
Abstract
Since the discovery of endocannabinoids and their receptors, two major members of the endocannabinoid family, anandamide (AEA) and 2-arachidonoylglycerol (2-AG), have been regarded almost as twin brothers. Pharmacological properties were initially considered to be similar, as these molecules were believed mutually exchangeable and almost indistinguishable in the regulation of synaptic functions, such as long- and short-term synaptic plasticity, and in behavioral aspects, such as learning and memory, reward and addiction, antinociception, and anxiety. In recent years, however, endocannabinoid signaling specificity began to emerge, in particular, due to the production of genetically engineered mice lacking key enzymes in endocannabinoid synthesis or degradation, together with the development of selective inhibitors of AEA or 2-AG catabolic enzymes. Evidence now suggests that AEA and 2-AG possess specific pharmacological properties, are engaged in different forms of synaptic plasticity, and take part in different behavioral functions. In this review, we provide an overview on similarities and specificities of the two endocannabinoids in the CNS and on the unresolved questions concerning their role in synaptic signaling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
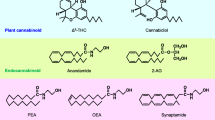
Among recent advances in pharmacology, the isolation of arachidonoylethanolamide (anandamide, AEA) and 2-arachidonoylglycerol (2-AG), which followed cloning and functional expression of the first type of cannabinoid receptors (CB1-R) [1], have provided a starting point to uncover the structure, chemistry, and functionality of the endocannabinoid (eCb) system. This system comprises a family of lipid molecules, receptors, and enzymes which role in the physiology and pathophysiology in the central and peripheral nervous systems has been spotlighted over the past 20 years.
As a consequence, the discovery of lipid mediators like AEA in 1992 [2] and 2-AG [3, 4] in 1995 has been the foremost evidence that the nervous system possessed its own endocannabinoid molecules, resembling in terms of the structure the main active principle of Cannabis plants, Δ9-tetrahydrocannabinol (THC) [5, 6]. AEA and 2-AG are lipid molecules derived from arachidonic acid (AA), which are conjugated with ethanolamide and glycerol, respectively (Fig. 1). Although similar in structure and general function, these eCbs exhibit large differences in terms of biochemical steps, receptor affinity, and breakdown pathways. AEA can be generated via a specific N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD)-dependent mechanism [7], whereas 2-AG is mainly synthesized by the combined action of a phospholipase C and two isoforms (α and β) of diacylglycerol-lipase (DAGL) [8, 9], although different alternative pathways are involved (Fig. 1) (see [10] for a recent excellent review). In addition, although AEA and 2-AG both bind to CB1-R and CB2-R, they have different affinities [3, 11, 12] and it is now clear that some AEA actions are also driven by receptor activation other than CB1 or CB2 [13]. Finally, it has been demonstrated that baseline levels of AEA and 2-AG are regulated by two main families of hydrolyzing enzymes, the fatty acid amide hydrolase (FAAH) and a monoacylglycerol lipase (MAG-L), respectively [14] (Fig. 1). As in their synthesis, the enzymatic machinery involved in AEA and 2-AG inactivation is far more complex, as summarized in Fig. 1. Besides FAAH, AEA can be hydrolyzed by N-acylethanolamine-hydrolyzing acid amidase (NAAA) [15] and oxygenated by cyclooxygenase-2 (COX-2) [16], lipooxigenase isoenzymes (LOX) [17], and by the P-450 cytochrome [18]. In addition to MAG-L, 2-AG can be hydrolyzed by a series of serine hydrolase α-β-hydrolase domain 6 or 12 (ABHD6, ABHD12) [19, 20].

Graphical depiction showing the main pathways involved in AEA and 2-AG formation and catabolism. Although both AEA and 2-AG are derivatives of AA-containing phospholipids, their production requires different enzymes and precursors. In fact, AEA is mainly synthesized by the activity of N-acyltransferase, which converts phospholipids into NAPE, and by the recruitment of several different enzymes. In fact, although the principal route of AEA biosynthesis involves a conversion form NAPE to AEA by a specific NAPE-PLD, recent studies have reported the existence of at least three other alternative pathways for AEA synthesis. In particular, it has been shown that: (a) NAPE can be converted into pAEA and, by PTPN22, into AEA; (b) a specific PLA2 is involved in lysoNAPE synthesis, which is then converted into AEA by a lysoPLD; and (c) ABHD4 hydrolyzes NAPE in lysoNAPE and the same enzyme yields GP-AEA. Ultimately by the activity of GDE1, GP-AEA is converted into AEA (top left panel). In line with multiple pathways for AEA production, 2-AG biosynthesis involves different routes, requiring the activity of either PLC or α and β isoforms of DAGL or PLA1 and PLC (bottom left panel). The main pathways involved in AEA and 2-AG breakdown also differs, requiring the activity of FAAH (top right panel) and MAG-L (bottom right panel), respectively. Furthermore, AEA and 2-AG catabolism might occur by the activity of other enzymes (e.g., NAAA, COX-2, and several LOX isoenzymes). Among the catabolic enzymes involved in 2-AG degradation, recent studies have reported a role of two hydrolases named ABHD6 and ABHD12, which activity dissociate 2-AG in AA and glycerol. AA arachidonic acid, ABHD4 6, 12, alpha, beta hydrolase 4, 6 and 12, COX-2 cyclooxygenase-2, DAGL alpha and beta diacylglycerol lipase alpha and beta, FAAH fatty acid amide hydrolase, GDE1 glycerolphosphodiesterase 1, GP-AEA glycerol-phosphoanandamide, LOX lipoxygenase, LysoNAPE lyso N-acyl phosphatidylethanolamine, LysoPLD lyso phospholipase D, MAG-L monoacylglycerol lipase, NAAA N-acylethanolamine-hydrolyzing acid amidase, NAPE N-arachidonoylphosphatidylethanolamine, NAPE-PLD N-acyl phosphatidylethanolamine phospholipase D, pAEA phosphoanandamide, PLA1 phospholipase A1, PLC phospholipase C, PTPN22 protein tyrosine phosphatase N22
Although early studies have regarded AEA and 2-AG as congeners with mutually exchangeable properties in the modulation of synaptic signaling, this notion has been overcome by recent discoveries about their sophisticated specificity in the regulation of several brain functions. To clarify this differential aspect, a pivotal role has been played by both the development of mouse lines lacking genes encoding for either FAAH or MAG-L [21, 22] and drugs that inhibit AEA and 2-AG catabolic enzymes (e.g., URB597 and JZL184) [23–25]. By either genetic or pharmacological FAAH and MAG-L inhibition, we have clarified the presumed interchangeable roles of AEA and 2-AG in synaptic plasticity, learning, memory, and reward, and interesting cues have been provided to drive new therapeutic applications for specific brain disorders. For this reason, in this review we present evidence that specific eCb molecules, through diverse biochemical pathways and mechanisms, may differentially modulate or coregulate physiological or pathological brain functioning.
AEA and 2-AG in synaptic plasticity: toward and beyond 2-AG
Synaptic plasticity modulation by CB1-R activation and the upstream involvement of eCbs in this process have attracted the interest of several research groups. Two decades of observations in this field have provided evidence that AEA and 2-AG are not similar in their functioning and that they perform highly specific roles in synaptic remodeling regulation [26].
eCbs have been regarded as main characters in synaptic plasticity since their discovery. Pioneer studies carried out by different groups have strongly supported this notion. In neurons, the intracellular cascade triggered by cannabinoid agonist binding to CB1-Rs depresses neurotransmitter release by activating presynaptic K+ channels [27] and inhibiting N − and P/Q-type Ca2+ channels [28–31]. A form of synaptic plasticity attributed to eCbs was the so-called “depolarization-induced suppression of inhibition” (DSI) [32, 33], which, occurring in a Ca2+-dependent fashion, was first observed in the hippocampus [33]. In 2001, eCbs were discovered as DSI mediators, being mobilized as needed (on-demand) by postsynaptic cells to inhibit GABA release from presynaptic neurons [34, 35] (Fig. 2). Stimuli leading to eCb synthesis are the activation of specific metabotropic receptors (metabotropic glutamate receptors, mGluRs; D2 dopamine, muscarinic), depolarization of the postsynaptic cell, or stimulation of excitatory afferents (see [10, 26, 36] for excellent reviews). The discovery of eCb-mediated DSI was soon followed by the description of eCb involvement in “depolarization-induced suppression of excitation” (DSE) [37] (Fig. 2) and by findings that AEA and/or 2-AG mediated long-term forms of synaptic plasticity (e.g., long-term depression, LTD, and heterosynaptic long-term potentiation, LTP) [38–41].

