
Abstract
The intermediate filament A- and B-type lamins are key architectural components of the nuclear lamina, a proteinaceous meshwork that lies underneath the inner nuclear membrane. In the past decade, many different monogenic human diseases have been linked to mutations in various components of the nuclear lamina. Mutations in LMNA (encoding lamin A and C) cause a variety of human diseases, collectively called laminopathies. These include cardiomyopathies, muscular dystrophies, lipodystrophies and progeroid syndromes. In addition, elevated levels of lamin B1, attributable to genomic duplications of the LMNB1 locus, cause adult-onset autosomal dominant leukodystrophy. The molecular mechanism(s) enabling the mutations and perturbations of the nuclear lamina to give rise to such a wide variety of diseases that affect various tissues remains unclear. The composition of the nuclear lamina changes dynamically during development, between cell types and even within the same cell during differentiation and ageing. Here, we discuss the functional and cellular aspects of lamina remodelling and their implications for the tissue-specific nature of laminopathies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Nuclear lamina
The nucleus is the innermost sanctuary of every eukaryotic cell. It is encapsulated by the nuclear envelope, a double membrane structure consisting in the outer and inner nuclear membranes. Transport between the cytoplasm and the nucleus is facilitated by nuclear pore complexes (NPC), multimeric structures that span both layers of the nuclear envelope. The architectural integrity of the nucleus is maintained by the nuclear lamina, a thick (20–50 nm) proteinaceous meshwork that consists in type V intermediate filament proteins: the A- and B-type lamins. Expression of A-type lamins is limited to most somatic lineages, whereas B-type lamins are expressed in pluripotent stem cells and in their differentiated progeny.
A- and B-type lamins have been identified only in metazoans, a taxon including all multicellular animals arising around 700–800 million years ago (Peter et al. 2012; Melcer et al. 2007). Although plants harbour a nuclear lamina that is similar in its organisation and structure to the lamina found in multicellular animals, it is debatable whether its components are lamin analogues (Ciska and Moreno Diaz de la Espina 2013). Nevertheless, even in unicellular eukaryotes such as Dictyostelium or Trypanosomes, a lamina-type structure lines the nuclear periphery, highlighting the importance of this structural component in eukaryotic nuclei (Krüger et al. 2012; Batsios et al. 2012; DuBois et al. 2012). In addition to supporting the structural integrity of the nucleus, the components of the nuclear lamina play fundamental roles in sustaining cellular physiology.
A-type lamins
Lamin A, lamin AΔ10, lamin C and lamin C2 are all generated by alternative splicing of a single transcript from the LMNA gene. A-type lamins are absent in undifferentiated pluripotent stem cells (Constantinescu et al. 2006) but are ubiquitously expressed in most somatic lineages, in particular those of mesenchymal origin (Röber et al. 1989; Broers et al. 1997). Exceptions to this rule are the various neuronal and hematopoietic lineages in which A-type lamins can be expressed at low, or undetectable, levels (Röber et al. 1990; Jung et al. 2012). Lamin AΔ10 has been detected at low levels in cancer cells (Machiels et al. 1996), whereas the expression of lamin C2 is restricted to germ cells (Furukawa et al. 1994). In human fibroblasts, both lamin A and C are expressed at roughly equal levels but the relative amount of each isoform can vary greatly between tissues. For instance, neuronal lineages express little to no lamin A, whereas lamin C can readily be detected (Jung et al. 2012).
Lamin A (but not lamin C) undergoes extensive post-translational processing: the C-terminal CaaX motif of lamin A is farnesylated, cleaved, methylated and finally cleaved again to form the mature form of lamin A. Deletion of the protease involved in lamin A processing or mutations impairing the cleavage site can lead to the accumulation of permanently farnesylated, uncleaved pre-lamin A, and have been associated with restrictive dermopathy (RD) and Hutchinson-Gilford progeria syndrome (HGPS), respectively (see below).
B-type lamins
B-type lamins are subjected to similar post-translational processing. Both lamin B1 and B2 are farnesylated and cleaved at the –aaX motif but, in contrast to lamin A, B-type lamins undergo no further cleavage. Furthermore, lamin B1 and B2 are expressed in all cell types including embryonic stem cells (ESC), whereas lamin A/C expression is restricted to somatic lineages. Lamin B3 is a minor splice variant of lamin B2 and is exclusively expressed in male germ cells (Furukawa and Hotta 1993). B-type lamins are involved in various cellular processes, including DNA replication (Moir et al. 1994), cell cycle progression, chromatin remodelling and chromosome organisation (Moir et al. 1994; Guelen et al. 2008; Solovei et al. 2013), mitotic spindle assembly (Tsai et al. 2006; Kim et al. 2011) and, as most recently found, senescence and ageing (Dreesen et al. 2013a, 2013b; Shimi et al. 2011; Freund et al. 2012; Shah et al. 2013; Sadaie et al. 2013).
Diseases associated with mutations in nuclear lamina proteins
The number of known mutations in components of the nuclear lamina and lamina-associated factors is currently well over 400, resulting in a multitude of different diseases collectively termed laminopathies. Although partial overlaps occur in the clinical symptoms of the various laminopathies, they are extremely heterogeneous. This is particularly striking as many of them are caused by single point mutations in the LMNA gene. Mutations in LMNA can cause various muscular dystrophies (Emery-Dreifuss muscular dystrophy [EDMD], limb girdle muscular dystrophy [LGMD]), as well as lipodystrophy (familial partial lipodystrophy [FPLD]), dilated cardiomyopathy (DCM), neuropathy (Charcot-Marie-Tooth [CMT], autosomal dominant leukodystrophy [ADLD]), skin pathology (restrictive dermopathy [RD]), bone disease (mandibuloacral dysplasia [MAD]) and accelerated ageing/progeroid syndromes (Hutchinson Gilford progeria syndrome [HGPS] and atypical Werner syndrome) (Worman and Bonne 2007; Burke and Stewart 2014).
