
Abstract
The nuclear lamina is an intermediate filament meshwork composed largely of four nuclear lamins — lamins A and C (A-type lamins) and lamins B1 and B2 (B-type lamins). Located immediately adjacent to the inner nuclear membrane, the nuclear lamina provides a structural scaffolding for the cell nucleus. It also interacts with both nuclear membrane proteins and the chromatin and is thought to participate in many important functions within the cell nucleus. Defects in A-type lamins cause cardiomyopathy, muscular dystrophy, peripheral neuropathy, lipodystrophy, and progeroid disorders. In contrast, the only bona fide link between the B-type lamins and human disease is a rare demyelinating disease of the central nervous system — adult-onset autosomal-dominant leukoencephalopathy, caused by a duplication of the gene for lamin B1. However, this leukoencephalopathy is not the only association between the brain and B-type nuclear lamins. Studies of conventional and tissue-specific knockout mice have demonstrated that B-type lamins play essential roles in neuronal migration in the developing brain and in neuronal survival. The importance of A-type lamin expression in the brain is unclear, but it is intriguing that the adult brain preferentially expresses lamin C rather than lamin A, very likely due to microRNA-mediated removal of prelamin A transcripts. Here, we review recent studies on nuclear lamins, focusing on the function and regulation of the nuclear lamins in the central nervous system.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The nuclear lamina is an intermediate filament meshwork located adjacent to the inner nuclear membrane. In vertebrates, it is composed largely of four proteins called nuclear lamins — lamins A, C, B1, and B2 [1]. The lamins have been classified as A- and B-type based on their biochemical properties [2–4]. The expression of the A-type lamins (lamins A and C) starts during the later stages of embryonic development and peaks postnatally; B-type lamins (lamins B1 and B2) are expressed in all cell types from the earliest stages of development [5, 6].
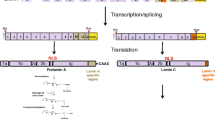
Lamins A and C are alternatively spliced products of the same gene, LMNA (Fig. 1) [7]. They are identical through the first 566 amino acids but diverge in their carboxyl-terminal domains. Lamin C terminates with exon 10 sequences and has six unique amino acids (not found in lamin A) at its carboxyl terminus. Prelamin A, the precursor to mature lamin A, includes sequences from two additional exons (exons 11 and 12), resulting in 98 unique amino acids at its carboxyl terminus. The 3′ UTRs of lamin C and prelamin A transcripts are also distinct. Lamin C’s 3′ UTR, formed by intron 10 sequences, is short (~100 bp); prelamin A’s 3′ UTR is ~1 kb in length and includes many sequences that have been conserved through mammalian evolution.

Schematic diagram of nuclear lamin genes and nuclear lamin structure. All nuclear lamins have α-helical central rod domains (in grey) flanked by globular head and tail domains. The tail domains contain a nuclear localization signal (NLS, in red). Prelamin A (664 amino acids) and lamin C (572 amino acids) are alternatively spliced products of a single gene, LMNA. Lamin C terminates with exon 10 sequences and contains six unique amino acids at its carboxyl terminus (in green); prelamin A contains sequences from exons 11 and 12 and contains 98 unique amino acids at its carboxyl terminus (in sky blue). The 3′ UTR for lamin C is yellow; the 3′ UTRs for the other nuclear lamins are purple. The 5′ UTRs are colored orange. Lamin A (646 amino acids) is formed from prelamin A by a series of four posttranslational processing events and involves the removal of the final 18 amino acids of prelamin A. Lamin B1 (586 amino acids) and lamin B2 (600 amino acids) are produced from independent genes, LMNB1 and LMNB2, respectively. All lamins, except lamin C, contain a carboxyl-terminal CaaX motif, which triggers protein farnesylation. The farnesylated segment of prelamin A (in sky blue with blue stripes) is removed during the biogenesis of mature lamin A
Lamins B1 and B2, are encoded by distinct genes, LMNB1 [8] and LMNB2 [9], respectively. These genes have a similar intron–exon structure, suggesting that they arose from a gene-duplication event [1, 10]. Lamin B1 and lamin B2 proteins are ~60 % identical at the amino acid level [11].
Like other intermediate filament proteins, all nuclear lamins have a highly conserved α-helical central rod domain (Fig. 1) [12, 13]. The rod domain is critical for lamin dimerization and for the formation of higher-order polymers [14]. Lamins also have globular head and tail domains, with the tail domain containing a nuclear localization signals (NLS) (Fig. 1) [12, 13].
Physiologic Importance of A-Type Lamins
To assess the importance of the A-type lamins, Sullivan and coworkers [15] generated Lmna knockout mice (Lmna −/−). Lmna −/− mice do not manifest developmental abnormalities and are indistinguishable from wild-type littermates at birth [15]. Soon thereafter, however, Lmna −/− mice develop a cardiac and skeletal myopathy, and they die by 6–8 weeks of age. The absence of A-type lamins compromises the structural integrity of the nuclear envelope, resulting in misshapen nuclei, which likely plays a part in the pathogenesis of the muscle disease [16]. The absence of lamins A and C also results in mislocalization of emerin, an inner nuclear membrane protein, to the endoplasmic reticulum. Emerin mutations cause muscular dystrophy in humans [2, 17–19].
Thus far, unique roles for lamin A and lamin C remain obscure. Fong et al. [20] created “lamin C–only mice” (Lmna LCO/LCO) that do not produce prelamin A transcripts and make approximately twice-normal amounts of lamin C transcripts. Surprisingly, the absence of prelamin A and lamin A in Lmna LCO/LCO mice does not appear to have adverse consequences; these mice are normal in size and vitality and have no obvious disease phenotypes. Subtle abnormalities in nuclear shape can be detected in cultured Lmna LCO/LCO fibroblasts [16, 20], but it is unclear whether any such abnormalities actually occur in parenchymal cells of Lmna LCO/LCO mice. Later, Coffinier et al. [21] generated “mature lamin A–only mice” (Lmna LAO/LAO) and Davies et al. [22] created “prelamin A–only mice” (Lmna PLAO/PLAO). Both mouse models are incapable of producing lamin C transcripts, and all of the output of the Lmna gene is channeled into prelamin A transcripts (encoding mature lamin A in the case of Lmna LAO/LAO mice or full-length prelamin A in Lmna PLAO/PLAO mice). The Lmna LAO/LAO and Lmna PLAO/PLAO mice are healthy and free of pathology. Misshapen nuclei can be observed in cultured Lmna LAO/LAO fibroblasts but were never observed in the tissues of Lmna LAO/LAO mice [21].
