
Abstract
The four mammalian phosphatidylinositol 4-kinases, together with the PI(4,5)P2 depleting 5-phosphatases of the oculocerebrorenal syndrome of Lowe and synaptojanin families, modulate neuronal pools of PI4P lipid and regulate intracellular membrane trafficking in the endocytic and secretory pathways. Dysfunctions in these enzymes have been associated with a broad spectrum of disorders including schizophrenia, bipolar disorder, Lowe syndrome, age-related neurodegeneration, Alzheimer’s disease and Down syndrome. Recent work has shown that reduced expression of individual phosphatidylinositol 4-kinase isozymes is associated with impaired survival of specific neuronal populations within the CNS. Furthermore, alterations to the concentrations of different phosphoinositide lipid species in the brain and, in particular, the ratio of PI4P to PI(4,5)P2 can have deleterious effects on clathrin-dependent membrane trafficking both in the Golgi–endosomal pathway and at the plasma membrane. In this article, we focus on the cell biology, biochemistry and neuronal functions of the phosphatidylinositol 4-kinases and their emerging roles in psychiatric and neurological pathologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
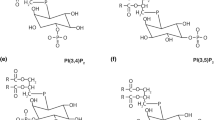
Phosphoinositide lipids regulate key cellular functions including endocytosis, signalling and secretion. In neuronal systems, well-studied areas include the roles of phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) in synaptic vesicle recycling [1, 2] and ion channel regulation (for examples, see [3–8]) and the function of phosphatidylinositol (3,4,5)-trisphosphate (PI(3,4,5)P3) in neuronal cell survival mediated through Akt activation [9]. Both PI(4,5)P2 generation by phosphatidylinositol 4-phosphate 5-kinases (PIPK) and the subsequent generation of PI(3,4,5)P3 by the phosphoinositide 3-kinases are dependent on an initial phosphorylation of phosphatidylinositol (PI) on the D4 position by one of the four mammalian PI 4-kinase enzymes synthesising phosphatidylinositol 4-phosphate (PI4P) [10, 11]. Indeed, there are now numerous examples showing a requirement for PI 4-kinase activity in the maintenance of PI(4,5)P2 pools necessary for ion channel regulation [12–22]. Furthermore, PI4P can function on intracellular membranes in the recruitment of clathrin adaptor proteins such as AP-3 [23], AP-1 [24] and GGAs [25] during Golgi–endosomal trafficking. In this way, PI 4-kinases have the potential to regulate many phosphoinositide-dependent trafficking and signalling functions. Yet despite being the first committed phosphorylation step in the pathway that synthesises PI(4,5)P2 and PI(3,4,5)P3 (Fig. 1), the neurobiological roles of PI4P are only slowly emerging.

PI kinases and phosphatases involved in the metabolism of PI4P in the CNS
In addition to its synthesis by PI 4-kinases, neuronal PI4P levels can potentially be augmented by the action of phosphoinositide 5-phosphatases such as the synaptojanins [2, 26–29] and oculocerebrorenal syndrome of Lowe (OCRL) [30] which dephosphorylate PI(4,5)P2 on the D5 position to produce PI4P. Synaptojanin also contains a sac1 PI4P phosphatase domain [28, 31, 32] and thus has the potential to also decrease PI4P levels in neuronal tissue. Recent evidence suggests that PI4P levels on intracellular membranes, and particularly at the trans-Golgi network and endosomes, have important neuronal functions and that deregulated PI4P generation can have neuropathological consequences. In this article, we evaluate and discuss recent trends in this newly emerging area.
Four mammalian PI 4-kinases have been cloned and characterised; they are the type III PI 4-kinases (PI4KIII) which are inhibited by high micromolar concentrations of the PI 3-kinase inhibitors wortmannin and LY 294002, and the type II PI 4-kinases (PI4KII) which are wortmannin-insensitive but can be inhibited by low micromolar concentrations of adenosine [11]. The two PI4KIIs, PI4KIIα (Fig. 2) and PI4KIIβ are highly homologous ~55-kDa enzymes [33], whereas the larger PI4KIIIs consisting of the 230 kDa PI4KIIIα and 92 kDa PI4KIIIβ isoforms form a distinct protein family with greater homology to the PI 3-kinase family of enzymes in both catalytic and non-catalytic domains [10]. Despite their structural and biochemical differences, all the four mammalian PI 4-kinases synthesise the same PI4P lipid product. However, as the PI 4-kinases are targeted to different subcellular membranes, there are also isoform-dependent differences in the rates of PI4P synthesis at different intracellular locations [34–37] and subsequently highly compartmentalised roles for individual isoforms in PI4P-dependent signalling and trafficking [34, 38].

Diagram illustrating the main structural features of the PI 4-kinases (adapted from [11]). a The PI4KII isoforms share homology in the conserved catalytic core. A conspicuous feature of the enzymes is a conserved cysteine-rich region in the kinase domain that undergoes palmitoylation and mediates membrane targeting to cholesterol-rich membrane microdomains. The N-terminal regions of the PI4KIIs are the least similar: whereas PI4KIIα is proline-rich, amphiphillic in character and contains an AP-3-binding motif 57ERQPLL62, the N-terminus of PI4KIIβ contains a large number of acidic residues. b Domain organisation of mammalian PI4KIIIs. The catalytic domains of PI4KIIIα and PI4KIIIβ display minimal homology to the PI4KIIs but are more homologous with the PI3K family of enzymes especially in the conserved lipid kinase unique (LKU) domain. PI4KIIIα contains an N-terminal proline-rich domain and an internal PH domain. Non-catalytic domains in PI4KIIIβ include a proline-rich sequence and a binding site for NCS-1
All of the PI 4-kinases are expressed in the brain and, with the exception of the PI4KIIβ isoform, detailed immunohistochemical analyses are available on their distributions within the CNS. While there is no single systematic study comparing the distributions of all the PI 4-kinases in the CNS, there are some trends in their neuroanatomical expression patterns that are worth considering.
Mapping of PI4KIII expression by in situ mRNA hybridisation [39], and by light and electron microscopy, [40] has revealed that PI4KIIIα and PI4KIIIβ are localised in neurons throughout the central nervous system. There are some differences in PI4KIII isoform distribution in that PI4KIIIα was more highly expressed in spinal cord and cerebral cortex neurons, whereas PI4KIIIβ was most intensely immunostained in Bergman glia in the molecular layer of the cerebellar cortex. In addition, the hippocampus expresses high levels of PI4KIIIα [40] and an mRNA differential display study has demonstrated that expression of this isozyme is selectively decreased in ischemia-induced delayed neuronal death in the CA1 area of this brain region [41]. Interestingly, relatively intense staining for PI4KIIIβ has also been reported in the CA1 subregion of the hippocampus although there is no evidence that levels of this protein are reduced following ischaemia [42]. Separately, there is evidence for a correlation between learning impairment and depressed G protein-coupled receptor (GPCR)-induced phosphoinositide signalling in the hippocampus of aged animals [43]. Since both PI4KIIIα and PI4KIIIβ are known to supply PI4P substrate during GPCR-activated phospholipase C signalling [34, 38], their hippocampal enrichments may indicate a potential role for these enzymes in muscarinic and metabotropic receptor signalling in this brain region.