Mutations in LMNA: HGPS
Arguably, one of the best-studied laminopathy is the early-onset accelerated ageing syndrome HGPS, initially described by Jonathan Hutchinson and Hastings Gilford in 1886-87. Children with progeria appear normal at birth but start to develop symptoms including thinning of the skin and alopecia after 2–3 years of age, and die in their mid-teens because of cardiovascular failure (De Sandre-Giovannoli et al. 2003; Eriksson et al. 2003). HGPS is caused by a de novo autosomal dominant mutation in LMNA (c.1824C→T) giving rise to an aberrantly spliced, truncated form of lamin A, called progerin. At the cellular level, the most obvious phenotype of HGPS is the presence of morphologically abnormal nuclei (Fig. 1; Goldman et al. 2004; Eriksson et al. 2003). Nuclear architecture defects can be corrected by introducing modified oligonucleotides that target the activated cryptic splice site, prevent aberrant splicing and thereby suppress expression of the mutant protein (Scaffidi and Misteli 2005). Nuclear shape can also be restored by treating HGPS-derived cells with farnesyltransferase inhibitors (FTIs), drugs that inhibit lamin A and progerin farnesylation (Capell et al. 2005; Toth et al. 2005). FTIs were used in the first clinical trial for progeria patients and some beneficial effects have been reported (Couzin-Frankel 2012; Gordon et al. 2014).

Three-dimensional rendering of the nuclear lamina in (a) wild-type and (b) HGPS fibroblast nuclei by three-dimensional structured-illumination super-resolution microscopy. Staining: lamin A/C antibody. Bar 6 μm
Recent studies have demonstrated that the inhibition of the N-acetyltransferase activity of NAT10 towards cytoplasmic microtubules, by a small molecule called “remodelin”, is also able to restore nuclear shape in HGPS or lamin-A-depleted cells. In addition, remodelin appears to improve the proliferation of HGPS cells (Larrieu et al. 2014). Interestingly, cells treated with both remodelin and FTI do not exhibit additional improvements in comparison with cells treated with either remodelin or FTI alone. This suggests that remodelin and FTIs do not act synergistically, but rather target a common pathway. In wild-type cells, FTIs have also been shown to affect microtubule dynamics and to cause nuclear abnormalities that can be prevented in the presence of remodelin (Suzuki et al. 1998; Verstraeten et al. 2011; Larrieu et al. 2014). Thus, both FTI and NAT10-modifying compounds might share parts of their mechanism of action with respect to nuclear shape.
Nuclear abnormalities are certainly a major phenotype of HGPS but there is more to progerin than meets the eye. Shortly after the HGPS mutation was identified, fibroblasts from HGPS patients were shown to exhibit increased DNA damage (Liu et al. 2005, 2006; Musich et al. 2009). Although treatment with FTIs or remodelin improves the aberrant nuclear shape of HGPS fibroblasts, only remodelin reduces DNA damage. These results suggest that nuclear morphology defects and DNA damage are independent phenotypes arising from progerin expression (Liu et al. 2005, 2006; Musich et al. 2009). Indeed, consistent with the persistent activation of DNA damage checkpoints, progeric fibroblasts exhibit a limited proliferative capacity and have significantly shorter telomeres than age-matched controls (Decker et al. 2009; Allsopp et al. 1992). In addition, the ectopic expression of progerin in wild-type fibroblasts inhibits their proliferation and triggers premature senescence (Benson et al. 2010). However, progerin-induced proliferative inhibition is alleviated by telomerase activation and, to some extent, by the inactivation of the p53 pathway (Kudlow et al. 2008). These results have provided the first evidence that progerin directly or indirectly damages telomeres, thereby activating p53, in a manner that can be alleviated by telomerase. More recently, Benson and colleagues extended these results by showing that the DNA damage foci in progerin-expressing cells co-localise with human TRF1, a component of the telomere-associated shelterin complex (Benson et al. 2010). Telomere-specific DNA damage is particularly detrimental for cells as it cannot be repaired by conventional DNA repair pathways and thus, triggers permanent growth arrest.
What is the physiological relevance of these findings and how does the accelerated ageing in HGPS patients relate to normal ageing? Increased DNA damage is a hallmark of cells undergoing senescence because of shortened or deprotected telomeres and shortened telomeres have been associated with human ageing (d’Adda di Fagagna et al. 2003; Takai et al. 2003; Canela et al. 2007). Nevertheless, one difference is that during normal ageing, telomere dysfunction is mainly a consequence of the end replication problem, which might also be enhanced by DNA damaging agents such as oxidative stress (Wang et al. 2009); whereas in HGPS, telomeres might be damaged more directly by progerin (Benson et al. 2010). However, the precise mechanism by which telomeres are damaged and whether only a subset of telomeres, such as those located at the nuclear periphery, are affected still needs to be established.
Mutations in lamin A versus lamin C
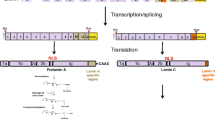
Although the HGPS mutation (c.1824C→T) is found exclusively in the lamin A splice isoform of LMNA, most other progeroid mutations (c.412G→ A, c.428C→T, c.433G→A, c.1583C→T, c.1619 T→C, c.1626G→C) and mutations involved in muscular dystrophies (EDMD, LGMD), lipodystrophy (FPLD), DCM, neuropathy (CMT), restrictive dermopathy (RD) and bone disease (MAD) simultaneously affect both lamin A and lamin C splice variants (Fig. 2). The subcellular localisation and similar domain structure of lamins A and C suggest that mutations in the LMNA locus affect cell physiology by equally altering both lamin A and C. Various mouse models have indeed shown that A-type lamin isoforms can behave independently of each other and fulfil similar functions (Fong et al. 2006). The finding that mice expressing exclusively lamin A or lamin C are disease-free as compared with Lmna knockout mice indicates a high level of functional redundancy (Sullivan et al. 1999; Kim and Zheng 2013; Kubben et al. 2011). However, LMNA mutations causing EDMD, DCM or lipodystrophy have been demonstrated to give rise to different effects when expressed in each isoform, including differential binding to other lamina components (Motsch et al. 2005; Sylvius et al. 2008). In particular, three EDMD-causing mutations (c.448A→C, c.1580G→C, c.1589T→C) disrupt lamin A binding to emerin, without affecting the association of emerin with lamin C (Motsch et al. 2005). Similarly, introducing the FPLD c.1444C→T mutation into lamin C impairs its localisation to the nuclear lamina, whereas the introduction of the same mutation into lamin A causes a much milder phenotype (Broers et al. 2005). Lastly, a decrease of lamin A protein abundance without affecting lamin C is also seen in the central nervous system in which only lamin A is specifically downregulated by miR-9 microRNA (Jung et al. 2012). Thus, despite an obvious functional redundancy between the two A-type lamins, mutations that affect both protein isoforms can impair cell function in a differential and/or synergistic manner.