Prelamin A contains a CaaX motif at its carboxyl terminus (Fig. 1), which triggers three sequential posttranslational CaaX processing steps: farnesylation of a carboxyl-terminal cysteine (the C of the CaaX motif), cleavage of the last three amino acids of the protein (i.e., the –aaX), and carboxyl methylation of the newly exposed farnesylcysteine [23, 24]. Prelamin A undergoes one additional processing step: cleavage of the last 15 amino acids of the protein, including the carboxyl-terminal farnesylcysteine methyl ester. This final step, carried out by the metalloprotease ZMPSTE24, releases mature lamin A. Lamin C does not have a CaaX motif and does not undergo any of these posttranslational processing steps.
Protein farnesylation and methylation have been assumed to be important for the association of the nuclear lamins with the inner nuclear membrane. This notion has been supported by in vitro evidence [25], but the in vivo relevance of these modifications remained uncertain until the development of Lmna LAO/LAO mice [21]. Lmna LAO/LAO mice, which produce mature lamin A directly — completely bypassing prelamin A synthesis and processing — are phenotypically normal. The levels of mature lamin A in cells and tissues are normal, and there is no significant defect in the ability of lamin A to reach the nuclear rim. Thus, the posttranslational processing of prelamin A is quite dispensable, at least in laboratory mice.
While the posttranslational processing of prelamin A appears to be dispensable, it is important to emphasize that defects in prelamin A processing can elicit severe disease. For example, ZMPSTE24 deficiency abolishes the final endoproteolytic cleavage event in lamin A biogenesis, leading to an accumulation of farnesyl–prelamin A in cells and progeria-like disease phenotypes — both in humans and in mice [26–29]. These disease phenotypes can be eliminated by reducing prelamin A synthesis with a single Lmna knockout allele [30] or a single Lmna LCO allele [20], clearly demonstrating that farnesyl-prelamin A was responsible for disease. Also, the classic progeroid syndrome of children, Hutchinson–Gilford progeria syndrome (HGPS), is caused by a LMNA mutation that prevents the conversion of farnesyl–prelamin A to mature lamin A [31, 32]. Finally, abolishing all prelamin A processing steps leads to cardiomyopathy, as illustrated by a gene-targeted mouse model by Davies et al. [22]. They created a “nonfarnesylated prelamin A–only” mouse (Lmna nPLAO/nPLAO) by replacing the cysteine in prelamin A’s CaaX motif with a serine. In Lmna nPLAO/nPLAO mice, prelamin A does not undergo farnesylation, nor does it undergo any of the subsequent posttranslational processing steps. The nonfarnesylated prelamin A in Lmna nPLAO/nPLAO mice is targeted quite normally to the nuclear rim, indistinguishable from mature lamin A in wild-type mice, but the Lmna nPLAO/nPLAO mice develop a dilated cardiomyopathy and die by 6–8 months of age [22].
Physiologic Importance of B-Type Lamins in Mammalian Cells
The B-type lamins have been assumed, based on in vitro studies [33–37], to be essential in eukaryotic cells, with unique and vital roles in DNA replication, the formation of the mitotic spindle, gene transcription, and a variety of other processes in the cell nucleus. However, until recently, the importance of B-type lamins had never been tested in vivo with appropriate mouse models. Yang et al. [38, 39] generated Lmnb1 and Lmnb2 conditional knockout alleles, making it possible to eliminate B-type lamins in specific cell types. Remarkably, the loss of both lamin B1 and B2 in skin keratinocytes has no perceptible effect on keratinocyte growth or on the complex developmental programs involved in the generation of the skin and adnexal structures. Misshapen nuclei can be observed in Lmnb1/Lmnb2–deficient keratinocytes in culture, but misshapen nuclei were never found within the skin of the keratinocyte-specific Lmnb1/Lmnb2–deficient mice [38]. Similarly, the loss of B-type lamins has no adverse effects on liver hepatocytes [39]. These studies suggest that B-type lamins are dispensable, at least in some cell types, and raise doubts about whether the B-type lamins truly have unique roles in mitosis, DNA replication, etc. Of note, skin keratinocytes and hepatocytes express A-type lamins; thus, it is possible that the nuclear lamin proteins have redundant roles in the cell nucleus and the A-type lamins are able to compensate for the loss of both B-type lamins.
Kim et al. [40] recently showed that B-type lamins are dispensable in embryonic stem cells. The authors derived ES cells lacking both Lmnb1 and Lmnb2 and found that the cells grew normally and could be differentiated to trophectoderm cells with an efficiency similar to that of wild-type ES cells. Lamin A and lamin C were undetectable in undifferentiated ES cells by routine western blots or conventional RT-PCR [40]. However, in our hands, low but easily detectable levels of Lmna transcripts are present in pluripotent undifferentiated ES cells, as judged by quantitative real-time RT-PCR (unpublished observations); hence, it remains possible that low levels of A-type lamins in ES cells compensate for the absence of B-type lamins.
Both lamin B1 and lamin B2 terminate with a CaaX motif (Fig. 1) and undergo farnesylation, endoproteolytic cleavage of the last three amino acids, and methylation of the farnesylcysteine. Unlike prelamin A, however, there is no additional endoproteolytic cleavage step; thus, the B-type lamins retain their carboxyl-terminal farnesylcysteine methyl ester. For both lamin B1 and lamin B2, the physiological relevance of the carboxyl-terminal posttranslational processing events is uncertain and needs to be investigated by creating knock-in mice that express nonfarnesylated versions of these proteins.
A Role for B-Type Lamins in Brain Development
The essential role of B-type lamins in the developing brain was first uncovered by Coffinier et al. [41] during the characterization of lamin B2-deficient mice (Lmnb2 −/−). Lmnb2 −/− mice die at birth; however, newborn Lmnb2 −/− mice are normal in size and there are no histopathological abnormalities in any organs, except for the brain. The brain in Lmnb2 −/− mice is slightly smaller than normal, and there is a striking defect in the layering of neurons within the cerebral cortex. Subsequent studies of Lmnb2 −/− embryos revealed a defect in the migration of neurons from the ventricular zone to the cortical plate. The neuronal migration defect was documented both by BrdU birthdating experiments and immunohistochemical studies with cortical layer–specific markers [41]. The development of the cerebellum is also profoundly abnormal in Lmnb2 −/− embryos, with a complete absence of foliation and an absence of a discrete Purkinje cell layer. The layering of neurons in the hippocampus is also perturbed in Lmnb2 −/− embryos [41].
Occasional neurons in the cortical plate of Lmnb2 −/− embryos have “comet-shaped” nuclei with detached centrosomes (located >20 μm from the cell nuclei) (Fig. 2a) [42]. These “stretched out” nuclei were never observed in wild-type brains. Lamin B1 remains uniformly distributed at the nuclear rim in Lmnb2 −/− neurons.