Synaptic mechanisms of action for anandamide (AEA) (top panel) and 2-arachidonoylglycerol (2-AG) (bottom panel). An important part of the knowledge about AEA and 2-AG functioning derives from studies where FAAH or MAG-L were pharmacologically blocked by their selective inhibitors, such as URB597 and JZL184, respectively. Top panel AEA is synthesized by postsynaptic NAPE-PLD or ABDH4 (or other enzymes, see Fig. 1) and hydrolyzed by FAAH. NAPE-PLD is expressed also presynaptically and AEA might act at postsynaptic TRPV1. Beside this mechanism, it was shown that AEA might retrogradely activate presynaptic TRPV1, or in an autocrinelike fashion, postsynaptic TRPV1. In particular, postsynaptic activation of TRPV1 leads to LTD. In vitro investigations have also reported a putative binding affinity of AEA on PPARα (dotted arrows). AEA functions as a classical retrograde messenger at presynaptic CB1-R at specific synapses in the BLA and in the DR, where it evokes LTDi and DSE, respectively. Bottom panel 2-AG is synthesized mainly by DAGL α and β isoforms (inhibited by THL or other selective compounds, see [10]). Synthesis involves a rapid production of 2-AG which, in this example, is triggered by activation of mGluRs in the postsynaptic cell. mGluRs, via aGq/11 protein, activate a phospholipase C (PLC) dependent increase in intracellular Ca2+ (mobilized from intracellular stores by IP3) and DAG mobilization. This intracellular pathway leads to 2-AG-release. 2-AG activates CB1-R-expressed on axon terminals, which ultimately suppress either GABA or glutamate release and trigger short- and long-term forms of synaptic plasticity (DSI, DSE, LTDi, or LTD). 2-AG is then hydrolyzed by MAG-L expressed in the presynaptic terminals or ABDH6, expressed postsynaptically. ABHD4 α,β-hydrolase-4, ABHD6 serine hydrolase α-β-hydrolase-6, BLA basolateral amygdala, CB1 cannabinoid type-1 receptor, DAG diacylglycerol, DR dorsal raphe, DSE depolarization-induced suppression of excitation, DSI depolarization-induced suppression of inhibition, FAAH fatty acid amide hydrolase, Glu glutamate, IP 3 inositol trisphosphate, LTD long-term depression at excitatory synapses, LTDi long-term depression at inhibitory synapses, MAG monoacylglycerol, NAPE-PLD N-acyl phosphatidylethanolamide phospholipase D, PLC phospholipase C, PPARα peroxisome proliferator-activated receptor type-α, THL tetrahydrolipstatin, TRPV1 transient potential vanilloid receptor type-1
Differential role of AEA and 2-AG in synaptic plasticity
Evidence suggests that 2-AG is the main eCb involved in synaptic plasticity regulation [36, 42], whereas AEA plays a minor role. Hence, studies performed with selective DAGL (i.e., tetrahydrolipstatin, THL) or MAG-L (i.e., JZL184) inhibitors and genetic ablation of either enzyme has underscored the critical 2-AG involvement in short- and long-term modulation of synaptic plasticity. Among brain areas where 2-AG exerts its properties as a neuromodulator, much research has been dedicated to the hippocampus and cerebellum. For instance, 2-AG mediates LTD at inhibitory synapses (LTDi) in the hippocampus CA1 region [38] and mGlu-dependent synaptic plasticity in hippocampal slices [43]. By using MAG-L−/− mice, in which 2-AG brain levels are considerably elevated due to the genetic ablation of 2-AG-degrading enzymes, Pan et al. [22] reported an augmented DSI in the hippocampus CA1 region, which was accompanied by a potentiated LTP and an increase in learning performance in knockout mice when compared to wild-type littermates. Other evidence comes from Mackie's group, who demonstrated that 2-AG application and the employment of MAG-L inhibitors modulated both DSE [44, 45] and DSI [46, 47] in autaptic hippocampal neurons. Interestingly, the latter effects do not seem to involve AEA, since the blockade of both excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs) by AEA [44, 46] could not be washed out, a profile inconsistent with AEA being a mediator of short-term synaptic depression.
Also synapses into Purkinje cells (PC) in the cerebellum have been identified as a specific site of action for 2-AG. For instance, studies in rodents have uncovered that 2-AG is required in mGluR-dependent plasticity [48] and LTD in PC [39]. Furthermore, separate subsets of experiments by Safo and Regehr [39] also showed that 2-AG is involved in DSI recorded in the cerebellar cortex, suggesting that this eCb might participate in the regulation of motor coordination and cognitive efficiency. Conversely, Szabo et al. [49] provided further evidence that AEA is not engaged in short-term forms of plasticity in the cerebellum, since FAAH manipulation did not affect DSI on PC, indicating that 2-AG likely plays a pivotal role in these eCb-mediated functions.
Among other synaptic remodeling aspects involving 2-AG, several authors focused on cortical, striatal, and limbic circuits. Laforucade et al. [50] reported a 2-AG-mediated effect in LTD expression in V/VI layers of the prefrontal cortex (PFC), and Yoshino et al. [51] recently showed that 2-AG modulates DSI in the same region. Overall, acting on both short- and long-term forms of plasticity, 2-AG might be a suitable candidate to explain synaptic modifications underlying cognitive and motivational functions mediated by the PFC.
Electrophysiological and molecular results by our group uncovered a prominent role of 2-AG in regulation of excitatory afferents to dopamine (DA) neurons in the ventral tegmental area (VTA), a brain region primarily involved in response to rewarding stimuli [52]. Furthermore, Seif et al. [53] described that 2-AG enabled subthreshold doses of DA D1 and D2 receptor agonists to increase firing activity of nucleus accumbens (NAc) neurons [53]. Taken together, these findings point to 2-AG as a neuromodulator involved in synaptic plasticity in the mesoaccumbens pathway.
2-AG involvement in retrograde signaling modulation in the brain has been investigated by genetic ablation of specific DAGL isoforms. In fact, the DAGLα isoform plays a prominent role in 2-AG synthesis [54], whereas the β isoform has a minor involvement in this process. In line with this evidence, it has been shown that DAGLα−/− mice display a dramatic reduction in 2-AG levels and a complete abolishment of the main forms of eCb-mediated retrograde signaling [54, 55]. In light of these findings, 2-AG activity is sufficient to explain eCb-driven synaptic plasticity. It must be pointed out that AEA also decreases in DAGLα−/− mice, leaving open the possibility that this eCb could also mediate synaptic plasticity. A possible explanation of the parallel decrease in 2-AG and AEA brain levels is provided in [10]. Here the author hypothesizes that, brain 2-AG levels being ~200-fold higher than those of AEA, 2-AG catabolism is a source of free ΑA [56], which can be reesterified into phospholipids. Reduced AA-containing phospholipids result in reduced brain AEA levels [10]. Nonetheless, recent studies are now complicating the scenario by involving different DAGLα pools to explain different 2-AG-mediated effects in response to distinct stimuli [57] and reporting that DAGL is not recruited in hippocampal DSI [58], suggesting other synthetic pathways for 2-AG regulated forms of short-term adaptation.
Beside the broad 2-AG effect in neuronal plasticity and retrograde signaling, AEA appears as a character of secondary importance; nonetheless, it exerts apparently finer and more discrete actions at specific synapses. In this regards, it must be pointed out that AEA binds not only to CB1- and CB2-Rs, but also to vanilloid (e.g., TRPV1) [59] and nuclear receptors (e.g., PPARs) [60, 61]. In particular, since many effects of AEA are mediated via TRPV1 receptors and prevail over those mediated by CB1-Rs or CB2-Rs, this molecule is more an endovanilloid than an endocannabinoid [62]. Noteworthy, these multiple AEA receptor targets are emerging as additional modulators of synaptic functions (see below).