Spatial distribution of LMNA mutations along A-type lamin cDNA. Representation of reported LMNA mutations (source: Universal Mutation Database LMNA). Mutations are represented by their position on the LMNA cDNA and categorised according to their phenotype(s): DCM (dilated cardiomyopathy), EDMD (Emery-Dreifuss muscular dystrophy), CMT (Charcot-Marie-Tooth), FPLD (familial partial lipodystrophy), LGMD (limb girdle muscular dystrophy), progeroid (HGPS and atypical Werner syndrome), MAD (mandibuloacral dysplasia), RD (restrictive dermopathy). A-type lamin protein domains are indicated: coil 1A (green), coil 1B (light blue), coil 2 (dark blue), nuclear localization signal (pink), lamin A-specific N-terminus (grey)
Diseases associated with perturbations of B-type lamins
In contrast to lamin A, no point mutations in lamin B1 have been linked to any diseases. However, elevated levels of lamin B1 have been observed in cells from patients with ataxia telangiectasia (AT) and adult-onset autosomal dominant leukodystrophy (ADLD), diseases whose clinical symptoms include neurological defects. Whilst the underlying cause of the elevated lamin B1 levels in AT remains unclear, ADLD is caused by a duplication of the lamin B1 locus and leads to symmetrical demyelination of the central nervous system. Similar to patients with multiple sclerosis (MS), ADLD patients progressively lose fine motor skills and suffer from autonomic symptoms including bowel/bladder dysfunction, orthostatic hypotension and male impotence. However, contrasting the neurodegenerative phenotype of MS, demyelination in ADLD is symmetrical and not associated with brain inflammation and loss of oligodendrocytes.
In the past two years, several in vitro and in vivo models have been used to investigate the way that elevated levels of lamin B1 affect cellular physiology. Heng et al. (2013) generated a bacterial artificial chromosome (BAC)-based transgenic ADLD mouse model by expressing lamin B1 under the control of its endogenous promoter. These lamin B1BAC mice exhibit several pathophysiological features of ADLD, including impaired cognitive function and age-dependent motor deficits (Heng et al. 2013). Ultrastructural analysis of 24-month-old ADLD mice revealed aberrant myelin formation, demyelination and axonal degeneration. Consistent with the non-inflammatory phenotype of ADLD patients, lamin B1BAC mice show no evidence of microglia activation or reactive astrocytes. Nevertheless, how do elevated levels of lamin B1 result in myelin loss? Analysis of the protein composition between lamin B1BAC and wild-type mice has revealed that lamin B1BAC mice exhibit a significant downregulation of proteolipid protein (PLP). PLP is a major component of the myelin sheet and has previously been implicated in other myelin-related diseases. In addition, elevated levels of lamin B1 in oligodendrocytes result in the transcriptional repression of the genes involved in myelin biosynthesis (myelin-basic protein, proteolipid protein and myelin oligodendrocyte glycoprotein; Lin and Fu 2009).
Although these results provide a link between lamin B1 overexpression and myelin abnormalities, the precise mechanism by which lamin B1 overexpression leads to reduced myelin remains unclear. Lamins have been shown to interact with DNA (Kind et al. 2013; Guelen et al. 2008) and, to some extent, might regulate gene expression (Finlan et al. 2008; Reddy et al. 2008; Shevelyov et al. 2009). Thus, elevated levels of lamin B1 might directly affect the transcriptional regulation of PLP.
Another possibility is that lamin B1 overexpression triggers a cellular program that in turn leads to myelin destabilisation. We and others have developed cell-based in vitro models to study the consequences of lamin B1 overexpression on cell proliferation and function (Barascu et al. 2012; Dreesen et al. 2013a, 2013b). However, in human fibroblasts, a 2– to 3-fold overexpression of lamin B1 only results in a moderate proliferation defect. These results have prompted us to investigate the reasons why human fibroblasts are relatively resistant to lamin B1 overexpression and why lamin B1 duplication in ADLD preferentially affects the central nervous system. We hypothesised that differences in the composition of the nuclear lamina in fibroblasts versus neuronal lineages render the latter more susceptible to aberrant lamin B1 levels. One such difference is that neuronal lineages express dramatically lower levels of lamin A than fibroblasts (Lehner et al. 1987; Röber et al. 1989; Zhang et al. 2011). We therefore increased lamin B1 in fibroblasts with reduced (50 % reduction) levels of lamin A/C. Strikingly, lamin B1↑lamin A/C↓ cells exhibited a pronounced proliferation defect, arrested at the G0/G1 stage of the cell cycle and stained positive for senescence-associated-β-gal activity. In addition, lamin B1↑lamin A/C↓ cells exhibited 53BP-1 DNA damage foci that were associated with telomeres (Dreesen et al. 2013a, 2013b). Reminiscent of the situation in progerin-expressing fibroblasts, the introduction of telomerase prevented the accumulation of telomeric DNA damage foci and restored the proliferation of lamin B1↑lamin A/C↓ cells (Dreesen et al. 2013a, b). Taken together, these results suggest that a reduction in lamin A/C levels dramatically potentiates the phenotypic consequences of lamin B1 overexpression. The question remains as to how perturbations in the nuclear lamina cause telomeric DNA damage.
Nuclear lamina remodelling
The nuclear lamina was at first thought to be a static meshwork but is now considered to be a highly plastic and dynamic structure. The composition of the nuclear lamina and the stoichiometry of lamina components varies between tissues and changes during development, the cell cycle and in different cell fates (Broers et al. 1997; Swift et al. 2013).
Remodelling during embryonic development and differentiation
Pioneering studies in the 1980s revealed that the composition of the nuclear lamina undergoes profound changes during embryonic development in chicken and mouse. Nigg and colleagues demonstrated that early chicken embryos contained substantial amounts of B-type lamins, whereas A-type lamins were absent and accumulated only during later stages of development (Lehner et al. 1987). A-type lamins appeared after ~8 days in the ectoplacental cone, the tissue that eventually forms the placenta and after 9–11 days in the embryo proper.