Studies on the PI4KIIs have shown that with the exception of white matter, PI4KIIα is expressed at varying levels in neurons and astrocytes throughout the brain with high levels in Purkinje cells and in Bergman glia of the cerebellar molecular layer [44]. A separate detailed immunocytochemical analysis of PI4KIIα expression in the hippocampus reported high expression of the enzyme in pyramidal cells and in the molecular layer of the dentate gyrus, the dentate hilius and the stratum lucidium of the CA3 region [45]. Immunohistochemical results from the Human Protein Atlas [46–48] have shown that PI4KIIβ distribution in the cerebellum mirrors that of both PI4KIIα and PI4KIIIβ with highest expression in the molecular layer. Overall, though, staining for PI4KIIβ is strongest in hippocampal neurons. It is worth noting that mRNA in situ hybridisation studies from the Allen Brain Atlas [49] also support hippocampal and cerebellar localisations for all the PI4K isoforms. Together these findings show that multiple PI 4-kinase isoforms can be expressed at different levels in a single neuroanatomical location (Fig. 3), and also that all of these enzymes can be localised to neurons.

In situ hybridisation images showing the distribution and expression of PI 4-kinase mRNAs in sagittal sections of mouse brain [49]. Note that mRNAs for all PI 4-kinase isoforms are highly expressed in the cerebellum and in the hippocampus. Data from Allen Mouse Brain Atlas, Seattle (WA): Allen Institute for Brain Science ©2009 (http://mouse.brain-map.org)
Subcellular Localisations of Pi 4-Kinases in Neurons of the CNS
Detailed ultrastructural studies on the localisation of the PI4KIIIs in neurons from the ventral horn of the spinal cord demonstrated that PI4KIIIα immunoreactivity was associated mainly with rough endoplasmic reticulum, mitochondrial outer membrane, occasionally on multivesicular bodies and close to synaptic specialisations [40]. PI4KIIIβ was also found associated with the rough endoplasmic reticulum, mitochondria and the Golgi complex [40]. In non-neuronal cells, confocal imaging studies have shown that PI4KIIIα is an endoplasmic reticulum enzyme while PI4KIIIβ is mainly found associated with Golgi membranes [50, 51]. Furthermore, a mitochondrial localisation for either PI4KIII isoform has not been reported in non-neuronal cells. Of the PI4KIIs, detailed neuronal subcellular localisation data is available only for the PI4KIIα isoform where it localises to dendrites, the Golgi and synaptic vesicles [45, 52]. Therefore, similar to non-neuronal cells, each PI 4-kinase isoform exhibits a distinct pattern of membrane localisation in neurons which also indicates that PI4P synthesis is likely to be highly compartmentalised within individual cells of the CNS (Fig. 4).

Intracellular localisations of PI 4-kinase isoforms in neurons. There is no systematic or comprehensive study detailing the subcellular distributions of the four mammalian PI 4-kinases in neurons and no data at all is available for PI4KIIβ. This schematic diagram therefore depicts proposed PI 4-kinase localisations using information derived from separate reports that used different methods and different neuronal cell types
Neuronal Functions and Dysfunctions of the PI 4-Kinases
Early work from the Martin laboratory identified PI 4-kinase activity on secretory vesicles involved in the supply of PI4P substrates to PI4P 5-kinases [53, 54]. In the intervening years, there has been some progress in understanding the functions of different PI 4-kinases in neuronal vesicle trafficking events.
In the absence of isoform-selective PI 4-kinase inhibitors, many early investigations used phenylarsine oxide (PAO) to inhibit PI4P synthesis. PAO is now known to react with vicinal thiol groups on a variety of enzymes to inhibit their catalytic activity [55–57], but nevertheless it was employed, albeit as a nonspecific intervention, to gain some initial insights into neurophysiological functions of the PI 4-kinases. As an example, sensitivity to PAO inhibition was used to infer a role for PI4P in the retrograde axonal transport of neurotrophin-4 [58] and nerve growth factor [59] in both sympathetic and sensory neurons, suggesting that PI 4-kinases may regulate multiple vesicle trafficking processes. It is important to bear in mind that there has been disagreement as to whether PAO is a more potent inhibitor of PI4KIIs or PI4KIIIs, particularly in light of earlier work which inferred a role for PI4KII activity on chromaffin granule membranes in regulated secretion [60]. This controversy has been more recently addressed by a detailed analysis [61] using purified recombinant PI 4-kinases which revealed that PAO was most selective for PI4KIIIα inhibition when added at low 1–5-μM concentrations. At higher PAO concentrations exceeding ~10 μM, there was pronounced inhibition of PI4P generation by three isoforms PI4KIIIα, PI4KIIIβ and PI4KIIα [61]. The recent discoveries of better isoform-selective small molecule inhibitors of the PI4KIIIs [62–64] have the potential to open up new avenues of research into the roles of these enzymes in the regulation of exocytosis and retrograde trafficking.
Studies on synaptic vesicle pools in synaptosomes derived from mature central nerve terminals determined that repetitive synaptic vesicle recycling did not require phosphoinositides, but that PI 4-kinases were required for the transfer of a non-releasable reserve pool of synaptic vesicles to a pool which was readily releasable in response to hypertonic stimulation [65]. Another interesting finding is there may also be cargo-specific requirements for PI 4-kinase activity in stimulated exocytosis, in that noradrenaline release requires phosphoinositide synthesis but glutamate or GABA release do not [66]. Therefore, PI 4-kinases and particularly PI4KIIIs have very specific functions restricted to particular points in the synaptic vesicle cycle, and these roles may be further restricted to particular neurotransmitters.
As regards the regulation of PI 4-kinases on secretory vesicles, PI4KIIIβ represents the best characterised example with insights being gained mainly from PC12 neuroendocrine cells [67, 68] but with some important observations also being derived from studies on non-neuronal cell lines [69–73]. On secretory vesicle membranes, the lipid kinase activity of PI4KIIIβ is stimulated by interaction with neuronal calcium sensor 1 (NCS-1) [67–83]. NCS-1 is a Ca2+-binding EF hand protein which can activate membrane-associated PI4KIIIβ in response to elevated cytosolic Ca2+ concentrations, thereby providing some rationalisation of the relationship between Ca2+-sensitive exocytosis and PI4P synthesis. Additionally, both NCS-1 and PI4KIIIβ are targeted to intracellular membranes via interactions with ADP-ribosylation factor (ARF) proteins [75, 84], and PI4KIIIβ interactions with ARF-1 are important for regulatory exocytosis [70].
While there has been significant progress in understanding the functions of PI4KIIIβ, NCS-1 and ARF-1 in the regulation of stimulated secretion, there is also some evidence that the PI4KIIα isozyme may also function on this pathway. Indeed, PI4KIIα activity is particularly enriched on synaptic [52] and secretory vesicles [85], and it is noteworthy that the enzyme was originally purified and cloned from a secretory vesicle membrane fraction as found by Barylko and colleagues [86]. PI4KIIα is required for the recruitment of AP-1 at the trans-Golgi network (TGN) and thus the formation of clathrin-coated vesicles [24]. A more recent work specifically investigating the role of PI4KIIα in neuronal vesicle trafficking has shown that the enzyme cooperates with the BLOC-1 and AP-3 complexes to regulate trafficking from the cell body to the nerve terminal [45]. In light of the recent precedent that both PI4KIIα and PI4KIIIβ isozymes are required for correct Golgi–lysosomal trafficking of the Gaucher disease enzyme β-glucocerebrosidase [87], it seems likely that different PI 4-kinases may control distinct biochemical steps in neuronal vesicle trafficking. Table 1 shows the emerging roles for PI 4-kinases in neuronal disease.