Studies performed by either genetic FAAH deletion or AEA administration showed that AEA in a CB1-dependent manner regulates LTDi in the basolateral amygdala (BLA) [63] and DSE evoked by glutamatergic afferent stimulation in serotonin neurons of the dorsal raphe [64]. In the former study, the possible coinvolvement of 2-AG in LTDi in the BLA was ruled out, and in the latter study, 2-AG contribution was not investigated. AEA might also modulate synaptic plasticity through activation of other receptor types, as demonstrated by Edwards et al. [65]. In fact, the authors showed that in a specific CA1 layer, the stratum radiatum, AEA triggered LTD at excitatory synapses on interneurons in a TRPV1-dependent fashion [65]. In 2010, Chavez et al. [66] and Grueter et al. [67] discovered that AEA can act in an autocrine-like fashion by activating postsynaptic TRPV1 in the hippocampus and NAc. Activated TRPV1 trigger a postsynaptically mediated LTD resulting from endocytosis of AMPA receptors. These results broaden the scenario about eCb modulation of synaptic plasticity in cognitive/subcortical areas, where the control by 2-AG had never been questioned before.
Overall, findings about AEA and 2-AG reported here support the notion that these molecules play highly specific roles in different forms of synaptic plasticity within the central nervous system (Fig. 2). Intriguingly, the possibility exists that AEA modulates synaptic transmission with mechanisms different from the classical retrograde action. This hypothesis is supported by studies showing that in hippocampal glutamatergic axon terminals, NAPE-PLD, the enzyme that synthesizes AEA, is expressed presynaptically rather than postsynaptically [68]. This localization, coupled with the absence of CB1-Rs on the same terminals and the presence of FAAH in somatodendritic domain of postsynaptic principal cells, suggests that this eCb, but possibly also other N-acylethanolamides synthesized by the same enzyme, might mediate both short- and long-term forms of synaptic plasticity through an anterograde mechanism. The targets of anterograde AEA were not identified by Nyilas at al., but postsynaptic TRPV1 receptors are likely candidates. Another mechanism, suggested by Cristino et al. [69], is that the postsynaptic NAPE-PLD localization in the cerebellum is compatible with an autocrine action of AEA and other N-acylethanolamides on synaptic plasticity via TRPV1 receptors [70].
Hence, this AEA feature might support and integrate 2-AG-mediated effects in forms of synaptic plasticity that require interactions between both eCbs. In line with this possibility, studies have shown that in the extended amygdala 2-AG and AEA modulate short- and long-term forms of synaptic plasticity through retrograde and autocrine-like mechanisms, respectively [71]. In fact, these authors showed that while 2-AG mediates short-term depression (STD) through CB1-R-dependent retrograde signaling, AEA released by a postsynaptic mGluR5-dependent mechanism modulates LTD through postsynaptic TRPV1 activation. In light of these findings, it appears that AEA and 2-AG cooperate by using different strategies to modulate synaptic plasticity in brain regions that govern highly dynamic processes [72]. Other examples of AEA and 2-AG interactions in the same form of synaptic plasticity within the same brain region have also been reported. For instance, in vitro studies by Sheinin et al. [73] demonstrated that both AEA and 2-AG are involved in STD on pyramidal neurons of the CA1, through a mechanism that requires postsynaptic mGluR5 activation leading to eCb release and a reversible decrease in IPSC amplitude form apposing GABA axons. Overall, recent literature supports the notion that AEA and 2-AG play mostly specialized roles to modulate complex forms of synaptic remodeling, which ultimately takes part in adaptive responses to environmental requests.
Endocannabinoids in neuroprotection: from physiology to injury
Neuronal damage caused by toxic or ischemic insults, such as energy or oxygen deprivation, or traumatic injury, is worsened by excitatory amino acid release, depolarization of cell membranes, and intracellular Ca2+ increases. The same stimuli are able to trigger release of eCbs from a variety of neurons. eCbs might reduce potential excitotoxic damage by depressing excitation strength, and therefore, they can be envisaged as neuroprotective agents [74]. Consistently, mice lacking the CB1 receptor gene are more susceptible to injury after stroke [75] or kainic-induced epileptic seizures [76]. Accordingly, exogenous cannabinoids exert neuroprotection in a variety of in vitro and in vivo neuronal injury models [77]. When specific eCbs are concerned, a number of in vivo and in vitro studies suggest that 2-AG prevails over AEA in neuronal repair and adaptation mechanisms induced by injury.
AEA and 2-AG in neuroprotection
The overwhelming interest in 2-AG, which has dominated the debate on AEA and 2-AG involvement in neuronal repair, is supported by a number of findings on its protective role in different injury models. Studies over the last 10 years have provided evidence that 2-AG is recruited as protective neuromodulator against traumatic brain injuries [78], cerebral focal ischemia in mice [79], and β-amyloid insults [80]. Electrophysiological studies carried out in our laboratory highlighted that 2-AG is the main eCb enrolled to protect DA neurons against hypoxia/ischemia [81]. Additionally, studies on underlying 2-AG mechanisms in neuroprotection revealed that not only CB1-Rs are involved. Experimental observations from different groups suggested multiple 2-AG-activated pathways, which include CB2-Rs and other atypical receptors. This is consistent with the fact that 2-AG is a full agonist at CB2-Rs, whereas AEA is a partial agonist and might even behave as a functional antagonist during elevated tonic activation of CB2-Rs [82, 83].
Based on these studies, 2-AG might induce neuroprotection through either a neuronal CB1-R-dependent mechanism [76, 81] or CB2-R activation [84–86]. CB2-R are expressed by immune cells and microglia and upregulated in activated microglia [87], and stimulation by CB2-R ligands, such as 2-AG, increases microglial cell migration and decreases the release of several factors, such as proinflammatory cytokines [85, 86]. These molecules are cytotoxic and can secondarily activate astrocytes leading to a further induction of the expression of inflammatory factors. These proinflammatory mediators promote influx into the CNS of immunocytes from peripheral non-neuronal sites that also express CB2-Rs [87, 88]. A recent paper by Kreutz et al. found that through the activation of an abnormal cannabidiol receptor, 2-AG was able to induce neuroprotection by modulating migration and proliferation of microglial cells after brain lesion in the hippocampal dentate gyrus [89].
Most of these studies did not take into account the possible synergy between 2-AG and AEA. Indeed, other studies revealed that AEA also mediates neuronal protection. In agreement with the latter consideration, molecular studies have reported that AEA levels vary significantly when the brain is exposed to a number of insults. For instance, early studies by Hansen et al. [90], who reported changes in eCb levels after mild to moderate brain injuries, observed augmented AEA levels, but not 2-AG. Moreover, other molecular findings extended the neuroprotective AEA effect to other brain damage models, such as the controlled in vivo blood flow interruption [91] and middle cerebral artery occlusion (MCAO) [92]. Interestingly, the latter effect might be likely due to FAAH activity reduction and increased NAPE-PLD levels [92], suggesting AEA as a protective agent in acute degeneration phases. Amantea et al. [92] reported that AEA levels in MCAO mice were threefold higher in the striatum compared to controls, with substantially no changes in the cortex, which indicated that AEA might be protective in specific brain areas. Additionally, in vitro assays in transfected cells by Iuvone et al. [93] showed that elevated AEA levels were protective against proliferation of activated glial cells in neural damage. In vivo studies have reported that AEA provided support to white matter during neurodevelopment, which protected the neonatal brain in a model of excitotoxic lesion induced by several agents (i.e., S-bromo-willardiine and AMPA-kainate receptor agonists) [94].
Studies carried out with FAAH inhibitors like URB597 and AM374 have broadened the range of AEA effects in neuroprotection. For instance, AEA catabolism inhibition appears to be protective in distinct injury models, such as retinal damage in ischemic/reperfusion studies [95], kainic acid-induced seizures in the hippocampus [96], and multiple sclerosis-induced spasticity [97]. This might represent an interesting frontier for the development of new pharmacological approaches against neuronal damage based on therapeutic efficacy of FAAH inactivation. However, beside this number of findings on protective roles of enhanced AEA levels, other studies have underscored pathological conditions where AEA might not be involved. In line with this possibility, De Lago et al. [98] demonstrated that UCM709, an AEA uptake inhibitor, does not prevent neurodegeneration nor improve neurological recovery in Huntington's disease (HD), autoimmune encephalitis (EAE), and Parkinson's disease models. On the other hand, UCM709 does improve hyperkinesia and spasticity of the hind limb in HD and EAE models, suggesting a strong involvement of AEA in some, but not all, symptoms of these degenerative disorders.