The differential expression of lamins during embryonic development can also be recapitulated at the cellular level in vitro: pluripotent ESC and undifferentiated teratocarcinoma stem cells exclusively express B-type lamins, whereas the expression of A-type lamins is confined to their differentiated progeny. Conversely, the reprogramming of somatic cells into induced pluripotent stem cells (iPSC) or even the exposure of somatic cells to ESC extracts results in the downregulation of lamin A/C (Zhang et al. 2011; Liu et al. 2011; Bru et al. 2008).
The differential expression of lamins during embryonic development in vivo and during differentiation in vitro raises the question as to whether the lamina is actively involved in regulating gene expression during development. At least in the mouse, neither A- nor B-type lamins appear to be necessary for early embryonic development: Lmna-/- mice develop to term and only exhibit impaired growth and muscular dystrophy after birth (Sullivan et al. 1999). Similarly, mice lacking both lmnb1 and lmnb2 also develop to term but die shortly thereafter. Perhaps most surprising is the fact that E12.5 and E18 embryos from Lmnb1+/- Lmnb2+/- intercrosses reveal a normal Mendelian distribution and that all internal organs form properly. In agreement with these findings, mESC derived from Lmnb1/2-/- mice retain their pluriopotency markers and differentiate into trophectoderm with a similar efficiency as wild-type mESC (Kim et al. 2011). Taken together, these results suggest that B-type lamins are not actively involved in regulating transcriptional programs during ES cell differentiation.
Haematopoietic system
Most cells of the haematopoietic system either do not express A-type lamins or express it at extremely low levels (Röber et al. 1990; Guilly et al. 1990). However, a recent study demonstrated that T-lymphocytes, which are generally devoid of A-type lamins, show a transient increase in lamin A/C expression upon T-cell activation (Gonzales-Granado et al. 2014). These results might be physiologically relevant as elevated levels of lamin A enhance T-cell activation both in vitro and in vivo, whereas lamin A-deficiency reduces T-cell activation. Do these results have implications for laminopathies? Children with progeria appear to have a normal haematopoietic system and respond normally to various infections. A detailed analysis of telomere length in haematopoietic lineages including T-cells, B-cells, natural killer cells and granulocytes, revealed that the median telomere length in three out of four HGPS patients is comparable with that of age-matched controls (Decker et al. 2009). In contrast, fibroblasts from the same patients exhibit dramatically shortened telomeres. Is it possible that the reactivation of lamin A and progerin in activated T-cells from HGPS patients is too transient or at too low a level to damage telomeres? The reactivation of lamin A in activated T-cells and the apparent resistance of the immune system to progerin is intriguing and would benefit from further investigation.
Central nervous system
In most adult tissues, lamins A and C are found in roughly equal amounts. An exception to this rule is the central nervous system: whilst lamin C is expressed in most cell types of the adult mouse brain, lamin A levels remain low or undetectable (Jung et al. 2012). As lamin A and lamin C are alternatively spliced isoforms of the LMNA gene, what could account for their dramatically different expression levels? Jung and colleagues found that the 3’untranslated region of pre-lamin A mRNA but not lamin C mRNA, contains a putative binding site for miR-9, a microRNA that is specifically expressed in the brain and is a key player in neural development (Leucht et al. 2008). Subsequent experiments by various groups have shown that miR-9 expression in human fibroblasts, HeLa cells and iPSC-derived mesenchymal stem cells leads to reduced levels of lamin A but not lamin C. Importantly, the miR-9-dependent removal of lamin A from neuronal tissues is of critical clinical importance to patients with HGPS: miR-9 prevents the accumulation of mutant lamin A (progerin) thereby protecting them from complications in the central nervous system (Nissan et al. 2012; Jung et al. 2012, 2014).
The lamina composition in the brain also appears to change over time. In mice, the temporal expression of lamin B1 is developmentally regulated and changes during brain development: lamin B1 protein and mRNA levels are highest at birth and subsequently decline throughout adult life (Lin and Fu 2009). Similarly, reduced lamin B1 levels can be observed in rats during oligodendrocyte maturation in vitro (Dugas et al. 2006). Interestingly, lamin B1 levels are inversely correlated with myelin-specific proteins, including myelin basic protein and myelin-associated glycoprotein. Therefore, the orchestration of the onset and levels of lamin B1 expression is important as excessive production of lamin B1 (because of genomic duplication of the LMNB1 locus) is associated with ADLD and demyelination in the central nervous system (Lin and Fu 2009).
Tissue rigidity
The central nervous system not only has a highly characteristic ratio of lamin A abundance compared with that of lamin C, it also has one of the lowest ratios of A-type to B-type lamins. Swift et al. (2013) showed that A:B lamin stoichiometry changes across tissues and is correlated with their elasticity. Thus, tissues such as cartilage, bone, muscle or heart have a high A:B ratio (high A-type lamin expression compared with B-type lamins), whereas softer tissues such as liver, kidney and brain are characterised by a low A:B ratio (low A-type lamin expression compared with B-type lamins). This variation of A:B-type lamin abundance is mainly dependent on the modulation of A-type lamins, as the levels of B-type lamins remain relatively constant across tissues. What could explain the diverse A:B-type ratio in various tissues and how is it regulated? In a series of papers, the Discher group demonstrated that the lamina composition dynamically changes in order to adapt to various tissue environments. This is facilitated by the phosphorylation of lamin A, which in turn changes the physical properties of the lamina. Lamin A phosphorylation occurs when cells are grown on soft matrices, whereas lamin A/C levels are elevated with increasing stiffness of the matrix. Thus, both lamin A/C levels and its phosphorylation status modify the mechanical properties of the nucleus and facilitate cell growth in various tissue environments (Swift et al. 2013; Buxboim et al. 2014).