Brain abnormalities in Lmnb1 and Lmnb2 knockout mice. a Immunostaining of cerebral cortex from wild-type (Lmnb2 +/+) and Lmnb2 knockout embryos (Lmnb2 −/−) at E16.5 with an antibody against lamin B1 (green). Arrowheads indicate “comet-shaped” nuclei. b Immunostaining of cerebral cortex from Lmnb1 +/+ and Lmnb1 knockout embryos (Lmnb1 Δ/Δ) at E16.5 with an antibody against lamin B2 (green). Arrowheads indicate solitary blebs and asymmetric distribution of lamin B2. Scale bars, a and b, 25μm c Hematoxylin and eosin staining of coronal brain section from control mouse (Lmnb1 fl/fl Lmnb2 fl/fl) and coronal (middle) and sagittal (bottom) brain sections from forebrain-specific Lmnb1/Lmnb2 double knockout mice (Emx1-Cre Lmnb1 fl/fl Lmnb2 fl/fl). In the double knockout brain, the hippocampus and the cortex are nearly absent. Reproduced with permission from Coffinier et al. [42]
We proposed that the “stretched out” nuclei in Lmnb2 −/− neurons are likely a consequence of the deformational stresses associated with nucleokinesis during neuronal migration [43, 44]. A crucial step in neuronal migration is pulling the cell nucleus forward (by cytoplasmic motors) toward the centrosome in the direction of the leading edge of the cell [43, 44]. We suspect that the structural integrity of the nuclear envelope is compromised by the absence of lamin B2 (particularly since there is little or no Lmna expression in neurons during development) [42]. We have suggested that the same forces that are meant to pull the nucleus forward are responsible for “stretching out” the nucleus (creating the distinctive comet-shaped nucleus) in Lmnb2 −/− neurons [42–44].
Lmnb2 −/− embryonic fibroblasts exhibit few abnormalities; Lmnb2 −/− fibroblasts grow normally and do not exhibit aneuploidy [41]. Staining of Lmnb2 −/− embryonic fibroblasts with antibodies against several nuclear envelope proteins did not uncover abnormalities in nuclear shape [41].
After uncovering a role for lamin B2 in the developing brain, Coffinier and coworkers [42] were interested in ascertaining the importance of lamin B1 in brain development. In approaching this problem, they built on earlier studies by Vergnes et al. [45], who had generated lamin B1–deficient mice (Lmnb1 Δ/Δ) with a gene-trap ES cell clone. Lmnb1 Δ/Δ mice synthesize a lamin B1–βgeo fusion protein that lacks a large fraction of the lamin B1 coding sequences, including two known phosphorylation sites, the nuclear localization signal, and the carboxyl-terminal CaaX motif [45]. Lmnb1 Δ/Δ mice were abnormally small during development and died shortly after birth, with abnormalities in lung and bones [45]. In the work by Vergnes and coworkers [45], the possibility of neuropathology was not specifically investigated, but the authors did note that the calvarium in Lmnb1 Δ/Δ mice was abnormally flattened and that these mice had abnormalities in the cranial sutures. Interestingly, Lmnb1 Δ/Δ embryonic fibroblasts in culture had misshapen nuclei and exhibited a reduced replication rate, polyploidy, and premature senescence [45].
When Coffinier et al. [42] examined the brains of Lmnb1 Δ/Δ embryos, they found that the characteristic layering of neurons in the cerebral cortex is markedly abnormal. Using a combination of BrdU birthdating and immunohistochemistry studies, they showed that the neuronal layering defect is due to defective neuronal migration. They also found that the cortical plate of Lmnb1 Δ/Δ embryos is abnormally thin, with fewer neuronal progenitor cells and more apoptotic cells, implying a defect in neuronal survival. The cerebellum of Lmnb1 Δ/Δ embryos is small and devoid of foliation.
In Lmnb1 Δ/Δ embryos, many of the neurons in the cortical plate contain a solitary nuclear bleb. Also, lamin B2 is asymmetrically distributed at the nuclear rim, with a large fraction of the lamin B2 being located at the rim of the nuclear bleb (Fig. 2b) [42]. The authors suggested that the solitary bleb might be the consequence of the deformational forces that occur with nucleokinesis during neuronal migration [42]. We have observed the same “solitary nuclear blebs” in cells of peripheral tissues of Lmnb1 Δ/Δ embryos (unpublished observations), raising the possibility that deformational forces on cell nuclei could exist in peripheral tissues as well.
To investigate the role of the B-type lamins in the postnatal brain, Coffinier et al. [42] took advantage of a forebrain-specific Cre transgene (Emx1-Cre) [46] and conditional knockout alleles for Lmnb1 and Lmnb2 [38] to create forebrain-specific Lmnb1 and Lmnb2 knockout mice (Emx1-Cre Lmnb1 fl/fl and Emx1-Cre Lmnb2 fl/fl, respectively). Immunohistochemical studies showed that the knockout mutations worked as planned; ~90 % of the cells in the cortical plate of Emx1-Cre Lmnb1 fl/fl and Emx1-Cre Lmnb2 fl/fl embryos have no lamin B1 or lamin B2, respectively. Both forebrain-specific knockout mouse strains are viable, and by 4 months of age, Emx1-Cre Lmnb1 fl/fl and Emx1-Cre Lmnb2 fl/fl mice exhibit a markedly smaller forebrain, suggesting that the postnatal survival of neurons is impaired (Fig. 2c). Mice lacking both lamin B1 and lamin B2 in the forebrain (Emx1-Cre Lmnb1 fl/fl Lmnb2 fl/fl) were also generated. In Emx1-Cre Lmnb1 fl/fl Lmnb2 fl/fl embryos, it is possible to identify neurons lacking both lamin B1 and lamin B2. However, in adult Emx1-Cre Lmnb1 fl/fl Lmnb2 fl/fl mice, the forebrain is completely atrophic (Fig. 2c); the thin layer of tissue representing the forebrain does not contain neurons, nor does it contain any cells lacking lamins B1 and B2. Thus, it would appear that neurons in the adult brain do not survive in the absence of both B-type lamins.
Misshapen cell nuclei are common in forebrain neurons of Emx1-Cre Lmnb1 fl/fl and Emx1-Cre Lmnb2 fl/fl embryos. Many neurons in Emx1-Cre Lmnb1 fl/fl embryos contain a solitary nuclear bleb, and comet-shaped nuclei are observed in neurons of Emx1-Cre Lmnb2 fl/fl embryos. Interestingly, however, few of the neurons in the forebrain of adult Emx1-Cre Lmnb1 fl/fl and Emx1-Cre Lmnb2 fl/fl mice are misshapen, perhaps because neurons of the adult brain (unlike the neurons of embryos) express lamin C [42, 47].
In a recent study, Kim et al. [40] suggested that B-type lamins might have redundant roles in interkinetic nuclear migration. They found that neuronal progenitor cells containing phospho-histone H3 (a marker for G2/M phase cells) in the brains of Lmnb1 and Lmnb2 single knockout mice (at E14.5) are located normally in the apical portion of ventricular zone, whereas the neuronal progenitor cells in the brains of Lmnb1/Lmnb2 double knockout mice are scattered along the basal side of ventricular zone.