Emerging Neuropathological Roles for the PI 4-Kinases
Schizophrenia
Even though there is still much to be learned about the role of the different PI 4-kinases in normal neuronal physiology, there are nevertheless a number of reports implicating these enzymes in psychiatric and neurological diseases. One emerging story is that PI4KIIIα and PI4KIIα may be important in schizophrenia. The gene for PI4KIIIα, termed PI4K3A, maps to chromosome 22q11, a locus which is of high interest in terms of understanding the genetic basis of mental illness. Individuals with a hemizygous deletion in this chromosomal region in what is known as 22q11.2 deletion syndrome are more susceptible to a number of psychiatric conditions including depression, autism, bipolar disorder and schizophrenia.
In a study involving a cohort of 310 Dutch patients, Jungerius and colleagues [88] identified a significant association between three intronic PI4K3A single nucleotide polymorphisms and schizophrenia. It is noteworthy that this association was later confirmed in a different study in patients with 22q11.2 deletion syndrome [89]. However, a similar study on a Japanese group of patients did not replicate these findings [90]. Therefore, compensatory mechanisms due to differences in genetic background and possibly related to ethnic variation may affect the linkage between PI4K3A and schizophrenia. In a separate study which investigated the frequencies of PI4K3A polymorphisms [91] in schizophrenia and bipolar disorder, there was some evidence for the occurrence of two rare but possibly functional variants in a few patients; the first could possibly result in the creation of a CREB transcription factor binding site in the promoter region and the second had the potential to disrupt splicing of the PI4K3A gene.
While most work has been focused on the possible role of the PI4KIIIα isoform in schizophrenia, there is some evidence that other PI 4-kinases may have a role in this disease. A potential association between the PI4KIIβ isoform and schizophrenia was suggested in a study centred on a large Scottish family cohort [92], and the PI4KIIα isoform is involved in vesicular transport of dysbindin, a schizophrenia susceptibility protein [45]. This leads to the now familiar conclusion that multiple PI 4-kinases may be involved in schizophrenia although it is not yet clear if such an association is only limited to a subset of patients.
PI4KIIIα in Chronic Alcohol Consumption
Altered PI4KIIIα expression has been reported in rat hippocampus following chronic ethanol treatment. Using cDNA microarrays to analyse gene expression, Saito et al. [93] found that the expression of PI4KIIIα was downregulated 1.6-fold in the brains of ethanol-treated rats. Several other proteins including profilin, synaptophysin, ARF-1 and dynamin-1 also exhibited a similar degree of downregulaltion, leading to the suggestion that cytoskeletal and vesicular trafficking processes may be particularly sensitive to perturbation by ethanol [93]. PI4KIIIα is not known to regulate post-Golgi vesicle trafficking, but emerging evidence from other experimental models suggesting a pro-survival signalling role for this isoform via the MAPK/ERK pathway may be worth investigating in a neuronal context. In particular, knockdown of PI4KIIIα in zebrafish morphilinos gave rise to anomalous brain development which seems likely to be due to defective growth factor-stimulated MAPK and PI 3-kinase signalling [94]. Also noteworthy in this regard is a siRNA kinase screen in medulloblastoma-derived cell lines which identified PI4KIIIα as a protein required to sustain cell proliferation and in underlying resistance to the chemotherapeutic reagent cisplatin [95]. These results combined with other emerging insights into the role of PI 4-kinases in cell survival [96] suggest that further work is warranted to investigate the possible targeting of these enzymes in brain cancers difficult to treat such as medulloblastomas which have poor survival rates.
Neuronal Cell Death Following Ischemia
In a rat model for transient forebrain ischemia, induced by the four-vessel occlusion method, PI4KIIIα expression is specifically downregulated 30–80 % in CA1 pyramidal neurons but not in other brain regions [41]. This event precedes the delayed neuronal apoptosis that occurs in these neurons following ischemic shock and correlated with a reduction in PI(4,5)P2 levels. Significantly, recombinant overexpression of wild-type but not catalytically inactive PI4KIIIα in neuroblastoma cells could rescue hypoxia-induced cell death, thus demonstrating that PI4KIIIα-catalysed PI4P generation was essential for cell survival under these conditions [41].
Alzheimer’s Disease
Both phosphoinositde levels [97] and PI 4-kinase activity are reduced by up to 50 % in the brains of patients with Alzheimer’s disease [98]. Intriguingly, pathologically relevant nanomolar concentrations of amyloid β (Aβ) protein can inhibit PI4KII activity in a neuronal plasma membrane preparation [99] with concomitant, augmented glutamate toxicity. In a later study, it was demonstrated that the suppression of PI4P synthesis and glutamate neurotoxicity by Aβ protein could be antagonised with a simple Ile-Gly-Leu tripeptide [100]. Similar to the key Alzheimer’s proteases BACE [101] and γ-secretase [102], the catalytic activity of PI4KIIα is highly sensitive to membrane cholesterol levels, and this enzyme is also targeted to cholesterol- and glycosphingolipid-rich microdomains of the TGN and endosomes [103–105]. Recently, it has been reported that γ-secretase activity is strongly inhibited by phosphatidylinositol in vitro [106]. Furthermore, single nucleotide polymorphisms in PICALM, a gene encoding phosphatidylinositol clathrin assembly lymphoid myeloid leukaemia (PICALM), are strongly associated with Alzheimer’s disease [107, 108]. PICALM binds PI(4,5)P2 and regulates clathrin-mediated endocytosis of amyloid precursor protein and its subsequent trafficking to endosomes where it is proteolytically processed into Aβ [109, 110]. Thus PICALM functionally links PI4P metabolism with endocytic trafficking and amyloid plaque formation [109, 110]. Together these observations suggest a common sterol-sensitive pathway that may link amyloid protein processing with PI4P metabolism and possibly in the upstream production of PI(4,5)P2 [26].
Neuropathology in PI4KIIα Knock-out Mice
Acute RNAi-induced inhibition of any of the PI 4-kinase isozymes in cell lines can result in defective phosphoinositide signalling and aberrant intracellular trafficking. Thus, it could have been expected that PI 4-kinase knock-out animals would exhibit multiple abnormalities. To date, only the pi4k2a gene encoding the PI4KIIα isozyme has been knocked out in mice [44]. Surprisingly however, generation of a PI4KIIα gene trap knock-out mouse showed that homozygous−/− animals were viable and initially developed normally [44]. As the animals aged, a progressive neurological phenotype developed, with the mice exhibiting a spastic gait, nodding tremor and incontinence. These characteristics resemble the progression of autosomal recessive hereditary spastic paraplegia. Histological analysis of aged animals revealed a marked decrease in the number of Purkinje cells, along with axonal defects in both the ascending and descending tracts of the spinal cords [44]. The appearance of lipofuscin deposits in affected mice suggests that loss of PI4KIIα expression induces a cumulative failure in endo-lysosomal trafficking, since defects in this pathway characteristically give rise to endosomal storage diseases [87]. However, further investigations are required to establish whether there exists a link between early onset neuropathy and defective, PI4KIIα-dependent, intracellular trafficking.
The progressive nature of the defects in PI4KIIα knock-out mice suggests that there is initial compensation for the loss of the enzyme perhaps through functional redundancy of the other PI 4-kinase isotypes. While it is not known whether pro-survival neuronal signalling was inhibited or if membrane trafficking was defective in the knock-out mouse model, it is clear that, over time, the continued expression of PI4KIIα is essential for the viability of particular cell populations in the CNS.