Nuclear lamina dissolution during mitosis
The most drastic remodelling of the nuclear lamina occurs during cell division, when the nuclear envelope and the lamina are broken down (Güttinger et al. 2009). During cell division, A- and B-type lamin filaments are depolymerised following their phosphorylation by kinases including protein kinase C (PKC), Aurora A and polo-like kinase 1 (Gerace and Blobel 1980; Güttinger et al. 2009; Mall et al. 2012). Phosphorylation of A-type lamins induces their solubilisation and release, first into the nucleoplasm and, after nuclear envelope breakdown, into the cytosol. In contrast, phosphorylated B-type lamins remain associated with mitotic endoplasmic reticulum (ER) membranes (Georgatos et al. 1997). Reassembly of the nuclear lamina after mitosis starts by the binding of the ER tubules to chromatin (Anderson and Hetzer 2008), assembly of NPC prepores (Sheehan et al. 1988), remodelling of the ER tubules into flattened nuclear envelope patches and the binding of inner nuclear membrane proteins to chromatin (Anderson and Hetzer 2007). Dephosphorylation of lamins by protein phosphatases and inactivation of cyclin-dependent kinase 1 is required to reintegrate the nuclear lamina proteins into a new nuclear lamina; this will occur only once nuclear import has been restored (Newport et al. 1990).
Viruses take advantage of this mechanism to modulate the nuclear lamina structurally and to egress from the nucleus into the cytoplasm. Herpes simplex virus type 1 and the murine cytomegalovirus achieve this by recruiting the cellular PKC to the nuclear periphery (Park and Baines 2006). This in turn triggers phosphorylation of nuclear envelop proteins, leads to irregular lamina distribution and dissolution, and facilitates the exit of virions from the nucleus (Muranyi et al. 2002; Camozzi et al. 2008).
Lamina remodelling in cancer cells
The Papanicolaou smear test is routinely used to identify squamous cell carcinomas, one of the most common and malignant neoplasms among women. The test typically includes the search for nuclear abnormalities, such as enlarged and irregularly shaped nuclei (Bengtsson and Malm 2014). Such alterations of the nuclear lamina and the variable levels of A-type lamins have been recurrently observed in cancer cells, although their exact significance and relevance remain unclear (de Las Heras et al. 2013). Down-regulation of A-type lamins has been reported in gastrointestinal neoplasms (Moss et al. 1999), gastric carcinoma (Wu et al. 2009), colon (Belt et al. 2011), breast (Capo-chichi et al. 2011a, 2011b) and ovarian cancers (Capo-chichi et al. 2011a, 2011b), whereas others have shown the up-regulation of lamins in colorectal (Willis et al. 2009) and prostate cancer (Kong et al. 2012). Thus, although different lamin A expression levels can be correlated with the prognosis of various cancers, it remains unclear whether lamin A/C fluctuations are causal or simply a consequence of the widespread genomic abnormalities that occur in cancer cells. Some recent studies have suggested that lower lamin A levels confer a selective advantage to some cancer cells, as the decreased expression of lamin A improves nuclear deformability and cell migration through three-dimensional environments (Rowat et al. 2013; Wolf et al. 2013). In contrast, Kong et al. (2012) modulated lamin levels in prostate cancer cells and found that the overexpression of lamin A/C enhances cell proliferation, migration and invasion, suggesting a causal role of A-type lamins on cancer cell propagation. In conclusion, a clear consensus for the role of A-type lamins in cancer cells might be difficult to find: the diverse types of cancers might respond differently to aberrant lamin A levels, depending on the selective advantage (proliferation, invasiveness) that is required.
Nuclear lamina remodelling during cellular senescence
Cellular senescence is an irreversible cell cycle exit that can be triggered by various forms of cellular stress, including irreparable DNA damage, oxidative, mitotic or oncogenic stress. Senescent cells are characterised by the activation of p53 and Rb tumour suppressor pathways and can be found in premalignant tumours, including benign nevi. Senescent cells also accumulate with age in various tissues, thereby limiting their regenerative potential. The entry into senescence is associated with morphological changes, the expression of senescence-associated β-galactosidase activity (SA-β–gal) and changes in gene expression and chromatin organisation, including the generation of senescence-associated heterochromatin foci and the senescence-associated secretory phenotype (Rodier and Campisi 2011). In addition, the nuclear lamina remodels dramatically as cells senesce (Dreesen et al. 2013a, 2013b; Freund et al. 2012; Shimi et al. 2011). These changes include a reduction of lamin B1 and the lamina associated polypeptide 2α (LAP2α). The loss of lamin B1 levels is specific to senescent cells, whereas LAP2α levels also decline in quiescent cells. Loss of lamin B1 can additionally be used to identify senescent cells in vivo: lamin B1 levels decline in mice treated with a senescence-inducing dose of ionising radiation (Freund et al. 2012), in a mouse model for the accelerated ageing syndrome progeria (McKenna et al. 2014) and during the chronological ageing of human skin (Dreesen et al. 2013a, 2013b). In addition to the loss of lamin B1, a reduced expression of lamin B1 receptor and a relocalisation of lamin A/C from the perinuclear region suggest that the nuclear envelope is structurally compromised in senescent cells (Ivanov et al. 2013). Taken together, these results demonstrate that the nuclear envelope is dramatically remodelled in senescent cells and that loss of lamin B1 can be used as a marker to identify senescent cells in vitro and in vivo.
These data raise the question as to whether the down-regulation of lamin B1 causes the widespread chromatin changes observed in senescent cells, i.e., whether the reduction of lamin B1 is a cause or a consequence of cellular senescence. Several papers have addressed this question with differing results. Whilst some groups reported that small-hairpin-RNA-mediated downregulation of lamin B1 triggers cellular senescence and apoptosis (Harborth et al. 2001; Shimi et al. 2011; Shah et al. 2013), other studies suggested that merely reducing lamin B1 levels is not sufficient to trigger senescence (Dreesen et al. 2013a, 2013b; Sadaie et al. 2013). Undoubtedly, lamin B1 levels decline dramatically during senescence but the causality of this reduction on the senescence phenotype remains debated. Mice that lack lamin B1 and lamin B2 specifically in epidermal keratinocyte or hepatocytes do not show any overt problems in the development and maintenance of skin, or any defects in liver development, histology or function. Similarly, most organs of lamin B1 knockout mice develop to term and are apparently normal, despite the fact that these mice die shortly after birth and exhibit bone, lung and cranial abnormalities. These results demonstrate that lamin B1 is not essential for cell proliferation during organ development in mice. In contrast, when grown under in vitro cell culture conditions, lamin-B1-deficient mouse embryonic fibroblasts exhibit nuclear abnormalities, impaired proliferation and premature senescence. This therefore suggests that lamin B1 deficiency impairs cellular function(s), which in conjunction with additional stress, such as growth under sparse conditions or in vitro culture, triggers senescence (Vergnes et al. 2004; Dreesen et al. 2013a, 2013b).