At this point, we know that both lamin B1 and lamin B2 are crucial for brain development and postnatal neuron survival [41, 42]. We also know that the neuropathology in the lamin B2-deficient mice is distinct from that in lamin B1-deficient mice [41, 42]. Lamin B1 deficiency has a greater impact on the size and cellularity of the cortical plate than lamin B2 deficiency [42]. Also, the nuclear shape abnormalities in lamin B1- and lamin B2-deficient neurons are distinct. However, these observations do not provide clear-cut insights into whether lamin B1 and lamin B2 have functionally redundant roles in the development of the brain; additional knock-in mouse models will be required to address this issue. For example, it would be informative to generate a mutant mouse in which the lamin B1 coding sequences are “knocked in” to the Lmnb2 locus, thereby replacing the production of lamin B2 with lamin B1. In these mice, it would be possible to ask: Does the production of lamin B1 from the Lmnb2 locus prevent the neuropathology that is normally associated with lamin B2 deficiency? If the answer is “no,” that would imply that the two B-type lamins have distinct and nonredundant roles in brain development.
Recently, Yoon et al. [48] found that lamin B2, but not lamin A or lamin B1, is translated in Xenopus retinal ganglion cell (RGC) axons and that association of exonuclear lamin B2 with mitochondria is essential for RGC axon maintenance. The role of lamin B2, if any, in axon maintenance in mammals is unknown and should be investigated.
Regulation of the A-Type Lamins
LMNA expression is developmentally regulated; high levels of LMNA expression appear late in embryonic development and in differentiated cells. For this reason, the expression of lamins A and C has been used as a marker of stem cell differentiation [5, 49]. However, surprisingly little is known about mechanisms controlling the expression of LMNA during development. Lin et al. [50] observed that the proximal promoter of LMNA drives transcription in many cell types, including Burkitt lymphoma Raji cells (where A-type lamins are not expressed). These findings suggested that the LMNA promoter might be active in all cells and that distinct regulatory elements serve to silence LMNA expression in undifferentiated cells. Later, Tiwari et al. [51] showed that AP1 and SP1 sites in the 5′ promoter of rat Lmna are sufficient for the active transcription, and suggested that conformational changes in chromatin might underlie cell-type specific repression of Lmna expression.
DNase I hypersensitive sites, indicators of open chromatin structure, have been observed in the first intron of mouse Lmna [52]. When reporter constructs containing cell-type specific Lmna DNase I hypersensitive sites were stably integrated into the chromosomal DNA, transcription increased [52]. The methylation status of a CpG island in the LMNA promoter also appears to correlate with inactivation of LMNA in the setting of hematologic malignancies [53]. Agrelo et al. [53] found hypermethylation of a promoter CpG island of LMNA (as judged by bisulfite sequencing) in tumor cells from leukemias and lymphomas patients, and they suggested that methylation could be responsible for the minimal expression of A-type lamins in those cells. Thus, the expression of A-type lamins may depend, at least in part, on epigenetic regulation of LMNA.
The fact that the expression of A-type lamins is activated late in development has prompted speculation that lamins A and C could play unique roles in differentiated cells [5, 6]. However, it is also conceivable that the expression of A-type lamins is suppressed in undifferentiated cells simply because these lamins are deleterious for embryonic development. At least in Drosophila, ectopic expression of lamin C leads to stage-specific lethality [54]. It would be worthwhile determining if ectopic expression of lamin A/C in early phases of development adversely affects mammalian development. For example, it would be interesting to express A-type lamins in undifferentiated cells at early stages of development by “knocking in” lamin A coding sequences into the Lmnb1 locus. If mouse embryos heterozygous for this sort of knock-in allele failed to develop normally, it would imply that lamin A expression adversely affects embryonic development.
Regulation of Lamin A Expression in the Brain by a MicroRNA
The health and vitality of Lmna LCO/LCO [20], Lmna LAO/LAO [21], and Lmna PLAO/PLAO [22] have demonstrated that the loss of one of the two Lmna isoforms does not cause significant pathology. However, the vitality of these “single isoform” mouse models should not be interpreted as proof that the two isoforms lack unique functions. The preservation of the two protein isoforms during mammalian evolution represents strong evidence that the two proteins are important, and we are confident that unique functions for the two isoforms will eventually be uncovered.
Another reason to believe that the two protein isoforms have distinct properties comes from recent studies by Jung et al. [47], showing that the relative amounts of lamin A and lamin C can be quite different in different tissues. In most peripheral tissues, the levels of lamin A and C are similar, but the situation is quite different in the brain. By performing western blots of protein extracts from different tissues, Jung et al. [47] showed that the expression of lamin A is far lower in the central nervous system than in peripheral tissues, while the level of lamin C expression is similar (Fig. 3a). By immunohistochemistry, lamin A expression is nearly absent in neurons and glia of the mouse brain, and the only cells that produce significant amounts of lamin A within the brain are capillary endothelial cells and meningeal cells (Fig. 3b). Lamin C is expressed in all cell types within the adult brain. Similar findings were observed with Zmpste24-deficient mice, where the conversion of farnesyl–prelamin A to mature lamin A is blocked. The brains of those mice contain abundant amounts of lamin C but almost no prelamin A — except in capillary endothelial cells and the meninges. Studies of lamin C and prelamin A transcript levels yielded concordant results: lamin C transcript levels in the brain are high, while prelamin A transcript levels are low.

Exclusive expression of lamin C in the mouse brain and downregulation of lamin A expression by miR-9, a brain-specific microRNA. a Western blot of tissue extracts from a WT (Lmna +/+) mouse and a Lmna knockout (Lmna −/−) mouse. Lamins A and C are expressed highly in the peripheral tissues (liver, heart, and kidney) of wild-type mice, whereas the brain (cerebellum and cortex) expresses mainly lamin C and little lamin A. b Immunostaining of the liver and the cerebellum from WT mouse with antibodies against lamin A (red) and lamin C (green). DNA was stained with DAPI (blue). The liver expresses both lamin A and lamin C, whereas expression of lamin A in the brain is largely restricted to vascular endothelium cells. Neurons in the granular layers of cerebellum expresses lamin C. c Reduced expression of lamin A in HeLa cells transfected with a miR-9 expression vector. Lamin C expression was unaffected. Transfected cells were readily identified by GFP expression (arrowheads). Expression of lamin A was not affected when cells were transfected with a control expression vector. Reproduced with permission from Jung et al. [47]
To understand the differential expression of lamin A and lamin C in the brain, Jung et al. [47] first examined the most likely explanation — a distinct pattern of mRNA splicing in the brain. They reasoned that if alternative splicing were the mechanism, then lamin A expression should be abundant in the central nervous system of lamin A-only mice (Lmna LAO/LAO) [21] — where alternative splicing is absent and all of the output of the Lmna gene is channeled into prelamin A transcripts. But this was not the case! The amount of lamin A in the central nervous system of Lmna LAO/LAO mice is extremely low, as judged by western blots analysis. Also, by immunohistochemistry, lamin A can be found in capillary endothelial cells within the brain of Lmna LAO/LAO mice but is almost absent in neurons and glia. The same sorts of findings are observed in gene-targeted mice carrying a mutant Lmna allele that yields progerin, the mutant prelamin A found in patients with HGPS. In that mouse model, high levels of progerin are found in peripheral tissues, but very little is found in the brain. Both of these mouse models show that the low levels of lamin A in the brain cannot be due to alternative splicing and instead must be due to another posttranscriptional mechanism.