Neuronal Dysfunction Controlled by other PI4P Modulators
While perturbations of the PI 4-kinase isoforms themselves are sufficient to induce neuronal dysfunction, PI4P concentrations can also be modulated by PI4P 5-kinase phosphorylation to generate PI(4,5)P2 by, or conversely via phosphatase-mediated D5 dephosphorylation of PI(4,5)P2 to generate PI4P. There is now strong evidence that deregulation of either pathway of PI4P metabolism can lead to neuronal dysfunction and disease.
OCRL
OCRL is a phosphoinositide D5 phosphatase, capable of producing PI4P through the dephosphorylation of PI(4,5)P2 (reviewed in [111]). OCRL is deleted or mutated in individuals suffering from Lowe syndrome and in Dent’s disease [112]. This is an X-linked disorder and, along with the associated ophthalmological and renal symptoms, there is a distinct neuronal phenotype, with affected boys suffering from varying degrees of intellectual impairment, seizures and maladaptive behavioural issues. OCRL is typically found on endosomes and at the Golgi of non-neuronal cell lines. Defective OCRL leads to PI(4,5)P2 accumulating on early endosomes and consequently enhanced N-WASP-mediated F-actin accumulation and defective trafficking [113], thus indicating that alterations to the PI4P:PI(4,5)P2 balance on intracellular membranes can have pathological consequences. Very little is known about the role of PI4P in the endosomal pathway, and this may be partly due to the technical difficulties in imaging non-Golgi PI4P pools with currently available anti-PI4P antibodies and PI4P-specific PH domain-binding proteins [114]. Nevertheless, work involving the expression of recombinant, catalytically inactive PI4KIIα, has implicated PI4P generation by this isoform in the recruitment of AP-3 to late endosomes in non-neuronal cells [23]. More recently, Larimore and colleagues have shown that PI4KIIα operating in conjunction with the AP-3 and the BLOC-1 complexes, mediates trafficking of synaptic-like microvesicles from the cell body to both neurites and nerve terminals [45]. These new insights suggest that changes to PI4P concentrations on intracellular membranes have the potential to alter the dynamics of cargo delivery to synaptic membranes, but further work is needed to evaluate the degree to which compartmentalised changes to PI 4-kinase catalytic activity can alter localised PI4P:PI(4,5)P2 ratios and to establish if this impacts on intra-neuronal vesicular trafficking.
OCRL also contains a clathrin-binding motif within a non-phosphoinositide-binding PH domain [115], a non-functional Rho-GAP domain, and domains which can interact with endosomal trafficking proteins such as APPL1 and Rab GTPases (reviewed in [116]), suggesting that the protein functionally integrates PI4P generation with intracellular vesicle trafficking. A splice variant of OCRL, termed OCRLa, is only expressed in the brain, has a higher affinity for clathrin binding than the more ubiquitously expressed OCRLb variant, and is found associated with clathrin-coated intermediates [117], again suggesting an important linkage between clathrin-dependent trafficking and PI4P generation in the CNS. In concordance with this, OCRL has been co-purified with neuronal clathrin-coated vesicles from synaptosomal preparations [118] and has been imaged in association with late stage clathrin-coated endocytic pits [119]. Further work is needed to evaluate the relative contributions of altered PI4P metabolism and the non-catalytic endosomal functions of OCRL in neurological disease particularly since missense mutations within the APPL1 binding region, situated outside the phosphatase domain, are sufficient to induce the OCRL neuropathology [120].
Synaptojanin 1
Synaptojanin 1, the main neuronal PI(4,5)P2 D5 phosphatase, which also contains a PI4P phosphatase sac1 domain, is essential for synaptic vesicle endocytosis in neurons and is thus essential for maintenance of neuronal transmission. Knock-out mice deficient in Synaptojanin 1 die shortly after birth and exhibit numerous neurological defects including severe weakness, ataxia and convulsions. These mice have elevated levels of PI(4,5)P2, and accumulate a large number of clathrin-coated intermediates [2] which are infrequently observed in wild-type animals.
Synaptojanin is one of the genes present on chromosome 21, the trisomy of which results in excess production of synaptojanin 1 in individuals suffering from Down syndrome [121]. In a mouse model, increased gene dosage of synaptojanin 1 led to a 15–20 % increase in PI(4,5)P2 mass in the brains of affected mice [122] and impaired cognitive performance when assessed by the Morris water maze task. Neurons cultured from mouse models of Down syndrome, trisomic for synaptojanin 1, were found to possess significantly enlarged early endosomes [123]. This effect was recapitulated in neuroblastoma cell lines through the overexpression of tagged synaptojanin 1 [123]. Since synaptojanin possesses both D5 and D4-phosphatase activities, excesses of this enzyme have the potential to increase either PI4P or PI levels in cells. Indeed, both activities are required for synaptic vesicle recycling, indicating a requirement for both PI4P and PI(4,5)P2 in this event [32]. However, the membrane concentrations of different phosphoinositide species present on the enlarged early endosomes have not yet been determined.
Synaptojanin is also thought to play a role in the progression of synaptic dysfunction in Alzheimer’s disease [26]. Through their action on synaptojanin 1, Aβ peptides can acutely and chronically destabilise the metabolism of PI(4,5)P2 in primary cortical cultures, implicating synaptojanin and Aβ oligomers in pathophysiological progression of this disease. This implies that normal synaptojanin levels of expression are essential for both the highly specialised, neuron-specific, synaptic vesicle cycle, as well as the ubiquitous membrane trafficking pathways found in other tissues. As mentioned previously, Aβ proteins have also been shown to inhibit PI4KIIα activity [99, 100] in the nanomolar range all of which suggests that alterations to the PI:PI4P:PI(4,5)P2 ratio in neurons may be an important factor in the aetiology of Alzheimer’s disease.
PIPKIγ
Three PIPK enzymes are capable of generating PI(4,5)P2 through phosphorylation of PI4P on the D5 position. Of these three isoforms, PIPKIγ is the dominant form found in the nervous system, where it plays a critical role in synaptic transmission [124, 125], embryonic neural tube closure, adherens junction formation and neuronal migration [126]. Indeed murine genetic studies have revealed that loss of PIPKIγ results in either embryonic [126] or early postnatal lethality [1, 127], and also that a single allele of PIPKIγ is sufficient to ensure development to adulthood and to maintain neuronal PI(4,5)P2 levels [127].
A lipid kinase-inactivating, single point mutation in PIPKIγ resulting in the substitution of aspartic acid with asparagine at amino acid 253 was found to be the cause of lethal congenital contractural syndrome type 3 (LCCS3), a disease which causes foetal or neo-natal mortality [128]. Interestingly, the PIPKIγ knock-out mouse did not replicate the muscle wasting and joint contracture of LCCS3, although it did cause early postnatal lethality [1]. Detailed studies of the presynapse of these knock-out mice revealed a significant defect and delay in the reformation of synaptic vesicles following exocytosis, in conjunction with pronounced rapid exocytic depression during periods of intense stimulation [1]. It is noteworthy that these PIPKIγ-deficient synapses produced significantly more and larger endosomes in response to elevated stimulation. These are reminiscent of activity-dependent bulk endocytosis profiles, which are induced in central nervous synapses in response to strong synaptic stimulus [129]. The resulting smaller recycling pool and delayed synaptic vesicle recycling of PIPKIγ-deficient synapses may indicate a defect in the generation of single synaptic vesicles which bud from these large bulk endosomes. This implicates a hitherto uninvestigated role for PI4P-derived phosphoinositides in the generation of synaptic vesicle membranes from bulk endocytic structures.