Concluding remarks
Originally considered as a static and rigid piece of hardware, the nuclear lamina is now known as a highly plastic, modular and versatile part of the nucleus. The sheer number of the known different diseases and mutations linked to the nuclear lamina reveals the complexity of its cellular functions (Worman and Bonne 2007). In addition, as discussed here, the relative abundance of the various lamina components appears to play an essential role in controlling the tissue-specific nature of some laminopathies.
One of the cellular hallmarks of HGPS is the presence of abnormally shaped nuclei, coupled with the accumulation of a mutant lamin A (progerin) within the nuclear lamina. Despite progerin being expressed in nearly all somatic cells, it is a segmental ageing disorder in which the brain is not affected. A major breakthrough has been the discovery of the brain-specific expression of miR-9, which targets LMNA transcripts and therefore prevents progerin expression (Nissan et al. 2012; Jung et al. 2012, 2014). These results highlight the importance of nuclear lamina composition, the way that it varies among different tissues and the manner in which this can affect disease etiology. The peculiar lamina composition in the brain might also account for the phenotypic specificity of ADLD: our studies suggest that the lower abundance of A-type lamin renders the central nervous system particularly vulnerable to elevated levels of lamin B1 (Dreesen et al. 2013a, b). These investigations emphasise the essential role of the nuclear lamina during development and cell differentiation and the highly complex chain of events leading from a single point mutation to the disease phenotype. Importantly, they indicate that, whereas mutation dictates the starting point of this detrimental chain of events, the widely diverse composition of the nuclear lamina in each tissue might ultimately define the tissue-specific nature of laminopathies.
References
Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, Greider CW, Harley CB (1992) Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A 89:10114–10118
Anderson DJ, Hetzer MW (2007) Nuclear envelope formation by chromatin-mediated reorganization of the endoplasmic reticulum. Nat Cell Biol 9:1160–1166
Anderson DJ, Hetzer MW (2008) Reshaping of the endoplasmic reticulum limits the rate for nuclear envelope formation. J Cell Biol 182:911–924
Barascu A, Le Chalony C, Pennarun G, Genet D, Imam N, Lopez B, Bertrand P (2012) Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J 31:1080–1094
Batsios P, Peter T, Baumann O, Stick R, Meyer I, Gräf R (2012) A lamin in lower eukaryotes? Nucleus 3:237–243
Belt EJ, Fijneman RJ, Berg EG van den, Bril H, Delis-van Diemen PM, Tijssen M, Essen HF van, Lange-de Klerk ES de, Beliën JA, Stockmann HB, Meijer S, Meijer GA (2011) Loss of lamin A/C expression in stage II and III colon cancer is associated with disease recurrence.Eur J Cancer 47:1837–1845
Bengtsson E, Malm P (2014) Screening for cervical cancer using automated analysis of PAP-smears. Comput Math Methods Med 2014:842037
Benson EK, Lee SW, Aaronson S (2010) Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci 123:2605–2612
Broers JL, Machiels BM, Kuijpers HJ, Smedts F, Kieboom R van den, Raymond Y, Ramaekers FC (1997) A- and B-type lamins are differentially expressed in normal human tissues. Histochem Cell Biol 107:505–517
Broers JLV, Kuijpers HJ, Ostlund C, Worman HJ, Endert J, Ramaekers FC (2005) Both lamin A and lamin C mutations cause lamina instability as well as loss of internal nuclear lamin organization. Exp Cell Res 304:582–592
Bru T, Clarke C, McGrew MJ, Sang HM, Wilmut I, Blow JJ (2008) Rapid induction of pluripotency genes after exposure of human somatic cells to mouse ES cell extracts. Exp Cell Res 314:2634–2642
Burke B, Stewart CL (2014) Functional architecture of the cell’s nucleus in development, aging, and disease. Curr Top Dev Biol 109:1–52
Buxboim A, Swift J, Irianto J, Spinler KR, Dingal PC, Athirasala A, Kao YR, Cho S, Harada T, Shin JW, Discher DE (2014) Matrix elasticity regulates lamin A, C phosphorylation and turnover with feedback to actomyosin. Curr Biol 24:1909–1917
Camozzi D, Pignatelli S, Valvo C, Lattanzi G, Capanni C, Dal Monte P, Landini MP (2008) Remodelling of the nuclear lamina during human cytomegalovirus infection: role of the viral proteins pUL50 and pUL53. J Gen Virol 89:731–740
Canela A, Vera E, Klatt P, Blasco M (2007) High-throughput telomere length quantification by FISH and its application to human population studies. Proc Natl Acad Sci U S A 104:5300–5305
Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS (2005) Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 102:12879–12884
Capo-chichi CD, Cai KQ, Simpkins F, Ganjei-Azar P, Godwin AK, Xu XX (2011a) Nuclear envelope structural defects cause chromosomal numerical instability and aneuploidy in ovarian cancer. BMC Med 9:28
Capo-chichi CD, Cai KQ, Smedberg J, Ganjei-Azar P, Godwin AK, Xu XX (2011b) Loss of A-type lamin expression compromises nuclear envelope integrity in breast cancer. Chin J Cancer 30:415–425
Ciska M, Moreno Diaz de la Espina S (2013) NMCP/LINC proteins: putative lamin analogs in plants? Plant Signal Behav 8:e26669
Constantinescu D, Gray HL, Sammak PJ, Schatten GP, Csoka AB (2006) Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells 24:177–185
Couzin-Frankel J (2012) Medicine. Drug trial offers uncertain start in race to save children with progeria. Science 337:1594–1595
d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426:194–198
De Las Heras JI, Batrakou DG, Schirmer EC (2013) Cancer biology and the nuclear envelope: a convoluted relationship. Semin Cancer Biol 23:125–137
De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Lévy N (2003) Lamin a truncation in Hutchinson-Gilford progeria. Science 300:5
Decker ML, Chavez E, Vulto I, Lansdorp PM (2009) Telomere length in Hutchinson-Gilford progeria syndrome. Mech Ageing Dev 130:377–383
Dreesen O, Chojnowski A, Ong PF, Zhao TY, Common JE, Lunny D, Lane EB, Lee SJ, Vardy L, Stewart CL, Colman A (2013a) Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J Cell Biol 200:605–617
Dreesen O, Ong PF, Chojnowski A, Colman A (2013b) The contrasting roles of lamin B1 in cellular aging and human disease. Nucleus 4:283–290