Next, Jung et al. [47] examined prelamin A’s 3′ UTR and identified a putative binding site for miR-9, a brain specific microRNA [55–58]. To determine if miR-9 might downregulate lamin A expression, they expressed miR-9 in fibroblasts and HeLa cells. The expression of miR-9 reduced lamin A expression but had no effect on lamin C expression (Fig. 3c). They [47] also showed that when the putative miR-9 binding motif in prelamin A’s 3′ UTR was mutated or deleted, miR-9 no longer downregulated prelamin A expression. Moreover, when the critical RNA-binding motif in the miR-9 expression vector was mutated, the ability of the mutant miR-9 to downregulate prelamin A expression was abolished [47]. Finally, Jung et al. [47] reported circumstantial evidence suggesting that microRNAs play a significant role in regulating lamin A expression in vivo. They generated forebrain-specific Dicer knockout mice [59, 60] (DICER is essential for the synthesis of microRNAs). As predicted, the level of lamin A protein in the forebrain-specific Dicer knockout mice increased in the forebrain but remained low in the cerebellum (where DICER is expressed normally) [47].
Reduced expression of lamin A in central nervous system neurons is not a peculiarity of the mouse. In a recent study, Wakabayashi et al. [61] examined the retina of the rat and found that retinal neurons express exclusively lamin C, while some of the surrounding cell types express both lamin A and lamin C. They speculated that the exclusive expression of lamin C in retinal neurons could be relevant to neuronal differentiation or gene expression.
Jung et al. [47] proposed that the elimination of prelamin A transcripts by miR-9 could explain why children with HGPS are spared from central nervous system disease. This explanation is both attractive and plausible, particularly since progerin levels are extremely low in the brains of Lmna knock-in mice expressing progerin [47]. However, one could argue that the brain might be spared from disease — even if it were to produce large amounts of progerin. Children with HGPS exhibit little or no pathology in the liver or kidney, even though those organs produce large amounts of progerin. Whether progerin expression is toxic to the brain needs to be tested with knock-in mice that produce progerin, rather than lamin C, in the brain.
In a recent paper, Nissan and coworkers [62] confirmed that miR-9 expression downregulates prelamin A expression in cultured cells, including neurons derived from induced pluripotent stem cells. When miR-9 was expressed in HGPS mesenchymal stem cells, progerin levels fell (relative to lamin C), and the percentage of cells with misshapen nuclei was slightly lower [62].
The studies by Jung et al. [47] and the subsequent studies by Nissan et al. [62] are certainly consistent with the idea that miR-9 is a key regulator of prelamin A/lamin A expression in the brain. However, it is important to remember that this conclusion has been supported mainly by in vitro studies in cultured cells. Also, the forebrain-specific Dicer knockout observations, while suggesting a role for microRNAs in prelamin A regulation, did not prove that miR-9 is solely responsible for prelamin A regulation. Mice lacking both miR-9-2 and mir-9-3 were recently characterized; these mice have severe neurodevelopmental abnormalities and die within 1 week [63]. Unfortunately, the expression of lamin A in these mice was not investigated. However, even if miR-9-2/3 knockout mice had elevated levels of prelamin A transcripts and lamin A protein in the brain, the interpretation would not be straightforward, given that miR-9 targets many other genes, and some of those changes might lead to secondary changes in lamin A expression.
At this point, it would be highly desirable to test the significance of miR-9 with Lmna knock-in mice with a mutation in the miR-9 binding site in prelamin A’s 3′ UTR. The analysis of such a mouse model should reveal whether miR-9 is the only relevant factor in prelamin A regulation, or whether other factors are also involved.
The discovery that lamin A expression in the brain is regulated by a microRNA raises a number of issues for future research. One is whether the expression of lamin C in the brain serves an important function during postnatal life. The Lmna LAO/LAO and Lmna PLAO/PLAO mice have negligible levels of lamin A expression in neurons and glia yet have no obvious pathology in the brain. However, it is possible that in-depth characterization of those mice would uncover behavioral and cognitive abnormalities. Another issue is why the mouse, and apparently the rat also, have evolved a mechanism to limit the expression of lamin A in the brain. Would the expression of lamin A in neurons and glia, rather than lamin C, be associated with neuropathology or defects in cognition? To address that issue, it would be necessary to generate a mouse model where neurons produce lamin A. Presumably, one could accomplish this goal by creating Lmna knock-in mice where prelamin A’s 3′ UTR was replaced with lamin C’s 3′ UTR. A final issue is whether robust downregulation of prelamin A transcripts occurs in the human brain. Indirect observations suggest that this could be the case: the miR-9 binding site is conserved in the human LMNA gene, and the levels of lamin A are far lower than the levels of lamin C in neuronal cells derived from human induced pluripotent stem cells [64]. In the future, it would be very interesting to assess lamin A and lamin C expression, along with miR-9 expression levels, in the brains of human fetuses, infants, children, and adults of various ages.
Nuclear Lamins and Human Disease
A-type lamins have been linked to multiple human genetic diseases, including cardiomyopathy, several forms of muscular dystrophy, peripheral neuropathy, partial lipodystrophy, mandibuloacral dysplasia, and HGPS [2, 3, 19]. Thus far, more than 200 clinically significant missense, nonsense, frameshift, and splicing mutations in LMNA have been described (http://www.umd.be) [65]. The “A-type lamin diseases” largely affect mesenchymal tissues, and none of them, as far as we are aware, causes primary disease within the central nervous system.
The sole bona fide link of B-type lamins to human disease is an adult-onset autosomal dominant leukodystrophy (ADLD), which is caused by a duplication of LMNB1 [66–68]. This demyelinating disorder, which is sometimes mistaken for a progressive form of multiple sclerosis, was first identified in 1984 in a large American-Irish family but has since been found in multiple kindreds around the world [69]. The disease generally presents in middle-aged years with autonomic dysfunction, followed by the development of progressive cerebellar and pyramidal tract abnormalities [70]. MRI scans show widespread symmetrical demyelination in the white matter, beginning in the frontoparietal area and extending to the brain stem and cerebellum [71–73]. Light microscopy shows vacuolization of the white matter with preservation of oligodendrocytes, and no evidence of inflammation. Abnormalities of the neurons or axons are minimal or absent [72, 73]. The locus for this disease was mapped to the long arm of chromosome 5 (near 5q31) by the laboratory of Dr. Ying-Hui Fu at the University of California, San Francisco. Later, the same group showed that ADLD is caused by a duplication of LMNB1 on chromosome 5 [66]. The size of the duplicated stretch of genomic DNA is variable in different kindreds. Increased expression of lamin B1 in ADLD patients has been documented both in the brain [66] and in peripheral blood leukocytes [74].