Interplay between AEA and 2-AG as protective agents
In contrast to studies that described AEA and 2-AG as separate neuromodulators involved in neural protection, the literature lacks observations about synergistic/cooperative effects between these eCbs in the same topic. One of the few studies that investigated this interplay showed that in a cerebral focal ischemia model, electroacupuncture pretreatment induced neuroprotection, upregulated the neuronal expression of rat brain CB1-Rs, and elevated the brain tissue content of AEA and 2-AG [99]. The same study also showed that pretreatment with both AEA and 2-AG reduced infarct size and improved neurological outcome in rodents through CB1-Rs.
Role of eCbs in learning and memory: toward and beyond AEA
THC, an exogenous counterpart of eCbs, has been consistently regarded as a molecule with detrimental effects on memory and learning. In contrast to studies focused on synaptic plasticity, those on learning and memory have traditionally spotlighted AEA as a principal eCb. However, recent findings have also taken into account the role of 2-AG in specific plasticity forms associated with mnemonic and learning functions.
AEA and 2-AG in learning and memory
Among compartments in which memory can be divided, AEA has been often regarded as a strong modulator of acquisition phases. In fact, early observations by Murillo-Rodriguez et al. [100] reported that AEA regulates several processes that ultimately drive memory acquisition. In this study, by taking advantage of inhibitory avoidance paradigms, the authors showed that AEA interacts in memory acquisition only in late phases of testing (24 h after training), being ineffective in such modulation shortly after (15 min) the training session. This result is even more interesting, since they reported AA effects in both parameters (15 min and 24 h). Phospholipids containing AA participate in biochemical pathways to produce both AEA and 2-AG; thus, eCbs might play a role in the early stages of memory acquisition by switching competencies in modulation of cognitive functioning. However, further studies should better characterize 2-AG involvement in acquisition of memory traces.
A wealth of preclinical data underscored that AEA microinfusions in the BLA or the administration of the methylated, metabolically stable form of AEA (methAEA) prevent memory retrieval (24 h after training) during aversive memory acquisition tasks [101] and in encoding phases of memory acquisition under delayed nonmatch-to-sample tasks [102], respectively. These studies focused on specific brain areas that encode for emotional learning and memory such as the BLA and hippocampus and provided evidence on AEA involvement in specific phases of memory formation. The latter study also ruled out TRPV1 in AEA-induced effects on hippocampal neuron inhibition, indicating that CB1-Rs are likely main players [102]. Moreover, by performing experiments using FAAH−/− mice, Wise et al. [103] showed that AEA plays a critical role in the improved acquisition of specific subsets of emotional memory. In this study, the authors highlighted that this eCb only modulates aversively motivated memory formation, being ineffective on the appetitive counterpart and broadened the scenario on AEA participation in early memory performance.
Other observations have provided evidence that AEA effects on memory might be multidirectional. For instance, a study reported that FAAH−/− mice have faster acquisition during the first session of working memory tasks compared to wild-type littermates [104]. Nonetheless, no difference between FAAH−/− and FAAH+/+ have been reported in subsequent sessions of the same task. The same authors showed that AEA exogenous application disrupted working memory performance only in FAAH −/− mice and was ineffective in controls. Further, methAEA impaired performance both in knockout and wild-type littermates. These latter results suggested that elevated AEA levels were responsible for worsening or improvement in different phases of memory performance depending on brain concentrations of this eCb. Finally, among other possible explanations of paradoxical AEA effects in memory performance modulation, studies have taken into account the possibility that receptors other than CB1-R might be involved. For example, Mazzola et al. [105] have uncovered an URB597-induced effect in memory acquisition that appeared to be mediated by the stimulation of a family of nuclear receptor transcription factors, known as α-type peroxisome proliferator-activated receptors (PPARα). However, since this study did not clarify whether AEA or other FAAH substrates, such as noncannabinoid N-acylethanolamides, were recruited in this effect, further studies should better analyze this possibility.
Other than memory acquisition, AEA is apparently involved in memory consolidation and extinction, expanding the range of action of this eCb in different phases of mnemonic mechanisms. In fact, recent studies have found that FAAH inactivation by URB597 worsened memory consolidation by producing amnesialike effects during the object recognition test [106] and enhanced the extinction of aversive memories [107]. Interestingly, the first observation was not replicated using JZL184; thus, this specific task most likely does not involve 2-AG [106]. These data have shed some light on the precise AEA and 2-AG functions in memory-relevant brain regions. Besides AEA, the role of 2-AG in the regulation of mnemonic functions has been investigated. In fact, since 2-AG plays a prominent role as a retrograde messenger in the hippocampus, where it modulates short- and long-term forms of plasticity (see above), it is highly likely that it is involved in memory-related synaptic mechanisms. Studies are now taking advantage of new selective MAG-L inhibitors and genetic tools. Among them, Pan et al. [22] showed an improved performance in Morris' water maze and novel object recognition test in MAG-L−/− mice compared to MAG-L+/+ littermates, providing new cues to evaluate the role of 2-AG in learning and memory tasks. On the contrary, other studies have found that augmented PFC 2-AG levels might explain memory and cognitive disruptions in an animal schizophrenia-like model [108]. Therefore, the role of 2-AG in these complex behavioral functions remains controversial. Finally, a recent work by Yoshida et al. [109] assessed 2-AG-regulated synaptic plasticity underlying extinction of fear memory. In fact, in this study, results from morphological and electrophysiological experiments suggested that biosynthetic pathways involved in 2-AG formation play a prominent role in the DSI of afferents from cholecystokinin-positive GABA interneurons to pyramidal cells in the BLA basal nucleus. To conclude, it appears that AEA and 2-AG have segregated roles in memory and learning regulation, with the former exerting broader effects, but with growing evidence that 2-AG is recruited to some functional subcompartments.
Interplay between 2-AG and AEA in learning and memory
Evidence is lacking in the literature that 2-AG and AEA cooperate in memory modulation. To our knowledge, only Cuellar and Isokawa [110] provided data about an interplay between AEA and 2-AG in memory and learning. They described an involvement of both eCbs in the ghrelin-induced inhibition of CREB activity and NR1 function in the hippocampus. According to this study, AEA and 2-AG might cooperate at different levels to produce neurobiological modifications relevant for cognitive functions through direct action on NMDA receptors and CB1-Rs, respectively. Further studies are needed to assess differential involvement of the two major eCbs in memory-related functions.
Role of AEA and 2-AG in nociception: crosstalk and specificities
THC is antinociceptive when administered to humans and animals and the employment of Cannabis derivatives as therapeutic analgesics is still debated [111, 112]. Early studies have proposed spinal and supraspinal mechanisms through which THC exerts analgesic effects in rodents [113], and this effect is now well characterized. In addition to THC, eCbs possess analgesic properties and exerts differential and similar effects in specific aspects of pain perception.
Differential involvement of AEA and 2-AG in nociception
Although interactions between AEA and 2-AG in pain modulation have been proposed, several studies have highlighted a differential involvement of these eCbs in specific nociception aspects. For instance, Starowicz et al. [114] reported that local infusion of AEA or URB597 produced antiallodynic and antihyperalgesic effects in an animal model of spinal neuropathic pain. This study underscored that spinal AEA produces antihyperalgesic effects by acting either at CB1 or TRPV1 receptors depending on the local concentration. In particular, CB1-mediated effects prevailed at low concentrations, whereas the TRPV1-mediated ones at high concentrations.
On the other hand, 2-AG likely played a role in pain modulation. According to Guindon et al. [115], local injection of 2-AG dose-dependently reduced nociception in the late phase of the formalin-induced pain test. These results have been replicated by the same group by administering the MAG-L inhibitor URB602. They observed an additive antinociceptive effect when both 2-AG and URB602 were coinfused at their EC50, suggesting that 2-AG might be a potential substrate for novel pharmacological approaches against nociception. Additionally, recent investigations described AEA and 2-AG effects in distinct regions of the dorsolateral (dl) and the ventrolateral (vl) periaqueductal gray (PAG). In fact, Olango et al. [116] used formalin nociception tests and reported that physiological enhancements of AEA levels in dlPAG was involved in fear-conditioning induced by nociception, whereas Liao et al. [117] described antinociceptive properties of 2-AG in vlPAG. Notably, the latter results provided a logic of action of 2-AG. The authors propose that 2-AG formation was triggered by TRPV1 activation on glutamatergic terminals in vlPAG. Released glutamate, in turn, activated postsynaptic mGluR5 receptors, which, via Gq protein, stimulated phospholipase C to yield DAG which is then deacylated by DAGL to 2-AG [117].
