was supported by a grant from the Israeli Ministry of Science, Technology and Space to D.F.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Nash, Y., Frenkel, D. (2016). The Role of Chronic Inflammation in the Etiology of Parkinson’s Disease. In: Buhlman, L. (eds) Mitochondrial Mechanisms of Degeneration and Repair in Parkinson's Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-42139-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-42139-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42137-7
Online ISBN: 978-3-319-42139-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)