Conclusions and Future Perspectives
PI 4-kinases and PI4P are beginning to be implicated across a wide range of neuronal functions and pathologies, but this remains a very underdeveloped field of study. Future work, perhaps on transgenic models with conditional CNS expression of PI 4-kinase structural variants may be key to understanding the catalytic and non-catalytic functions of these enzymes in neuronal vesicle trafficking and synaptic transmission. The recent availability of small molecule inhibitors particularly of the PI4KIIIs [62–64] may facilitate a more meaningful analysis of the neuronal functions of this class of enzymes and facilitate the design of novel chemotherapeutics with potential applications in the treatment of neurological and psychiatric diseases. On the other hand, the current dearth of isoform-specific inhibitors directed against the PI4KIIs, PIPKs and PI(4,5)P2 phosphatases is reflected in a continued reliance on recombinant and genetic strategies to understand the enzymology and regulation of neuronal PI4P pools. Finally, new approaches to manipulate and detect levels of PI4P in different subcellular compartments [8, 20, 38, 114] are likely to be extremely important in dissecting specific roles for this phospholipid in the CNS.
Abbreviations
- Aβ:
-
Amyloid β peptide
- ARF:
-
ADP-ribosylation factor
- NCS-1:
-
Neuronal calcium sensor-1
- TGN:
-
Trans-Golgi network
- PAO:
-
Phenylarsine oxide
- PI:
-
Phosphatidylinositol
- PI4P:
-
Phosphatidylinositol 4-phosphate
- PI(4,5)P2 :
-
Phosphatidylinositol (4,5)-bisphosphate
- PI(3,4,5)P3 :
-
Phosphatidylinositol (3,4,5)-trisphosphate
- PI 3-kinase:
-
Phosphoinositide 3-kinase
- PI4K:
-
Phosphatidylinositol 4-kinase
- PI4KIII:
-
Type III PI 4-kinase
- PICALM:
-
Phosphatidylinositol clathrin assembly lymphoid myeloid leukaemia
- PIPK:
-
PI4P 5-kinase
References
Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P (2004) Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431:415–422
Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, McCormick DA, De Camilli P (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99:179–188
Hilgemann DW, Ball R (1996) Regulation of cardiac Na+, Ca2+ exchange and KATP potassium channels by PIP2. Science 273:956–959
Hilgemann DW, Feng S, Nasuhoglu C (2001) The complex and intriguing lives of PIP2 with ion channels and transporters, Sci STKE 2001 re19
Huang CL, Feng S, Hilgemann DW (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391:803–806
Suh BC, Hille B (2008) PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 37:175–195
Suh BC, Kim DI, Falkenburger BH, Hille B (2012) Membrane-localized beta-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc Natl Acad Sci U S A 109:3161–3166
Lindner M, Leitner MG, Halaszovich CR, Hammond GR, Oliver D (2011) Probing the regulation of TASK potassium channels by PI4,5P(2) with switchable phosphoinositide phosphatases. J Physiol 589:3149–3162
Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275:661–665
Balla A, Balla T (2006) Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol 16:351–361
Minogue S, Waugh MG (2012) The phosphatidylinositol 4-kinases: don’t call it a comeback. Subcell Biochem 58:1–24
Zaika O, Zhang J, Shapiro MS (2011) Combined phosphoinositide and Ca2+ signals mediating receptor specificity toward neuronal Ca2+ channels. J Biol Chem 286:830–841
Suh BC, Hille B (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35:507–520
Lopes CM, Rohacs T, Czirjak G, Balla T, Enyedi P, Logothetis DE (2005) PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J Physiol 564:117–129
Trebak M, Lemonnier L, DeHaven WI, Wedel BJ, Bird GS, Putney JW Jr (2009) Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflugers Arch 457:757–769
Karashima Y, Prenen J, Meseguer V, Owsianik G, Voets T, Nilius B (2008) Modulation of the transient receptor potential channel TRPA1 by phosphatidylinositol 4,5-biphosphate manipulators. Pflugers Arch 457:77–89
Bernier LP, Ase AR, Tong X, Hamel E, Blais D, Zhao Q, Logothetis DE, Seguela P (2008) Direct modulation of P2X1 receptor-channels by the lipid phosphatidylinositol 4,5-bisphosphate. Mol Pharmacol 74:785–792
Bernier LP, Ase AR, Chevallier S, Blais D, Zhao Q, Boue-Grabot E, Logothetis D, Seguela P (2008) Phosphoinositides regulate P2X4 ATP-gated channels through direct interactions. J Neurosci 28:12938–12945
Chen X, Zhang X, Jia C, Xu J, Gao H, Zhang G, Du X, Zhang H (2011) Membrane depolarization increases membrane PtdIns(4,5)P2 levels through mechanisms involving PKC beta II and PI4 kinase. J Biol Chem 286:39760–39767
Hammond GR, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, Irvine RF (2012) PI4P and PI(4,5)P2 Are essential but independent lipid determinants of membrane identity. Science 337(6095):727–730
Korzeniowski MK, Popovic MA, Szentpetery Z, Varnai P, Stojilkovic SS, Balla T (2009) Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J Biol Chem 284:21027–21035
Zhang X, Chen X, Jia C, Geng X, Du X, Zhang H (2010) Depolarization increases phosphatidylinositol (PI) 4,5-bisphosphate level and KCNQ currents through PI 4-kinase mechanisms. J Biol Chem 285:9402–9409
Craige B, Salazar G, Faundez V (2008) Phosphatidylinositol-4-kinase type II alpha contains an AP-3 sorting motif and a kinase domain that are both required for endosome traffic. Mol Biol Cell 19(4):1415–1426
Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, Yin HL (2003) Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 114:299–310
Wang J, Sun HQ, Macia E, Kirchhausen T, Watson H, Bonifacino JS, Yin HL (2007) PI4P promotes the recruitment of the GGA adaptor proteins to the trans-Golgi network and regulates their recognition of the ubiquitin sorting signal. Mol Biol Cell 18:2646–2655
Berman DE, Dall’Armi C, Voronov SV, McIntire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G (2008) Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci 11:547–554
Chang-Ileto B, Frere SG, Chan RB, Voronov SV, Roux A, Di Paolo G (2011) Synaptojanin 1-mediated PI(4,5)P2 hydrolysis is modulated by membrane curvature and facilitates membrane fission. Dev Cell 20:206–218
McPherson PS, Garcia EP, Slepnev VI, David C, Zhang X, Grabs D, Sossin WS, Bauerfeind R, Nemoto Y, De Camilli P (1996) A presynaptic inositol-5-phosphatase. Nature 379:353–357
Woscholski R, Finan PM, Radley E, Parker PJ (1998) Identification and characterisation of a novel splice variant of synaptojanin1. FEBS Lett 432:5–8
Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, McInnes RR, Nussbaum RL (1992) The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 358:239–242
Guo S, Stolz LE, Lemrow SM, York JD (1999) SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J Biol Chem 274:12990–12995
Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P, Ryan TA (2007) The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron 56:1004–1018
Balla A, Tuymetova G, Barshishat M, Geiszt M, Balla T (2002) Characterization of type II phosphatidylinositol 4-kinase isoforms reveals association of the enzymes with endosomal vesicular compartments. J Biol Chem 277:20041–20050
Balla A, Tuymetova G, Tsiomenko A, Varnai P, Balla T (2005) A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol Cell 16:1282–1295
Waugh MG, Minogue S, Blumenkrantz D, Anderson JS, Hsuan JJ (2003) Identification and characterization of differentially active pools of type IIalpha phosphatidylinositol 4-kinase activity in unstimulated A431 cells. Biochem J 376:497–503
Waugh MG, Minogue S, Chotai D, Berditchevski F, Hsuan JJ (2006) Lipid and peptide control of phosphatidylinositol 4-kinase IIalpha activity on Golgi-endosomal rafts. J Biol Chem 281:3757–3763
Waugh MG, Chu KM, Clayton EL, Minogue S, Hsuan JJ (2011) Detergent-free isolation and characterization of cholesterol-rich membrane domains from trans-Golgi network vesicles. J Lipid Res 52:582–589
Szentpetery Z, Varnai P, Balla T (2010) Acute manipulation of Golgi phosphoinositides to assess their importance in cellular trafficking and signaling. Proc Natl Acad Sci U S A 107:8225–8230
Zolyomi A, Zhao X, Downing GJ, Balla T (2000) Localization of two distinct type III phosphatidylinositol 4-kinase enzyme mRNAs in the rat. Am J Physiol Cell Physiol 278:C914–C920
Balla A, Vereb G, Gulkan H, Gehrmann T, Gergely P, Heilmeyer LM Jr, Antal M (2000) Immunohistochemical localisation of two phosphatidylinositol 4-kinase isoforms, PI4K230 and PI4K92, in the central nervous system of rats. Exp Brain Res 134:279–288
Furuta Y, Uehara T, Nomura Y (2003) Correlation between delayed neuronal cell death and selective decrease in phosphatidylinositol 4-kinase expression in the CA1 subfield of the hippocampus after transient forebrain ischemia. J Cereb Blood Flow Metab 23:962–971
Nicolay NH, Hertle D, Boehmerle W, Heidrich FM, Yeckel M, Ehrlich BE (2007) Inositol 1,4,5 trisphosphate receptor and chromogranin B are concentrated in different regions of the hippocampus. J Neurosci Res 85:2026–2036
Nicolle MM, Gallagher M, McKinney M (2001) Visualization of muscarinic receptor-mediated phosphoinositide turnover in the hippocampus of young and aged, learning-impaired Long Evans rats. Hippocampus 11:741–746
Simons JP, Al-Shawi R, Minogue S, Waugh MG, Wiedemann C, Evangelou S, Loesch A, Sihra TS, King R, Warner TT, Hsuan JJ (2009) Loss of phosphatidylinositol 4-kinase 2alpha activity causes late onset degeneration of spinal cord axons. Proc Natl Acad Sci U S A 106:11535–11539
Larimore J, Tornieri K, Ryder PV, Gokhale A, Zlatic SA, Craige B, Lee JD, Talbot K, Pare JF, Smith Y, Faundez V (2011) The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol Biol Cell 22:4854–4867
Nilsson P, Paavilainen L, Larsson K, Odling J, Sundberg M, Andersson AC, Kampf C, Persson A, Al-Khalili Szigyarto C, Ottosson J, Bjorling E, Hober S, Wernerus H, Wester K, Ponten F, Uhlen M (2005) Towards a human proteome atlas: high-throughput generation of mono-specific antibodies for tissue profiling. Proteomics 5:4327–4337
Ponten F, Schwenk JM, Asplund A, Edqvist PH (2011) The Human Protein Atlas as a proteomic resource for biomarker discovery. J Intern Med 270:428–446
Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Bjorling L, Ponten F (2010) Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 28:1248–1250
Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176
Wong K, Meyers DDR, Cantley LC (1997) Subcellular locations of phosphatidylinositol 4-kinase isoforms. J Biol Chem 272:13236–13241
Weixel KM, Blumental-Perry A, Watkins SC, Aridor M, Weisz OA (2005) Distinct Golgi populations of phosphatidylinositol 4-phosphate regulated by phosphatidylinositol 4-kinases. J Biol Chem 280:10501–10508
Guo J, Wenk MR, Pellegrini L, Onofri F, Benfenati F, De Camilli P (2003) Phosphatidylinositol 4-kinase type IIalpha is responsible for the phosphatidylinositol 4-kinase activity associated with synaptic vesicles. Proc Natl Acad Sci U S A 100:3995–4000
Hay JC, Fisette PL, Jenkins GH, Fukami K, Takenawa T, Anderson RA, Martin TF (1995) ATP-dependent inositide phosphorylation required for Ca(2+)-activated secretion. Nature 374:173–177
Hay JC, Martin TF (1993) Phosphatidylinositol transfer protein required for ATP-dependent priming of Ca(2+)-activated secretion. Nature 366:572–575
Foley TD, Melideo SL, Healey AE, Lucas EJ, Koval JA (2011) Phenylarsine oxide binding reveals redox-active and potential regulatory vicinal thiols on the catalytic subunit of protein phosphatase 2A. Neurochem Res 36:232–240
Frost SC, Schwalbe MS (1990) Uptake and binding of radiolabelled phenylarsine oxide in 3 T3-L1 adipocytes. Biochem J 269:589–595
Oustrin ML, Belenguer P, Leroy D, Hoffmann I, Ducommun B (1995) Effect of phenylarsine oxide on the fission yeast Schizosaccharomyces pombe cell cycle. Biochimie 77:279–287
Bartlett SE, Reynolds AJ, Weible M 2nd, Hendry IA (2002) Phosphatidylinositol kinase enzymes regulate the retrograde axonal transport of NT-3 and NT-4 in sympathetic and sensory neurons. J Neurosci Res 68:169–175
Reynolds AJ, Heydon K, Bartlett SE, Hendry IA (1999) Evidence for phosphatidylinositol 4-kinase and actin involvement in the regulation of 125I-beta-nerve growth factor retrograde axonal transport. J Neurochem 73:87–95
Wiedemann C, Schafer T, Burger MM (1996) Chromaffin granule-associated phosphatidylinositol 4-kinase activity is required for stimulated secretion. EMBO J 15:2094–2101
Balla A, Tuymetova G, Toth B, Szentpetery Z, Zhao X, Knight ZA, Shokat K, Steinbach PJ, Balla T (2008) Design of Drug-resistant alleles of type-III phosphatidylinositol 4-kinases using mutagenesis and molecular modeling. Biochemistry 47:1599–1607
Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H (2011) Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J Virol 85:2364–2372
Bianco A, Reghellin V, Donnici L, Fenu S, Alvarez R, Baruffa C, Peri F, Pagani M, Abrignani S, Neddermann P, De Francesco R (2012) Metabolism of phosphatidylinositol 4-kinase IIIalpha-dependent PI4P Is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog 8:e1002576
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM (2006) A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125:733–747
Ashton AC, Ushkaryov YA (2005) Properties of synaptic vesicle pools in mature central nerve terminals. J Biol Chem 280:37278–37288
Khvotchev M, Sudhof TC (1998) Newly synthesized phosphatidylinositol phosphates are required for synaptic norepinephrine but not glutamate or gamma-aminobutyric acid (GABA) release. J Biol Chem 273:21451–21454