Further studies have evaluated the dual, yet specific, involvement of AEA and 2-AG in pain perception. Among them, Spradley et al. [118] recently reported a nonoverlapping effect of enhanced levels of AEA and 2-AG, induced by respective catabolism inhibitors in the modulation of capsaicin-induced nociception. Here, the authors found that either AEA or 2-AG regulated specific nociceptive features such as nocifensive behavior, thermal hyperalgesia, and mechanical allodynia, and reported that FAAH inhibition was only able to counteract the latter parameter, whereas JZL184 (the MAG-L inhibitor) affected the others. These results underscored that AEA and 2-AG may be separately recruited to exert antinociception in different brain subregions involved in pain.
Interplay between AEA and 2-AG in nociception
AEA and 2-AG synergy in modulation of pain have been reported in several studies. Early results showed a coordinated effect of AEA and 2-AG in prolonged footshock modulation in stress-induced analgesia [119] and indicated that both eCbs have augmented levels in the ipsilateral lumbar V dorsal root ganglion in an animal model of spinal nerve ligation [120]. The employment of specific FAAH and MAG-L inhibitors facilitated the investigation of eCb effects on pain. In fact, pharmacological studies carried out by Kinsey et al. [121] have shown that FAAH and MAG-L inhibition might be suitable targets to develop new pharmacological strategies to treat nociception. In this study, they showed that enhanced levels of AEA or 2-AG attenuates mechanical and acetone-induced cold allodynia but through different receptor-dependent mechanism. In fact, FAAH inactivation was effective through either CB1-R- or CB2-R-dependent mechanisms, whereas effects due to augmented 2-AG levels were counteracted only by CB1-R antagonism [121]. Another example of interplay has been shown by other studies where either AEA or 2-AG local injection or the administration of FAAH and MAG-L inhibitors were reported to reduce mechanical hyperalgesia in a mouse model of bone cancer [122, 123]. These studies are more interesting in light of the finding that AEA and 2-AG act through clearly separate mechanisms, involving dorsal root ganglion CB1-R and peripheral CB2-Rs, respectively [122, 123]. This would suggest that besides a switch in molecular pathways involved in pain, AEA and 2-AG may synergistically strengthen their role in nociception. Petrosino et al. [124] provided evidence of an interesting cooperation between AEA and 2-AG, which levels were enhanced after 3 and 7 days of chronic constriction injury of the sciatic nerve in the rat. Interestingly, after 3 days, AEA and 2-AG levels were increased only in the spinal cord and PAG, whereas after 7 days, augmented concentrations were detected also in the rostral ventral medulla.
An example of how AEA and 2-AG can cause different effects in the vl-PAG was provided also by Maione et al. [125]. These authors suggested that while AEA can produce antinociceptive effects via TRPV1 activation, 2-AG, like the synthetic agonist WIN55,212-2, may cause both antinociceptive effects by acting at CB1 on presynaptic GABAergic terminals and pronociceptive effects by acting at CB1 on presynaptic excitatory axons innervating vl-PAG output neurons. These results suggest that combined modulation induced by eCbs is present in both spinal and supraspinal levels and that AEA and 2-AG participate in chronic pain modulation.
AEA and 2-AG in anxiety
The role of cannabinoids in anxiety is highly debated. Exogenous cannabinoids display both anxiolytic and anxiogenic properties [126]. A likely explanation is that this bidirectional effect might depend on the dose administered (e.g., high doses of synthetic cannabinoids are anxiogenic and low doses are anxiolytic) [127] and by other parameters [128]. Nonetheless, a number of studies have reported an anxiolytic eCb effect. At first glance, AEA was studied in more detail in anxiety, when compared with other eCbs.
Do AEA and 2-AG differentially counteract anxiety?
AEA involvement in anxiety has been studied for a long time, and interest has not yet faded. In 2003, Kathuria et al. provided the first evidence on URB597 anxiolytic properties [24, 129], likely mediated by AEA through CB1-R activation. Subsequent studies have confirmed this idea using FAAH inhibitors or FAAH−/− mice. For instance, it was shown that anxiety-like behavioral responses were reduced by URB597 or by FAAH knockout through a CB1-R-dependent mechanism [130]. Additionally, among possible mechanisms by which both genetic and pharmacological FAAH blockade counteract anxiety, Rossi et al. [131] showed that enhanced AEA levels might act on striatal CB1-Rs, which levels in anxious phenotypes were dramatically downregulated. Finally, a recent paper by Cippitelli et al. [132] has proposed that AEA may be implicated in the modulation of nicotine withdrawal-induced anxiety responses, as measured by elevated plus maze and shock-probe defensive burying paradigms [132]. These authors observed AEA fluctuations with unaltered 2-AG concentrations in brain regions including amygdala, PFC, hippocampus, and hypothalamus during nicotine withdrawal in dependent rats. Further, they showed a protective effect of URB597 in the expression of withdrawal syndrome in these animals but not in the reduction of withdrawal-induced somatic signs.
Other studies have focused on AEA in anxiety by taking advantage of different tools, such as membrane transport inhibitors or local AEA injections. AM404, an AEA transport blocker, reportedly enhanced AEA levels to mediate anxiolytic responses in three animal anxiety models through PFC CB1-Rs [133] and low doses augmented the time spent in open arms of the elevated plus maze test [134]. Studies with AEA transport blockers have the limitation that these drugs might also interfere with 2-AG membrane transport [135, 136]; therefore, interpretation of results is difficult.
Local administration of AEA catabolism inhibitors has also confirmed the role of this eCb in anxiety. For example, the ventral hippocampus (vHIP) was investigated in a study where in situ URB597 administration improved mice responses using the elevated plus maze paradigm [137]. Nonetheless, AEA as antianxiety agent is questioned by conflicting evidence, which reported that AEA-induced effects may vary according to different parameters. For instance, studies have proposed that the AEA effects as an antianxiety molecule strongly depended on environmental conditions, since a change in environmental aversiveness in the experimental setup allowed a rapid AEA shift from anxiolytic to ineffective [138]. Further observations by Scherma et al. [139] reported that low AEA and URB597 administered together more likely exerted anxiolytic effects in rats under place-conditioning procedures and that, conversely, relatively high doses of AEA facilitated anxiety.
In accordance with these latter results, intriguing observations by Rubino et al. [140] reported that while microinjection of low URB597 doses in the PFC exerted anxiolytic effects, the administration of the same compound at high doses did not produce any effect or even increased anxiety in animal models [140]. The same authors also showed that virally mediated overexpression of FAAH in the PFC was anxiogenic. Finally, a study by Campos et al. [141] demonstrated that AEA microinjections in the vHIP in nonstressed animals induced anxiogenic behavior as measured by the elevated plus maze test, but under restrained conditions before the test (24 h), animals that received a local injection of the same eCb in the vHIP showed reduced anxiety behaviors. It is important to point out that in these studies, TRPV1 have a role in AEA effects on anxiety, to the extent that AEA act as endocannabinoid or endovanilloid depending on its concentrations. This dual function of AEA, together with environmental conditions, might indeed explain paradoxical results in animal models of anxiety, as well as in fear and compulsive behaviors [142–144].
Recent studies have also investigated the role of 2-AG in anxiety. In fact, 2-AG effects apparently converge into specific aspects of anxious experiences, such as those emerging from complex and unpredictable environmental requests. In line with this consideration, Sumislawski et al. [145] showed 2-AG enhancement during chronic, stress-induced, anxiety-like behavior in the BLA. The authors showed that 2-AG acts by potentiating LTD on GABAergic synapses and that this effect was likely due to reduced synthesis of MAG-L. Finally, additional results have also demonstrated a specific effect of MAG-L inactivation in anxiety response modulation under high, but not low, levels of environmental aversiveness [146]. Overall, AEA and 2-AG effects described here indicated a prominent involvement of AEA in such processes, which can be implemented by 2-AG under specific conditions.