In one family with ADLD but without autonomic disease, the disease locus was mapped to 5q23.2–q23.3, which contains 11 genes including LMNB1 [75]. No mutations were found in the coding regions of these genes, nor were there any gene duplications; nevertheless, the expression of lamin B1 in lymphoblastoid cell lines from affected patients was higher than in cell lines from healthy controls. The authors speculated that the ADLD in this kindred might be due to a mutation in a regulatory element that leads to increased LMNB1 expression [75].
Thus far, no mouse model of ADLD has been generated. However, important clues regarding disease pathogenesis have been uncovered with cell culture studies. Overexpression of lamin B1 in neuronal, astrocytic, and oligodendrocytic cell lines results in misshapen cell nuclei, and fibroblasts that overexpressed lamin B1 have abnormalities in the localization of heterochromatin protein 1β and methylated histone 3 [76]. Also, overexpression of LMNB1 in an oligodendrocyte cell line represses transcription of oligodendrocyte-specific genes such as myelin basic protein, proteolipid protein, and myelin oligodendrocyte glycoprotein [76]. Finally, expression of human LMNB1 in the eye of Drosophila leads to degenerative abnormalities in the eye, including pigment loss, roughening of the eye, and reduced eye size [66].
The expression of lamin B1 is downregulated by a microRNA, miR-23, which binds to lamin B1’s 3′ UTR [76]. Therefore, miR-23 has the potential to reverse the adverse effects of lamin B1 on oligodendrocyte gene expression, and indeed the expression of miR-23 did enhance oligodendrocyte differentiation in cultures of glial cells [76]. Interestingly, the expression pattern of miR-23 is the opposite of that of lamin B1. Lamin B1 levels peak late in development and decline in postnatal life; miR-23 levels are low during embryonic development and higher in postnatal life [76].
Currently, it is unclear why overexpression of lamin B1 leads to a demyelinating disease with adverse effects on oligodendrocyte gene expression, whereas a deficiency of lamin B1 leads to defects in neuronal migration and neuronal survival. Presumably, altered expression of lamin B1 changes the composition of the nuclear lamina, leading to secondary changes in gene expression and protein–protein interactions. The consequences of these changes are likely to be distinct in different cell types. In any case, the finding of ADLD with LMNB1 gene duplications [71–73], together with the neuropathology in Lmnb1 and Lmnb2 knockout mice [41, 42], underscores the importance of B-type lamins in brain development and homeostasis.
There have been no reports of LMNB1 or LMNB2 nonsense, frameshift, splicing, or missense mutations. Heterozygosity for Lmnb1 or Lmnb2 knockout mutations does not appear to cause neurodevelopmental abnormalities. Whether half-normal amounts of lamin B1 or lamin B2 synthesis would alter gene expression in neurons or glia later in life has never been evaluated. Given the phenotypes of the knockout mice, homozygous loss of LMNB1 or LMNB2 in humans would likely lead to severe defects in the development of the brain. We predict that homozygous mutations in B-type lamins will eventually be uncovered in human fetuses with defects in neuronal migration and neuronal survival. Neuronal migration abnormalities have also been implicated in the pathogenesis of milder forms of neurological disease, such as epilepsy and autism [77, 78]. We would not be surprised if human geneticists ultimately uncover LMNB1 or LMNB2 missense mutations in patients with familial forms of those diseases.
Perspectives
Recent discoveries on the role of the B-type lamins in the brain [41, 42] open a new window on mechanisms underlying neuronal migration and brain development. Finding a role for nuclear lamins in brain development has raised the possibility that genetic defects in LMNB1 and LMNB2 could cause neurodevelopmental abnormalities in humans. Also, the discovery that prelamin A expression in the brain is regulated by a microRNA [47] suggests that lamins A and C could have distinct functions in the brain.
The study of nuclear lamins in the central nervous system is an exciting new area of research, but our understanding of this topic is still quite limited. To better define the function of nuclear lamins in the brain, more studies with knock-in mouse models are necessary (as has been suggested repeatedly in the course of this review article). Also, we believe that the human genetics of B-type lamins needs more attention. It seems likely, based on the findings in knockout mice [41, 42], that LMNB1 and LMNB2 mutations will eventually be uncovered in humans with neurodevelopmental abnormalities.
References
Melcer S, Gruenbaum Y, Krohne G (2007) Invertebrate lamins. Exp Cell Res 313:2157–2166. doi:10.1016/j.yexcr.2007.03.004
Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ (2006) Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 86:967–1008. doi:10.1152/physrev.00047.2005
Worman HJ (2012) Nuclear lamins and laminopathies. J Pathol 226:316–325. doi:10.1002/path.2999
Dittmer TA, Misteli T (2011) The lamin protein family. Genome Biol 12:222. doi:10.1186/gb-2011-12-5-222
Rober RA, Weber K, Osborn M (1989) Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105:365–378
Lebel S, Lampron C, Royal A, Raymond Y (1987) Lamins A and C appear during retinoic acid-induced differentiation of mouse embryonal carcinoma cells. J Cell Biol 105:1099–1104
Lin F, Worman HJ (1993) Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem 268:16321–16326
Lin F, Worman HJ (1995) Structural organization of the human gene (LMNB1) encoding nuclear lamin B1. Genomics 27:230–236. doi:10.1006/geno.1995.1036
Biamonti G, Giacca M, Perini G, Contreas G, Zentilin L, Weighardt F, Guerra M, Della Valle G, Saccone S, Riva S et al (1992) The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S-phase. Mol Cell Biol 12:3499–3506
Erber A, Riemer D, Hofemeister H, Bovenschulte M, Stick R, Panopoulou G, Lehrach H, Weber K (1999) Characterization of the Hydra lamin and its gene: a molecular phylogeny of metazoan lamins. J Mol Evol 49:260–271. doi:10.1007/PL00006548
Davies BS, Coffinier C, Yang SH, Jung HJ, Fong LG, Young SG (2011) Posttranslational processing of nuclear lamins. In: Tamanoi F, Hrycyna CA, Bergo MO (eds) The enzymes. Elsevier, Amsterdam, pp 21–41
Fisher DZ, Chaudhary N, Blobel G (1986) cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci U S A 83:6450–6454
McKeon FD, Kirschner MW, Caput D (1986) Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 319:463–468. doi:10.1038/319463a0