DuBois KN, Alsford S, Holden JM, Buisson J, Swiderski M, Bart JM, Ratushny AV, Wan Y, Bastin P, Barry JD, Navarro M, Horn D, Aitchison JD, Rout MP, Field MC (2012) NUP-1 is a large coiled-coil nucleoskeletal protein in trypanosomes with lamin-like functions. PLoS Biol 10:e1001287
Dugas JC, Tai YC, Speed TP, Ngai J, Barres B (2006) Functional genomic analysis of oligodendrocyte differentiation. J Neurosci 26:10967–10983
Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS (2003) Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423:293–298
Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore W (2008) Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4:e1000039
Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, Coté N, Gavino B, Qiao X, Chang SY, Young SR, Yang SH, Stewart CL, Lee RT, Bennett CF, Bergo MO, Young SG (2006) Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest 116:743–752
Freund A, Laberge R, Demaria M, Campisi J (2012) Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23:2066–2075
Furukawa K, Hotta Y (1993) cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells. EMBO J 12:97–106
Furukawa K, Inagaki H, Hotta Y (1994) Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice. Exp Cell Res 212:426–430
Georgatos SD, Pyrpasopoulou A, Theodoropoulos P (1997) Nuclear envelope breakdown in mammalian cells involves stepwise lamina disassembly and microtubule-drive deformation of the nuclear membrane. J Cell Sci 110:2129–2140
Gerace L, Blobel G (1980) The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 19:277–287
Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS (2004) Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 101:8963–8968
González-Granado JM, Silvestre-Roig C, Rocha-Perugini V, Cibrián D, Morlino G, Blanco-Berrocal M, Osorio G, Freije JMP, López-Otín C, Sánchez-Madrid F, Andrés V (2014) Nuclear envelope lamin-A couples actin dynamics with immunological synapse architecture and T cell activation. Sci Signal 7:ra37
Gordon LB, Massaro J, D’Agostino RB, Campbell SE, Brazier J, Brown WT, Kleinman ME, Kieran MW (2014) Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 130:27–34
Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, Klein A de, Wessels L, Laat W de, Steensel B van (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453:948–951
Guilly MN, Kolb JP, Gosti F, Godeau F, Courvalin JC (1990) Lamins A and C are not expressed at early stages of human lymphocyte differentiation. Exp Cell Res 189:145–147
Güttinger S, Laurell E, Kutay U (2009) Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol 10:178–191
Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 114:4557–4565
Heng MY, Lin S, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptáček LJ, Fu YH (2013) Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J Clin Invest 123:2719–2729
Ivanov A, Pawlikowski J, Manoharan I, Tuyn J van, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, Wu H, Berger SL, Adams PD (2013) Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202:129–143
Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BSJ, Yang SH, Barnes RH, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG (2012) Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A 109:E423–E431
Jung HJ, Tu Y, Yang SH, Tatar A, Nobumori C, Wu D, Young SG, Fong LG (2014) New Lmna knock-in mice provide a molecular mechanism for the “segmental aging” in Hutchinson-Gilford progeria syndrome. Hum Mol Genet 23:1506–1515
Kim Y, Zheng Y (2013) Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem Biophys Res Commun 440:8–13
Kim Y, Sharov AA, McDole K, Cheng M, Hao H, Fan C, Gaiano N, Ko MS, Zheng Y (2011) Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 334:1706–1710
Kind J, Pagie L, Ortabozkoyun H, Boyle S, Vries SS de, Janssen H, Amendola M, Nolen LD, Bickmore W, Steensel B van (2013) Single-cell dynamics of genome-nuclear lamina interactions. Cell 153:178–192
Kong L, Schäfer G, Bu H, Zhang Y, Zhang Y, Klocker H (2012) Lamin A/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis 33:751–759
Krüger A, Batsios P, Baumann O, Luckert E, Schwarz H, Stick R, Meyer I, Gräf R (2012) Characterization of NE81, the first lamin-like nucleoskeleton protein in a unicellular organism. Mol Biol Cell 23:360–370
Kubben N, Voncken JW, Konings G, Weeghel M van, Hoogenhof MM van den, Gijbels M, Erk A van, Schoonderwoerd K, Bosch B van den, Dahlmans V, Calis C, Houten SM, Misteli T, Pinto YM (2011) Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus 2:195–207
Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK (2008) Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol Biol Cell 19:5238–5248
Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP (2014) Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344:527–532
Lehner CF, Stick R, Eppenberger HM, Nigg E (1987) Differential expression of nuclear lamin proteins during chicken development. J Cell Biol 105:577–587
Leucht C, Stigloher C, Wizenmann A, Klafke R, Folchert A, Bally-Cuif L (2008) MicroRNA-9 directs late organizer activity of the midbrain-hindbrain boundary. Nat Neurosci 11:641–648
Lin S-T, Fu Y (2009) miR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis Model Mech 2:178–188
Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang J, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadiñanos J, López-Otín C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z (2005) Genomic instability in laminopathy-based premature aging. Nat Med 11:780–785
Liu G-H, Barkho BZ, Ruiz S, Diep D, Qu J, Yang S-L, Panopoulos AD, Suzuki K, Kurian L, Walsh C, Thompson J, Boue S, Fung HL, Sancho-Martinez I, Zhang K, Yates J 3rd, Izpisua Belmonte JC (2011) Recapitulation of premature ageing with iPSCs from Hutchinson–Gilford progeria syndrome. Nature 472:221–225
Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y (2006) DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci 119:4644–4649
Machiels BM, Zorenc HG, Endert JM, Kuijpers HJ, Eys GJ van, Ramaekers FC, Broers JL (1996) An alternative splicing product of the lamin A/C gene lacks exon 10. J Biol Chem 271:9249–9253
Mall M, Walter T, Gorjánácz M, Davidson IF, Nga Ly-Hartig TB, Ellenberg J, Mattaj IW (2012) Mitotic lamin disassembly is triggered by lipid-mediated signaling. J Cell Biol 198:981–990