Interplay between AEA and 2-AG in anxiety
Tools to deactivate catabolic enzymes for AEA and 2-AG did not only provide evidence of differential involvement of these molecules in anxiety modulation, but also interesting interactions between the two eCbs. A recent paper by Kinsey et al. [147] demonstrated that FAAH and MAG-L inhibitors prevented obsessive–compulsive (OC) behavior during a marble burying test. OC behavior represents an interesting aspect of anxiety disorders and this finding may drive future research on pharmacology for OC disorders, where therapeutic approaches remain poorly effective. Kinsey's results are even more intriguing, since he showed that the effects of FAAH and MAG-L inhibitors were due to the CB1-R activation. This finding was not unique though, since other studies have reported that anxiolytic properties of FAAH and MAG-L inhibitors in the elevated plus maze test and other paradigms were caused by the activation of CB1- and CB2-Rs, respectively [106], which complicated the scenario concerning receptors involved in anxiety modulation. In light of these findings, it is possible that synergies between AEA and 2-AG recruit a similar/different mechanism according to specific subtypes of anxiety manifestations and specific brain areas.
AEA and 2-AG in reward and drug addiction: the paradox of addictive molecules counteracting addiction
Since the discovery of AEA and 2-AG, much interest has focused on whether these molecules are involved in neurobiological processes underlying drug abuse and addiction. Similar with the psychoactive component of Cannabis, THC, both eCbs are self-administered by nonhuman primates [148–150]. This effect is also related to AEA and 2-AG involvement in synaptic plasticity in reward-related areas. In fact, neurobiological processes that ultimately drive drug addiction require an array of short- and long-term modifications in synaptic plasticity in motivational and emotional brain areas [151], where AEA and 2-AG might take part.
Studies over the last two decades have substantially confirmed this hypothesis. Nonetheless, some recent finding have complicated this scenario by providing evidence that FAAH inhibitors, i.e., URB597 or PF-3845, are not self-administered by nonhuman primates [149], do not produce generalization to the discriminative effects of THC in rats [152], do not cause cross-tolerance with cannabinoid agonists, and do not desensitize CB1-Rs upon chronic administration [153]. Inhibition of MAG-L, on the other hand, causes behavioral effects not observed following chronic FAAH blockade, such as hypomotility and hyperreflexia [25, 154], physical dependence, tolerance to CB1-R agonists, CB1-Rs desensitization, and downregulation [153], which suggest a broader impact on the brain cannabinoid system [153]. Although neither FAAH nor MAG-L inhibitors showed abuse liability, experiments with the recently developed dual FAAH/MAG-L blocker (JZL195) reported THC-like effects in drug discrimination tests [154].
Specificity of 2-AG and AEA in rewarding and addictive properties of drugs of abuse
Among the vast number of studies on the possible interaction between the eCb system and addictive drugs in the brain reward circuitry, one of the most explored areas is the role of AEA in nicotine addiction. For instance, using the AEA catabolism inhibitor, URB597, Scherma et al. [155] reported that FAAH inhibition prevents nicotine-induced behavioral and neurochemical effects. Consistently, our studies performed with electrophysiological techniques [156, 157] demonstrated that FAAH inhibition blocks nicotine-induced electrophysiological effects predictive of its rewarding properties, i.e., excitation of VTA DA neurons' firing rate [156] and inhibition of medium spiny neurons in the shell of the NAc (ShNAc) [157]. However, our evidence suggested that AEA was probably not involved. In fact, URB597, by blocking FAAH, does not only raise AEA levels, but also concentrations of other noncannabinoid N-acylethanolamides, i.e., oleoylethanolamide (OEA) and palmitoylethanolamide (PEA). These molecules are endogenous PPARα ligands and most URB597-mediated effects were blocked by the PPARα antagonist, MK886 [156, 157]. In a separate subset of experiments, we showed that both in vivo and in vitro administration of AEA and its hydrolysis-resistant analogue, methAEA, did not mimic URB597-mediated action on VTA DA neurons [156]. These results do not completely rule out AEA in nicotine-induced effects in the brain reward circuitry, since our further observations have discerned that URB597 effects on nicotine action in the ShNAc were prevented by both CB1-R and PPARα antagonists (e.g., rimonabant and MK886, respectively) [157]. Thus, this latter result might suggest interplay between AEA and other N-acylethanolamides.
In line with this possibility, recent studies by Scherma et al. [158] and Gamaleddin et al. [159] have clarified the role of AEA in nicotine rewarding and addictive behaviors. They found that the AEA transport inhibitors, AM404 and VDM11, which enhance AEA levels, but not those of OEA and PEA, block nicotine-induced conditioned place preferences, nicotine-induced elevation of DA levels to the ShNAc [158] and attenuate the reinstatement of seeking behavior induced by nicotine-associated cues and nicotine priming [159]. In neither study, however, the mechanism by which AEA transport inhibitors exerted their effects was investigated by using selective CB1 or TRPV1 antagonists. Furthermore, parallel observations by Cippitelli et al. [132] showed that AEA levels in brain areas involved in nicotine rewarding responses are physiologically altered in nicotine-abstinent mice, providing evidence that FAAH inhibition may also counteract withdrawal signs in animal models of nicotine addiction. No information on receptors involved is provided by this study. FAAH inhibitors represent suitable candidates for antismoking medications but other studies have reported contrasting results. For instance Merritt et al. [160] showed that FAAH−/− mice developed an augmented nicotine-induced condition place preference, enhanced withdrawal signs in a model of nicotine abstinence, and condition–place aversion in mecamylamine-induced nicotine withdrawal.
In contrast to the evidence on AEA and nicotine interactions in the brain reward circuitry, no studies have yet taken into account 2-AG involvement in the modulation of nicotine-rewarding and nicotine-addictive properties, with the exception of González et al. [161] who showed that chronic nicotine exposure elevates AEA and 2-AG levels in the brain stem and midbrain and decreases their levels in the hippocampus, striatum, and cortex. Regarding other addicting drugs, AEA is involved in several cocaine-, ethanol-, and opioid-induced rewarding effects. Among them, the role of the eCb system in cocaine reward and addiction is still questioned. In fact, Adamczyk et al. [162] reported that FAAH inhibition blocks cocaine-seeking behavior, but induced no changes in cocaine self-administration. Additionally, no changes in the AEA NAc levels after repeated administration of cocaine were found [163]. Our studies demonstrated that URB597 prevented cocaine-induced ShNAc responses, an effect not reverted by CB1-R antagonists, but by the PPARα antagonist MK886 [157], suggesting that OEA and PEA, rather than AEA, were responsible for this effect. On the other hand, this scenario is further complicated by the finding that AEA levels increase in the striatum after cocaine administration [164].
The possibility that AEA may modulate alcohol-induced effects is less controversial. Studies by our group showed that FAAH inhibition counteracted ethanol-induced ShNAc responses [165], and Serrano et al. [166] reported altered FAAH mRNA expression in the amygdala after ethanol withdrawal in animals exposed to either acute or intermittent administration. Furthermore, in the latter study, altered MAG-L mRNA expression was described and its levels were more pronounced only during intermittent alcohol exposure, suggesting a prominent role of AEA, rather than 2-AG, in the modulation of ethanol-induced responses. AEA might reduce ethanol intake by activating TRPV1 receptors, since a study illustrated that TRPV1−/− mice showed significantly higher preference for ethanol and consumed more ethanol in a two-bottle choice test as compared with wild type littermates [167]. This study and that by Adamczyk et al. [168] for cocaine addictive behavior underscore the emerging role of TRPV1 in addiction.
2-AG involvement in the modulation of rewarding and addictive responses is less clear. Studies with alcohol have reported that dialysate levels of 2-AG in the NAc were increased after ethanol self-administration [163], supporting the role of 2-AG in early steps of alcohol dependence. This notion has also been supported by other studies, which showed that alcohol intake and preference was increased by chronic treatment with neurotoxic doses of the psychostimulant, methamphetamine [169]. In this case, the authors showed that 7 days after methamphetamine administration, 2-AG levels were enhanced, whereas MAG-L activity was reduced in the limbic forebrain and that MAG-L inhibitors enhanced ethanol intake in treated and naïve mice [169].