Heitlinger E, Peter M, Lustig A, Villiger W, Nigg EA, Aebi U (1992) The role of the head and tail domain in lamin structure and assembly: analysis of bacterially expressed chicken lamin A and truncated B2 lamins. J Struct Biol 108:74–89. doi:1047-8477(92)90009-Y
Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B (1999) Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 147:913–920
Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT (2004) Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 113:370–378. doi:10.1172/JCI19670
Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D (1994) Identification of a novel X-linked gene responsible for Emery–Dreifuss muscular dystrophy. Nat Genet 8:323–327. doi:10.1038/ng1294-323
Ellis JA, Craxton M, Yates JR, Kendrick-Jones J (1998) Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery–Dreifuss muscular dystrophy phenotype. J Cell Sci 111(Pt 6):781–792
Worman HJ, Fong LG, Muchir A, Young SG (2009) Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest 119:1825–1836. doi:10.1172/JCI37679
Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, Cote N, Gavino B, Qiao X, Chang SY, Young SR, Yang SH, Stewart CL, Lee RT, Bennett CF, Bergo MO, Young SG (2006) Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest 116:743–752. doi:10.1172/JCI27125
Coffinier C, Jung HJ, Li Z, Nobumori C, Yun UJ, Farber EA, Davies BS, Weinstein MM, Yang SH, Lammerding J, Farahani JN, Bentolila LA, Fong LG, Young SG (2010) Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J Biol Chem 285:20818–20826. doi:10.1074/jbc.M110.128835
Davies BS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG (2010) An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet 19:2682–2694. doi:10.1093/hmg/ddq158
Davies BS, Coffinier C, Yang SH, Barnes RH 2nd, Jung HJ, Young SG, Fong LG (2011) Investigating the purpose of prelamin A processing. Nucleus 2:4–9. doi:10.4161/nucl.2.1.13723
Davies BS, Fong LG, Yang SH, Coffinier C, Young SG (2009) The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet 10:153–174. doi:10.1146/annurev-genom-082908-150150
Hennekes H, Nigg EA (1994) The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J Cell Sci 107(Pt 4):1019–1029
Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG (2002) Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A 99:13049–13054. doi:10.1073/pnas.192460799
Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C (2002) Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 31:94–99. doi:10.1038/ng871
Moulson CL, Go G, Gardner JM, van der Wal AC, Smitt JH, van Hagen JM, Miner JH (2005) Homozygous and compound heterozygous mutations in ZMPSTE24 cause the laminopathy restrictive dermopathy. J Invest Dermatol 125:913–919. doi:10.1111/j.0022-202X.2005.23846.x
Navarro CL, Cadinanos J, De Sandre-Giovannoli A, Bernard R, Courrier S, Boccaccio I, Boyer A, Kleijer WJ, Wagner A, Giuliano F, Beemer FA, Freije JM, Cau P, Hennekam RC, Lopez-Otin C, Badens C, Levy N (2005) Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet 14:1503–1513. doi:10.1093/hmg/ddi159
Fong LG, Ng JK, Meta M, Cote N, Yang SH, Stewart CL, Sullivan T, Burghardt A, Majumdar S, Reue K, Bergo MO, Young SG (2004) Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci U S A 101:18111–18116. doi:10.1073/pnas.0408558102
De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N (2003) Lamin a truncation in Hutchinson–Gilford progeria. Science 300:2055. doi:10.1126/science.1084125
Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS (2003) Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 423:293–298. doi:10.1038/nature01629
Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 114:4557–4565
Belmont AS, Zhai Y, Thilenius A (1993) Lamin B distribution and association with peripheral chromatin revealed by optical sectioning and electron microscopy tomography. J Cell Biol 123:1671–1685
Tsai MY, Wang S, Heidinger JM, Shumaker DK, Adam SA, Goldman RD, Zheng Y (2006) A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science 311:1887–1893. doi:10.1126/science.1122771
Moir RD, Montag-Lowy M, Goldman RD (1994) Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol 125:1201–1212
Tang CW, Maya-Mendoza A, Martin C, Zeng K, Chen S, Feret D, Wilson SA, Jackson DA (2008) The integrity of a lamin-B1-dependent nucleoskeleton is a fundamental determinant of RNA synthesis in human cells. J Cell Sci 121:1014–1024. doi:10.1242/jcs.020982
Yang SH, Chang SY, Yin L, Tu Y, Hu Y, Yoshinaga Y, de Jong PJ, Fong LG, Young SG (2011) An absence of both lamin B1 and lamin B2 in keratinocytes has no effect on cell proliferation or the development of skin and hair. Hum Mol Genet 20:3537–3544. doi:10.1093/hmg/ddr266
Yang SH, Jung HJ, Coffinier C, Fong LG, Young SG (2011) Are B-type lamins essential in all mammalian cells? Nucleus 2:562–569. doi:10.4161/nucl.2.6.18085
Kim Y, Sharov AA, McDole K, Cheng M, Hao H, Fan CM, Gaiano N, Ko MS, Zheng Y (2011) Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 334:1706–1710. doi:10.1126/science.1211222
Coffinier C, Chang SY, Nobumori C, Tu Y, Farber EA, Toth JI, Fong LG, Young SG (2010) Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc Natl Acad Sci U S A 107:5076–5081. doi:10.1073/pnas.0908790107
Coffinier C, Jung HJ, Nobumori C, Chang S, Tu Y, Barnes RH 2nd, Yoshinaga Y, de Jong PJ, Vergnes L, Reue K, Fong LG, Young SG (2011) Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol Biol Cell 22:4683–4693. doi:10.1091/mbc.E11-06-0504
Solecki DJ, Govek EE, Tomoda T, Hatten ME (2006) Neuronal polarity in CNS development. Genes Dev 20:2639–2647. doi:10.1101/gad.1462506
Salina D, Bodoor K, Eckley DM, Schroer TA, Rattner JB, Burke B (2002) Cytoplasmic dynein as a facilitator of nuclear envelope breakdown. Cell 108:97–107. doi:10.1016/S0092-8674(01)00628-6
Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K (2004) Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A 101:10428–10433. doi:10.1073/pnas.0401424101
Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22:6309–6314
Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BS, Yang SH, Barnes RH 2nd, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG (2012) Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A 109:E423–E431. doi:10.1073/pnas.1111780109
Yoon BC, Jung H, Dwivedy A, O'Hare CM, Zivraj KH, Holt CE (2012) Local translation of extranuclear lamin B promotes axon maintenance. Cell 148:752–764. doi:10.1016/j.cell.2011.11.064
Constantinescu D, Gray HL, Sammak PJ, Schatten GP, Csoka AB (2006) Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells 24:177–185. doi:10.1634/stemcells.2004-0159