Koizumi S, Rosa P, Willars GB, Challiss RA, Taverna E, Francolini M, Bootman MD, Lipp P, Inoue K, Roder J, Jeromin A (2002) Mechanisms underlying the neuronal calcium sensor-1-evoked enhancement of exocytosis in PC12 cells. J Biol Chem 277:30315–30324
Scalettar BA, Rosa P, Taverna E, Francolini M, Tsuboi T, Terakawa S, Koizumi S, Roder J, Jeromin A (2002) Neuronal calcium sensor-1 binds to regulated secretory organelles and functions in basal and stimulated exocytosis in PC12 cells. J Cell Sci 115:2399–2412
Gromada J, Bark C, Smidt K, Efanov AM, Janson J, Mandic SA, Webb DL, Zhang W, Meister B, Jeromin A, Berggren PO (2005) Neuronal calcium sensor-1 potentiates glucose-dependent exocytosis in pancreatic beta cells through activation of phosphatidylinositol 4-kinase beta. Proc Natl Acad Sci U S A 102:10303–10308
Haynes LP, Sherwood MW, Dolman NJ, Burgoyne RD (2007) Specificity, promiscuity and localization of ARF protein interactions with NCS-1 and phosphatidylinositol-4 kinase-III beta. Traffic 8:1080–1092
Kapp-Barnea Y, Melnikov S, Shefler I, Jeromin A, Sagi-Eisenberg R (2003) Neuronal calcium sensor-1 and phosphatidylinositol 4-kinase beta regulate IgE receptor-triggered exocytosis in cultured mast cells. J Immunol 171:5320–5327
Pan CY, Jeromin A, Lundstrom K, Yoo SH, Roder J, Fox AP (2002) Alterations in exocytosis induced by neuronal Ca2+ sensor-1 in bovine chromaffin cells. J Neurosci 22:2427–2433
Zhao X, Varnai P, Tuymetova G, Balla A, Toth ZE, Oker-Blom C, Roder J, Jeromin A, Balla T (2001) Interaction of neuronal calcium sensor-1 (NCS-1) with phosphatidylinositol 4-kinase beta stimulates lipid kinase activity and affects membrane trafficking in COS-7 cells. J Biol Chem 276:40183–40189
de Barry J, Janoshazi A, Dupont JL, Procksch O, Chasserot-Golaz S, Jeromin A, Vitale N (2006) Functional implication of neuronal calcium sensor-1 and phosphoinositol 4-kinase-beta interaction in regulated exocytosis of PC12 cells. J Biol Chem 281:18098–18111
Haynes LP, Thomas GM, Burgoyne RD (2005) Interaction of neuronal calcium sensor-1 and ADP-ribosylation factor 1 allows bidirectional control of phosphatidylinositol 4-kinase beta and trans-Golgi network-plasma membrane traffic. J Biol Chem 280:6047–6054
Kapp-Barnea Y, Ninio-Many L, Hirschberg K, Fukuda M, Jeromin A, Sagi-Eisenberg R (2006) Neuronal calcium sensor-1 and phosphatidylinositol 4-kinase beta stimulate extracellular signal-regulated kinase 1/2 signaling by accelerating recycling through the endocytic recycling compartment. Mol Biol Cell 17:4130–4141
Mora S, Durham PL, Smith JR, Russo AF, Jeromin A, Pessin JE (2002) NCS-1 inhibits insulin-stimulated GLUT4 translocation in 3T3L1 adipocytes through a phosphatidylinositol 4-kinase-dependent pathway. J Biol Chem 277:27494–27500
Rajebhosale M, Greenwood S, Vidugiriene J, Jeromin A, Hilfiker S (2003) Phosphatidylinositol 4-OH kinase is a downstream target of neuronal calcium sensor-1 in enhancing exocytosis in neuroendocrine cells. J Biol Chem 278:6075–6084
Taverna E, Francolini M, Jeromin A, Hilfiker S, Roder J, Rosa P (2002) Neuronal calcium sensor 1 and phosphatidylinositol 4-OH kinase beta interact in neuronal cells and are translocated to membranes during nucleotide-evoked exocytosis. J Cell Sci 115:3909–3922
Zheng Q, Bobich JA, Vidugiriene J, McFadden SC, Thomas F, Roder J, Jeromin A (2005) Neuronal calcium sensor-1 facilitates neuronal exocytosis through phosphatidylinositol 4-kinase. J Neurochem 92:442–451
Mikhaylova M, Reddy PP, Munsch T, Landgraf P, Suman SK, Smalla KH, Gundelfinger ED, Sharma Y, Kreutz MR (2009) Calneurons provide a calcium threshold for trans-Golgi network to plasma membrane trafficking. Proc Natl Acad Sci U S A 106:9093–9098
Haynes LP, Fitzgerald DJ, Wareing B, O’Callaghan DW, Morgan A, Burgoyne RD (2006) Analysis of the interacting partners of the neuronal calcium-binding proteins L-CaBP1, hippocalcin, NCS-1 and neurocalcin delta. Proteomics 6:1822–1832
Lim S, Strahl T, Thorner J, Ames JB (2011) Structure of a Ca2+ −myristoyl switch protein that controls activation of a phosphatidylinositol 4-kinase in fission yeast. J Biol Chem 286:12565–12577
Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, Iurisci C, Luini A, Corda D, De Matteis MA (1999) ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol 1:280–287
Panaretou C, Tooze SA (2002) Regulation and recruitment of phosphatidylinositol 4-kinase on immature secretory granules is independent of ADP-ribosylation factor 1. Biochem J 363:289–295
Barylko B, Gerber SH, Binns DD, Grichine N, Khvotchev M, Sudhof TC, Albanesi JP (2001) A novel family of phosphatidylinositol 4-kinases conserved from yeast to humans. J Biol Chem 276:7705–7708
Jovic M, Kean MJ, Szentpetery Z, Polevoy G, Gingras AC, Brill JA, Balla T (2012) Two phosphatidylinositol 4-kinases control lysosomal delivery of the Gaucher disease enzyme, beta-glucocerebrosidase. Mol Biol Cell 23:1533–1545
Jungerius BJ, Hoogendoorn ML, Bakker SC, Van’t Slot R, Bardoel AF, Ophoff RA, Wijmenga C, Kahn RS, Sinke RJ (2008) An association screen of myelin-related genes implicates the chromosome 22q11 PIK4CA gene in schizophrenia. Mol Psychiatry 13:1060–1068
Vorstman JA, Chow EW, Ophoff RA, van Engeland H, Beemer FA, Kahn RS, Sinke RJ, Bassett AS (2009) Association of the PIK4CA schizophrenia-susceptibility gene in adults with the 22q11.2 deletion syndrome. Am J Med Genet B Neuropsychiatr Genet 150B:430–433
Kanahara N, Iyo M, Hashimoto K (2009) Failure to confirm the association between the PIK4CA gene and schizophrenia in a Japanese population. Am J Med Genet B Neuropsychiatr Genet 150B:450–452
Saito T, Stopkova P, Diaz L, Papolos DF, Boussemart L, Lachman HM (2003) Polymorphism screening of PIK4CA: possible candidate gene for chromosome 22q11-linked psychiatric disorders. Am J Med Genet B Neuropsychiatr Genet 116:77–83
Houlihan LM, Christoforou A, Arbuckle MI, Torrance HS, Anderson SM, Muir WJ, Porteous DJ, Blackwood DH, Evans KL (2009) A case–control association study and family-based expression analysis of the bipolar disorder candidate gene PI4K2B. J Psychiatr Res 43:1272–1277
Saito M, Smiley J, Toth R, Vadasz C (2002) Microarray analysis of gene expression in rat hippocampus after chronic ethanol treatment. Neurochem Res 27:1221–1229
Ma H, Blake T, Chitnis A, Liu P, Balla T (2009) Crucial role of phosphatidylinositol 4-kinase IIIalpha in development of zebrafish pectoral fin is linked to phosphoinositide 3-kinase and FGF signaling. J Cell Sci 122:4303–4310
Guerreiro AS, Fattet S, Kulesza DW, Atamer A, Elsing AN, Shalaby T, Jackson SP, Schoenwaelder SM, Grotzer MA, Delattre O, Arcaro A (2011) A sensitized RNA interference screen identifies a novel role for the PI3K p110gamma isoform in medulloblastoma cell proliferation and chemoresistance. Mol Cancer Res 9:925–935