Recent evidence pointed toward a possible modulatory effect of 2-AG in opiate reward and addiction. In fact, it has been recently shown that MAG-L inhibitors suppress withdrawal symptoms in a model of naloxone-precipitated abstinence [170], suggesting that 2-AG might play a prominent role in opiate withdrawal. Conversely, FAAH inhibition only partially reduced some measures of withdrawal score, suggesting that AEA has modest beneficial effects under these circumstances. It has been demonstrated that 2-AG levels are strongly reduced in many limbic areas during morphine tolerance [171]. However, beside this intriguing role of 2-AG on chronic opiate effects, other studies have highlighted interactions between the latter eCb and AEA (see below), which suggested coinvolvement of 2-AG in the mechanisms of early and late phases of morphine and heroin addiction.
Interactions between endocannabinoids in rewarding and addictive properties of drugs of abuse
Few studies have specifically investigated the interactions between 2-AG and AEA. eCb levels were measured in brain tissue during drug intake and often found elevations/decreases of both AEA and 2-AG levels. For instance, Malinen et al. [172] found increased AEA and 2-AG levels in specific brain regions of alcohol-preferring male and female rats (AA rats) compared with nonpreferring counterparts. AEA and 2-AG changes were nonoverlapping and showed differential occurrences in accordance with gender and alcohol exposure sessions, suggesting that AEA and 2-AG might have a distinct modulatory role. The findings by Caillè et al. [163] provided further support to the notion that AEA and 2-AG display different profiles during drug intake. In this paper, the authors showed that in heroin self-administration, an increase in NAc AEA levels was accompanied by a slight, but significant, decrease in 2-AG concentrations in this brain area. Others reported that under chronic administration of drugs of abuse, changes in eCb contents may involve AEA and 2-AG [161]. For example, studies highlighted that AEA and 2-AG levels were decreased in the midbrain after chronic alcohol exposure and increased in the brain stem and decreased in hippocampus and cortex after chronic administration of nicotine [161]. In an interesting study, Viganò et al. [173] showed AEA and 2-AG interactions during different phases of morphine sensitization that might suggest a homeostatic adaptation of the whole eCb system induced by chronic opiate exposure. Here, the authors reported changes in AEA and 2-AG levels in opposite directions after acute morphine administration, prolonged withdrawal, and expression phase of sensitization, with increase in AEA and decrease in 2-AG concentrations in most of the areas analyzed (i.e., NAc, caudate–putamen, and hippocampus). These results are in line with the study by Caillè et al. [163] with heroin self-administration and might underscore AEA and 2-AG interactions to balance their contents during opiate sensitization, thereby likely preserving the integrity of this circuitry.
Concluding remarks
The present literature review draws a complex picture about AEA and 2-AG, which may interact or differentially compete in the modulation of specific CNS functions. It is now well established that AEA and 2-AG are not fully interchangeable, since they definitely take part in highly specialized compartments of physiological and pathophysiological functioning. Even when they cooperate, AEA and 2-AG often partake in distinct steps of the same function. Overall, it appears that 2-AG is more involved in diffuse physiological functions, such as synaptic plasticity and neuroprotection, whereas AEA might intervene to fine-tune those processes. Accordingly, 2-AG might be regarded as providing functional support and orientating physiological modifications necessary in homeostasis, development, and adaptive behavior. On the other hand, AEA apparently plays a prominent role in modulation of superior functions such as learning and memory. Interestingly, even when interacting, AEA and 2-AG preserve their specific functionality, for instance, by recruiting different mechanisms and acting through different pathways.
In spite of this reductive view, which does not account for circumstances where AEA and 2-AG act through the same mechanism, some issues need to be discussed. First of all, recent observations demonstrated that AEA catabolism inhibitors, while promoting increases of AEA levels in nervous tissue, may also reduce the 2-AG biosynthesis in specific brain regions, such as the striatum [72, 174]. In light of these findings, it becomes less clear whether effects observed in some functions by application of URB597 and congeners were actually mediated by increased AEA levels, or by reduced 2-AG levels. Moreover, the findings that FAAH hydrolyzes both AEA and other N-acylethanolamides (i.e., OEA and PEA) [24] make it difficult to isolate the AEA-mediated effects from those mediated by OEA and PEA. Additionally, it has been reported that AEA may have an affinity for PPARα ([175] but see [156, 176, 177]). In line with the multifaceted AEA profile, which goes beyond the affinity for classical CB1-Rs, studies have demonstrated that effects produced by this eCb are likely due to TRPV1 activation [59, 178] (see above). Interestingly, this effect may strictly link AEA activity with that mediated by 2-AG, since it has been observed that TRPV1 receptor activation may also trigger the 2-AG biosynthesis [117], or, conversely, reduce 2-AG levels [174, 179]. In conclusion, these combined circumstances make the scenario on AEA and 2-AG interaction as neuromodulators more complex and provide a challenge that is worth addressing in the future to specifically isolate the role of eCb-like molecules in brain physiology and pathophysiology.
References
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346(6284):561–564
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258(5090):1946–1949
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50(1):83–90
Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K (1995) 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 215(1):89–97
Milne GM Jr, Johnson MR (1981) Levonantradol: a role for central prostanoid mechanisms? J Clin Pharmacol 21(8–9 Suppl):367S–374S
Thomas BF, Adams IB, Mascarella SW, Martin BR, Razdan RK (1996) Structure–activity analysis of anandamide analogs: relationship to a cannabinoid pharmacophore. J Med Chem 39(2):471–479. doi:10.1021/jm9505167
Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279(7):5298–5305
Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163(3):463–468. doi:10.1083/jcb.200305129
Stella N, Schweitzer P, Piomelli D (1997) A second endogenous cannabinoid that modulates long-term potentiation. Nature 388(6644):773–778
Di Marzo V (2011) Endocannabinoid signaling in the brain: biosynthetic mechanisms in the limelight. Nat Neurosci 14(1):9–15. doi:10.1038/nn.2720
Reggio PH (2002) Endocannabinoid structure–activity relationships for interaction at the cannabinoid receptors. Prostaglandins Leukot Essent Fatty Acids 66(2–3):143–160. doi:10.1054/plef.2001.0343
Pertwee RG, Ross RA (2002) Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids 66(2–3):101–121
Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA (2010) International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2). Pharmacol Rev 62(4):588–631. doi:10.1124/pr.110.003004
Vandevoorde S, Lambert DM (2007) The multiple pathways of endocannabinoid metabolism: a zoom out. Chem Biodivers 4(8):1858–1881. doi:10.1002/cbdv.200790156
Ueda N, Tsuboi K, Uyama T (2010) N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog Lipid Res 49(4):299–315. doi:10.1016/j.plipres.2010.02.003
Yu M, Ives D, Ramesha CS (1997) Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem 272(34):21181–21186
Ueda N, Yamamoto K, Yamamoto S, Tokunaga T, Shirakawa E, Shinkai H, Ogawa M, Sato T, Kudo I, Inoue K et al (1995) Lipoxygenase-catalyzed oxygenation of arachidonylethanolamide, a cannabinoid receptor agonist. Biochim Biophys Acta 1254(2):127–134
Bornheim LM, Kim KY, Chen B, Correia MA (1993) The effect of cannabidiol on mouse hepatic microsomal cytochrome P450-dependent anandamide metabolism. Biochem Biophys Res Commun 197(2):740–746
Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N (2010) The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci 13(8):951–957. doi:10.1038/nn.2601
Savinainen JR, Saario SM, Laitinen JT (2012) The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol (Oxf) 204(2):267–276. doi:10.1111/j.1748-1716.2011.02280.x
Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH (2001) Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA 98(16):9371–9376. doi:10.1073/pnas.161191698