McKenna T, Rosengardten Y, Viceconte N, Baek J-H, Grochová D, Eriksson M (2014) Embryonic expression of the common progeroid lamin A splice mutation arrests postnatal skin development. Aging Cell 13:292–302
Melcer S, Gruenbaum Y, Krohne G (2007) Invertebrate lamins. Exp Cell Res 313:2157–2166
Moir RD, Montag-Lowy M, Goldman RD (1994) Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol 125:1201–1212
Moss SF, Krivosheyev V, Souza A de, Chin K, Gaetz HP, Chaudhary N, Worman HJ, Holt PR (1999) Decreased and aberrant nuclear lamin expression in gastrointestinal tract neoplasms. Gut 45:723–729
Motsch I, Kaluarachchi M, Emerson LJ, Brown C, Brown SC, Dabauvalle MC, Ellis J (2005) Lamins A and C are differentially dysfunctional in autosomal dominant Emery-Dreifuss muscular dystrophy. Eur J Cell Biol 84:765–781
Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH (2002) Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857
Musich R, Zou Y, Musich PR (2009) Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany N.Y.) 1:28–37
Newport JW, Wilson KL, Dunphy WG (1990) A lamin-independent pathway for nuclear envelope assembly. J Cell Biol 111:2247–2259
Nissan X, Blondel S, Navarro C, Maury Y, Denis C, Girard M, Martinat C, De Sandre-Giovannoli A, Levy N, Peschanski M (2012) Unique preservation of neural cells in Hutchinson-Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep 2:1–9
Park R, Baines JD (2006) Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80:494–504
Peter A, Stick R (2012) Evolution of the lamin protein family: what introns can tell. Nucleus 3:44–59
Reddy KL, Zullo JM, Bertolino E, Singh H (2008) Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452:243–247
Röber RA, Weber K, Osborn M (1989) Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105:365–378
Röber RA, Sauter H, Weber K, Osborn M (1990) Cells of the cellular immune and hemopoietic system of the mouse lack lamins A/C: distinction versus other somatic cells. J Cell Sci 95:587–598
Rodier F, Campisi J (2011) Four faces of cellular senescence.J Cell Biol 192:547-556
Rowat AC, Jaalouk DE, Zwerger M, Ung WL, Eydelnant I, Olins DE, Olins AL, Herrmann H, Weitz D, Lammerding J (2013) Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J Biol Chem 288:8610–8618
Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young AR, Narita MM, Pérez-Mancera P, Bennett DC, Chong H, Kimura H, Narita M (2013) Redistribution of the lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev 27:1800–1808
Scaffidi P, Misteli T (2005) Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med 11:440–445
Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks H, Rai TS, McBryan T, Gregory BD, Adams PD, Berger SL (2013) Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 27:1787–1799
Sheehan M, Mills D, Sleeman M, Laskey R, Blow JJ (1988) Steps in the assembly of replication-competent nuclei in a cell-free system from Xenopus eggs. J Cell Biol 106:1–12
Shevelyov YY, Lavrov S, Mikhaylova LM, Nurminsky ID, Kulathinal RJ, Egorova KS, Rozovsky YM, Nurminsky DI (2009) The B-type lamin is required for somatic repression of testis-specific gene clusters. Proc Natl Acad Sci U S A 106:3282–3287
Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, Goldman RD (2011) The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 25:2579–2593
Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, Herrmann H, Blum H, Engelkamp D, Stewart CL, Leonhardt H, Joffe B (2013) LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152:584–598
Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B (1999) Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 147:913–920
Suzuki N, Del Villar K, Tamanoi F (1998) Farnesyltransferase inhibitors induce dramatic morphological changes of KNRK cells that are blocked by microtubule interfering agents. Proc Natl Acad Sci U S A 95:10499–10504
Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PCDP, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE (2013) Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341:1240104
Sylvius N, Hathaway A, Boudreau E, Gupta P, Labib S, Bolongo PM, Rippstein P, McBride H, Bilinska ZT, Tesson F (2008) Specific contribution of lamin A and lamin C in the development of laminopathies. Exp Cell Res 314:2362–2375
Takai H, Smogorzewska A, Lange T de (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13:1549–1556
Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG (2005) Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A 102:12873–12878
Tsai MY, Wang S, Heidinger JM, Shumaker DK, Adam SA, Goldman RD, Zheng Y (2006) A mitotic lamin B matrix induced by RanGTP required for spindle assembly.Science 311:1887–1893
Vergnes L, Péterfy M, Bergo MO, Young SG, Reue K (2004) Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A 101:10428–10433
Verstraeten VLRM, Peckham LA, Olive M, Capell BC, Collins FS, Nabel EG, Young SG, Fong LG, Lammerding J (2011) Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc Natl Acad Sci U S A 108:4997–5002
Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, Zglinicki T von (2009) DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8:311–323
Willis ND, Cox TR, Rahman-Casañs SF, Smits K, Przyborski SA, Brandt P van den, Engeland M van, Weijenberg M, Wilson RG, de Bruïne A, Hutchison CJ (2008)Lamin A/C is a risk biomarker in colorectal cancer.PLoS One 3:e2988
Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P (2013) Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 201:1069–1084
Worman HJ, Bonne G (2007) “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res 313:2121–2133
Wu Z, Wu L, Weng D, Xu D, Geng J, Zhao F (2009) Reduced expression of lamin A/C correlates with poor histological differentiation and prognosis in primary gastric carcinoma. J Exp Clin Cancer Res 28:8
Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, Stewart CL, Colman A (2011) A human iPSC model of Hutchinson Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 8:31–45
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors acknowledge funding from the Agency of Science, Technology and Research A*STAR, Singapore.
Rights and permissions
About this article

Cite this article
Chojnowski, A., Ong, P.F. & Dreesen, O. Nuclear lamina remodelling and its implications for human disease. Cell Tissue Res 360, 621–631 (2015). https://doi.org/10.1007/s00441-014-2069-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-014-2069-4