Lin F, Worman HJ (1997) Expression of nuclear lamins in human tissues and cancer cell lines and transcription from the promoters of the lamin A/C and B1 genes. Exp Cell Res 236:378–384. doi:10.1006/excr.1997.3735
Tiwari B, Muralikrishna B, Parnaik VK (1998) Functional analysis of the 5′ promoter region of the rat lamin A gene. DNA Cell Biol 17:957–965
Nakamachi K, Nakajima N (2000) DNase I hypersensitive sites and transcriptional activation of the lamin A/C gene. Eur J Biochem 267:1416–1422. doi:10.1046/j.1432-1327.2000.01135.x
Agrelo R, Setien F, Espada J, Artiga MJ, Rodriguez M, Perez-Rosado A, Sanchez-Aguilera A, Fraga MF, Piris MA, Esteller M (2005) Inactivation of the lamin A/C gene by CpG island promoter hypermethylation in hematologic malignancies, and its association with poor survival in nodal diffuse large B-cell lymphoma. J Clin Oncol 23:3940–3947. doi:10.1200/JCO.2005.11.650
Stuurman N, Delbecque JP, Callaerts P, Aebi U (1999) Ectopic overexpression of Drosophila lamin C is stage-specific lethal. Exp Cell Res 248:350–357. doi:10.1006/excr.1999.4396
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T (2002) Identification of tissue-specific microRNAs from mouse. Curr Biol 12:735–739. doi:10.1016/S0960-9822(02)00809-6
Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS (2003) A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 9:1274–1281
Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V (2004) Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 5:R13. doi:10.1186/gb-2004-5-3-r13
Gao FB (2010) Context-dependent functions of specific microRNAs in neuronal development. Neural Dev 5:25. doi:10.1186/1749-8104-5-25
Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ (2005) Characterization of Dicer-deficient murine embryonic stem cells. Proc Natl Acad Sci U S A 102:12135–12140. doi:10.1073/pnas.0505479102
Kawase-Koga Y, Otaegi G, Sun T (2009) Different timings of Dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev Dyn 238:2800–2812. doi:10.1002/dvdy.22109
Wakabayashi T, Mori T, Hirahara Y, Koike T, Kubota Y, Takamori Y, Yamada H (2011) Nuclear lamins are differentially expressed in retinal neurons of the adult rat retina. Histochem Cell Biol 136:427–436. doi:10.1007/s00418-011-0853-8
Nissan X, Biondel S, Navarro C, Maury Y, Denis C, Girard M, Martinat C, De Sandre-Giovannoli A, Levy N, Peschanski M (2012) Unique preservation of neural cells in Hutchinson–Gilford progeria syndrome Is due to the expression of the neural-specific miR-9 microRNA. Cell Reports 1:1–9. doi:10.1016/j.celrep. 2012.05.015
Shibata M, Nakao H, Kiyonari H, Abe T, Aizawa S (2011) MicroRNA-9 regulates neurogenesis in mouse telencephalon by targeting multiple transcription factors. J Neurosci 31:3407–3422. doi:10.1523/JNEUROSCI.5085-10.2011
Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, Stewart CL, Colman A (2011) A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 8:31–45. doi:10.1016/j.stem.2010.12.002
Stewart CL, Kozlov S, Fong LG, Young SG (2007) Mouse models of the laminopathies. Exp Cell Res 313:2144–2156. doi:10.1016/j.yexcr.2007.03.026
Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH (2006) Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 38:1114–1123. doi:10.1038/ng1872
Brussino A, Vaula G, Cagnoli C, Mauro A, Pradotto L, Daniele D, Di Gregorio E, Barberis M, Arduino C, Squadrone S, Abete MC, Migone N, Calabrese O, Brusco A (2009) A novel family with Lamin B1 duplication associated with adult-onset leucoencephalopathy. J Neurol Neurosurg Psychiatry 80:237–240. doi:10.1136/jnnp. 2008.147330
Meijer IA, Simoes-Lopes AA, Laurent S, Katz T, St-Onge J, Verlaan DJ, Dupre N, Thibault M, Mathurin J, Bouchard JP, Rouleau GA (2008) A novel duplication confirms the involvement of 5q23.2 in autosomal dominant leukodystrophy. Arch Neurol 65:1496–1501. doi:10.1001/archneur.65.11.1496
Eldridge R, Anayiotos CP, Schlesinger S, Cowen D, Bever C, Patronas N, McFarland H (1984) Hereditary adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. N Engl J Med 311:948–953
Schwankhaus JD, Katz DA, Eldridge R, Schlesinger S, McFarland H (1994) Clinical and pathological features of an autosomal dominant, adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. Arch Neurol 51:757–766
Bergui M, Bradac GB, Leombruni S, Vaula G, Quattrocolo G (1997) MRI and CT in an autosomal-dominant, adult-onset leukodystrophy. Neuroradiology 39:423–426
Coffeen CM, McKenna CE, Koeppen AH, Plaster NM, Maragakis N, Mihalopoulos J, Schwankhaus JD, Flanigan KM, Gregg RG, Ptacek LJ, Fu YH (2000) Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum Mol Genet 9:787–793. doi:10.1093/hmg/9.5.787
Melberg A, Hallberg L, Kalimo H, Raininko R (2006) MR characteristics and neuropathology in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. AJNR Am J Neuroradiol 27:904–911
Schuster J, Sundblom J, Thuresson AC, Hassin-Baer S, Klopstock T, Dichgans M, Cohen OS, Raininko R, Melberg A, Dahl N (2011) Genomic duplications mediate overexpression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms. Neurogenetics 12:65–72. doi:10.1007/s10048-010-0269-y
Brussino A, Vaula G, Cagnoli C, Panza E, Seri M, Di Gregorio E, Scappaticci S, Camanini S, Daniele D, Bradac GB, Pinessi L, Cavalieri S, Grosso E, Migone N, Brusco A (2010) A family with autosomal dominant leukodystrophy linked to 5q23.2–q23.3 without lamin B1 mutations. Eur J Neurol 17:541–549. doi:10.1111/j.1468-1331.2009.02844.x
Lin ST, Fu YH (2009) miR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis Model Mech 2:178–188. doi:10.1242/dmm.001065
Deutsch SI, Burket JA, Katz E (2010) Does subtle disturbance of neuronal migration contribute to schizophrenia and other neurodevelopmental disorders? Potential genetic mechanisms with possible treatment implications. Eur Neuropsychopharmacol 20:281–287. doi:10.1016/j.euroneuro.2010.02.005
Wegiel J, Kuchna I, Nowicki K, Imaki H, Marchi E, Ma SY, Chauhan A, Chauhan V, Bobrowicz TW, de Leon M, Louis LA, Cohen IL, London E, Brown WT, Wisniewski T (2010) The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol 119:755–770. doi:10.1007/s00401-010-0655-4
Acknowledgments
This work was supported by the National Institutes of Health Grants HL86683 (LGF), HL089781 (LGF), HL76839 (SGY), and AG035626-09 (SGY).
Conflict of interest statement
None declared.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Jung, HJ., Lee, J.M., Yang, S.H. et al. Nuclear Lamins in the Brain — New Insights into Function and Regulation. Mol Neurobiol 47, 290–301 (2013). https://doi.org/10.1007/s12035-012-8350-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-012-8350-1