Chu K, Minogue S, Hsuan J, Waugh M (2010) Differential effects of the phosphatidylinositol 4-kinases, PI4KIIalpha and PI4KIIIbeta, on Akt activation and apoptosis. Cell Death Dis 1:e106
Stokes CE, Hawthorne JN (1987) Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J Neurochem 48:1018–1021
Zubenko GS, Stiffler JS, Hughes HB, Martinez AJ (1999) Reductions in brain phosphatidylinositol kinase activities in Alzheimer’s disease. Biol Psychiatry 45:731–736
Wu B, Kitagawa K, Liu B, Zhang NY, Xiong ZM, Inagaki C (2006) Attenuation of amyloid beta (Abeta)-induced inhibition of phosphatidylinositol 4-kinase activity by Abeta fragments, Abeta20-29 and Abeta31-35. Neurosci Lett 396:148–152
Xiong ZM, Kitagawa K, Nishiuchi Y, Kimura T, Inagaki C (2007) Protective effects of Abeta-derived tripeptide, Abeta(32–34), on Abeta(1–42)-induced phosphatidylinositol 4-kinase inhibition and neurotoxicity. Neurosci Lett 419:247–252
Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K (2005) Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem 280:36815–36823
Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ (2008) Direct and potent regulation of gamma-secretase by its lipid microenvironment, J Biol Chem
Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knolker HJ, Simons K (2008) Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science 320:520–523
Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, Sisodia SS, Thinakaran G (2005) Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem 280:25892–25900
Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G (2004) Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem 279:44945–44954
Holmes O, Paturi S, Ye W, Wolfe MS, Selkoe DJ (2012) Effects of membrane lipids on the activity and processivity of purified gamma-secretase. Biochemistry 51:3565–3575
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41:1088–1093
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43:436–441
Treusch S, Hamamichi S, Goodman JL, Matlack KE, Chung CY, Baru V, Shulman JM, Parrado A, Bevis BJ, Valastyan JS, Han H, Lindhagen-Persson M, Reiman EM, Evans DA, Bennett DA, Olofsson A, DeJager PL, Tanzi RE, Caldwell KA, Caldwell GA, Lindquist S (2011) Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 334:1241–1245
Xiao Q, Gil SC, Yan P, Wang Y, Han S, Gonzales E, Perez R, Cirrito JR, Lee JM (2012) Role of Phosphatidylinositol Clathrin Assembly Lymphoid-Myeloid Leukemia (PICALM) in Intracellular Amyloid Precursor Protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem 287:21279–21289
Ooms LM, Horan KA, Rahman P, Seaton G, Gurung R, Kethesparan DS, Mitchell CA (2009) The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem J 419:29–49
Hichri H, Rendu J, Monnier N, Coutton C, Dorseuil O, Poussou RV, Baujat G, Blanchard A, Nobili F, Ranchin B, Remesy M, Salomon R, Satre V, Lunardi J (2011) From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat 32:379–388
Vicinanza M, Di Campli A, Polishchuk E, Santoro M, Di Tullio G, Godi A, Levtchenko E, De Leo MG, Polishchuk R, Sandoval L, Marzolo MP, De Matteis MA (2011) OCRL controls trafficking through early endosomes via PtdIns4,5P(2)-dependent regulation of endosomal actin. EMBO J 30:4970–4985
Hammond GR, Schiavo G, Irvine RF (2009) Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem J 422:23–35
Mao Y, Balkin DM, Zoncu R, Erdmann KS, Tomasini L, Hu F, Jin MM, Hodsdon ME, De Camilli P (2009) A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. EMBO J 28:1831–1842
Pirruccello M, De Camilli P (2012) Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends Biochem Sci 37:134–143
Choudhury R, Noakes CJ, McKenzie E, Kox C, Lowe M (2009) Differential clathrin binding and subcellular localization of OCRL1 splice isoforms. J Biol Chem 284:9965–9973
Ungewickell A, Ward ME, Ungewickell E, Majerus PW (2004) The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc Natl Acad Sci U S A 101:13501–13506
Erdmann KS, Mao Y, McCrea HJ, Zoncu R, Lee S, Paradise S, Modregger J, Biemesderfer D, Toomre D, De Camilli P (2007) A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell 13:377–390
McCrea HJ, Paradise S, Tomasini L, Addis M, Melis MA, De Matteis MA, De Camilli P (2008) All known patient mutations in the ASH-RhoGAP domains of OCRL affect targeting and APPL1 binding. Biochem Biophys Res Commun 369:493–499
Arai Y, Ijuin T, Takenawa T, Becker LE, Takashima S (2002) Excessive expression of synaptojanin in brains with Down syndrome. Brain Dev 24:67–72
Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, Arancio O, Davisson MT, Antonarakis SE, Gardiner K, De Camilli P, Di Paolo G (2008) Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc Natl Acad Sci U S A 105:9415–9420
Cossec JC, Lavaur J, Berman DE, Rivals I, Hoischen A, Stora S, Ripoll C, Mircher C, Grattau Y, Olivomarin JC, de Chaumont F, Lecourtois M, Antonarakis SE, Veltman JA, Delabar JM, Duyckaerts C, Di Paolo G, Potier MC (2012) Trisomy for Synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes, Hum Mol Genet
Wenk MR, Pellegrini L, Klenchin VA, Di Paolo G, Chang S, Daniell L, Arioka M, Martin TF, De Camilli P (2001) PIP kinase Igamma is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron 32:79–88
Ishihara H, Shibasaki Y, Kizuki N, Wada T, Yazaki Y, Asano T, Oka Y (1998) Type I phosphatidylinositol-4-phosphate 5-kinases. Cloning of the third isoform and deletion/substitution analysis of members of this novel lipid kinase family. J Biol Chem 273:8741–8748
Wang Y, Lian L, Golden JA, Morrisey EE, Abrams CS (2007) PIP5KI gamma is required for cardiovascular and neuronal development. Proc Natl Acad Sci U S A 104:11748–11753
Volpicelli-Daley LA, Lucast L, Gong LW, Liu L, Sasaki J, Sasaki T, Abrams CS, Kanaho Y, De Camilli P (2010) Phosphatidylinositol-4-phosphate 5-kinases and phosphatidylinositol 4,5-bisphosphate synthesis in the brain. J Biol Chem 285:28708–28714
Narkis G, Ofir R, Landau D, Manor E, Volokita M, Hershkowitz R, Elbedour K, Birk OS (2007) Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKI gamma of the phophatidylinsitol pathway. Am J Hum Genet 81:530–539
Clayton EL, Evans GJ, Cousin MA (2008) Bulk synaptic vesicle endocytosis is rapidly triggered during strong stimulation. J Neurosci 28:6627–6632
Acknowledgments
The authors acknowledge support from the BBSRC (grant BB/G021163/1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clayton, E.L., Minogue, S. & Waugh, M.G. Phosphatidylinositol 4-Kinases and PI4P Metabolism in the Nervous System: Roles in Psychiatric and Neurological Diseases. Mol Neurobiol 47, 361–372 (2013). https://doi.org/10.1007/s12035-012-8358-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-012-8358-6