Pan B, Wang W, Zhong P, Blankman JL, Cravatt BF, Liu QS (2011) Alterations of endocannabinoid signaling, synaptic plasticity, learning, and memory in monoacylglycerol lipase knock-out mice. J Neurosci 31(38):13420–13430. doi:10.1523/JNEUROSCI.2075-11.2011
Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF (2009) Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol 16(4):411–420. doi:10.1016/j.chembiol.2009.02.013
Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D (2003) Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9(1):76–81
Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5(1):37–44. doi:10.1038/nchembio.129
Katona I, Freund TF (2012) Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. doi:10.1146/annurev-neuro-062111-150420
Daniel H, Crepel F (2001) Control of Ca(2+) influx by cannabinoid and metabotropic glutamate receptors in rat cerebellar cortex requires K(+) channels. J Physiol 537(Pt 3):793–800
Twitchell W, Brown S, Mackie K (1997) Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 78(1):43–50
Brown SP, Safo PK, Regehr WG (2004) Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci 24(24):5623–5631. doi:10.1523/JNEUROSCI.0918-04.2004
Hoffman AF, Lupica CR (2000) Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J Neurosci 20(7):2470–2479
Huang CC, Lo SW, Hsu KS (2001) Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol 532(Pt 3):731–748
Llano I, Leresche N, Marty A (1991) Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron 6(4):565–574
Pitler TA, Alger BE (1992) Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12(10):4122–4132
Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410(6828):588–592
Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29(3):729–738
Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89(1):309–380. doi:10.1152/physrev.00019.2008
Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29(3):717–727
Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38(3):461–472
Safo PK, Regehr WG (2005) Endocannabinoids control the induction of cerebellar LTD. Neuron 48(4):647–659. doi:10.1016/j.neuron.2005.09.020
Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA 99(12):8384–8388. doi:10.1073/pnas.122149199
Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5(5):446–451
Kim J, Alger BE (2010) Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci 13(5):592–600. doi:10.1038/nn.2517
Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M (2002) Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci 15(6):953–961
Straiker A, Mackie K (2005) Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol (Lond) 569(2):501–517. doi:10.1113/jphysiol.2005.091918
Straiker A, Hu SS, Long JZ, Arnold A, Wager-Miller J, Cravatt BF, Mackie K (2009) Monoacylglycerol lipase limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation in autaptic hippocampal neurons. Mol Pharmacol 76(6):1220–1227. doi:10.1124/mol.109.059030
Straiker A, Mackie K (2009) Cannabinoid signaling in inhibitory autaptic hippocampal neurons. Neuroscience 163(1):190–201. doi:10.1016/j.neuroscience.2009.06.004
Straiker A, Wager-Miller J, Hu SS, Blankman JL, Cravatt BF, Mackie K (2011) COX-2 and fatty acid amide hydrolase can regulate the time course of depolarization-induced suppression of excitation. Br J Pharmacol 164(6):1672–1683. doi:10.1111/j.1476-5381.2011.01486.x
Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M (2005) Synaptically driven endocannabinoid release requires Ca2 + -assisted metabotropic glutamate receptor subtype 1 to phospholipase Cbeta4 signaling cascade in the cerebellum. J Neurosci 25(29):6826–6835. doi:10.1523/JNEUROSCI.0945-05.2005
Szabo B, Urbanski MJ, Bisogno T, Di Marzo V, Mendiguren A, Baer WU, Freiman I (2006) Depolarization-induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2-arachidonoylglycerol. J Physiol (Lond) 577(Pt 1):263–280. doi:10.1113/jphysiol.2006.119362
Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ (2007) Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS One 2(1):e709. doi:10.1371/journal.pone.0000709
Yoshino H, Miyamae T, Hansen G, Zambrowicz B, Flynn M, Pedicord D, Blat Y, Westphal RS, Zaczek R, Lewis DA, Gonzalez-Burgos G (2011) Postsynaptic diacylglycerol lipase mediates retrograde endocannabinoid suppression of inhibition in mouse prefrontal cortex. J Physiol (Lond) 589(Pt 20):4857–4884. doi:10.1113/jphysiol.2011.212225
Melis M, Perra S, Muntoni AL, Pillolla G, Lutz B, Marsicano G, Di Marzo V, Gessa GL, Pistis M (2004) Prefrontal cortex stimulation induces 2-arachidonoyl-glycerol-mediated suppression of excitation in dopamine neurons. J Neurosci 24(47):10707–10715
Seif T, Makriyannis A, Kunos G, Bonci A, Hopf FW (2011) The endocannabinoid 2-arachidonoylglycerol mediates D1 and D2 receptor cooperative enhancement of rat nucleus accumbens core neuron firing. Neuroscience 193:21–33. doi:10.1016/j.neuroscience.2011.07.055
Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M (2010) The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 65(3):320–327. doi:10.1016/j.neuron.2010.01.021
Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, Samad TA, Doherty P (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30(6):2017–2024. doi:10.1523/JNEUROSCI.5693-09.2010
Nomura DK, Hudak CS, Ward AM, Burston JJ, Issa RS, Fisher KJ, Abood ME, Wiley JL, Lichtman AH, Casida JE (2008) Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorg Med Chem Lett 18(22):5875–5878. doi:10.1016/j.bmcl.2008.08.007
Alger BE, Kim J (2011) Supply and demand for endocannabinoids. Trends Neurosci 34(6):304–315. doi:10.1016/j.tins.2011.03.003
Min R, Testa-Silva G, Heistek TS, Canto CB, Lodder JC, Bisogno T, Di Marzo V, Brussaard AB, Burnashev N, Mansvelder HD (2010) Diacylglycerol lipase is not involved in depolarization-induced suppression of inhibition at unitary inhibitory connections in mouse hippocampus. J Neurosci 30(7):2710–2715. doi:10.1523/JNEUROSCI.BC-3622-09.2010
Di Marzo V (2010) Anandamide serves two masters in the brain. Nat Neurosci 13(12):1446–1448. doi:10.1038/nn1210-1446
Sun Y, Bennett A (2007) Cannabinoids: a new group of agonists of PPARs. PPAR Res 2007:23513
Pistis M, Melis M (2010) From surface to nuclear receptors: the endocannabinoid family extends its assets. Curr Med Chem 17(14):1450–1467
Starowicz K, Nigam S, Di Marzo V (2007) Biochemistry and pharmacology of endovanilloids. Pharmacol Ther 114(1):13–33. doi:10.1016/j.pharmthera.2007.01.005
Azad SC (2004) Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci 24(44):9953–9961. doi:10.1523/jneurosci.2134-04.2004
Haj-Dahmane S, Shen RY (2009) Endocannabinoids suppress excitatory synaptic transmission to dorsal raphe serotonin neurons through the activation of presynaptic CB1 receptors. J Pharmacol Exp Ther 331(1):186–196. doi:10.1124/jpet.109.153858
Edwards JG, Gibson HE, Jensen T, Nugent F, Walther C, Blickenstaff J, Kauer JA (2010) A novel non-CB1/TRPV1 endocannabinoid-mediated mechanism depresses excitatory synapses on hippocampal CA1 interneurons. Hippocampus. doi:10.1002/hipo.20884
Chavez AE, Chiu CQ, Castillo PE (2010) TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci 13(12):1511–1518. doi:10.1038/nn.2684
Grueter BA, Brasnjo G, Malenka RC (2010) Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci 13(12):1519–1525. doi:10.1038/nn.2685
Nyilas R, Dudok B, Urban GM, Mackie K, Watanabe M, Cravatt BF, Freund TF, Katona I (2008) Enzymatic machinery for endocannabinoid biosynthesis associated with calcium stores in glutamatergic axon terminals. J Neurosci 28(5):1058–1063. doi:10.1523/JNEUROSCI.5102-07.2008
Cristino L, Starowicz K, De Petrocellis L, Morishita J, Ueda N, Guglielmotti V, Di Marzo V (2008) Immunohistochemical localization of anabolic and catabolic enzymes for anandamide and other putative endovanilloids in the hippocampus and cerebellar cortex of the mouse brain. Neuroscience 151(4):955–968. doi:10.1016/j.neuroscience.2007.11.047
Maione S, Cristino L, Migliozzi AL, Georgiou AL, Starowicz K, Salt TE, Di Marzo V (2009) TRPV1 channels control synaptic plasticity in the developing superior colliculus. J Physiol 587(Pt 11):2521–2535. doi:10.1113/jphysiol.2009.171900
Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ (2011) Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci 14(12):1542–1547. doi:10.1038/nn.2974
Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agro A, Cravatt BF, Centonze D (2008) Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci 11(2):152–159
Sheinin A, Talani G, Davis MI, Lovinger DM (2008) Endocannabinoid- and mGluR5-dependent short-term synaptic depression in an isolated neuron/bouton preparation from the hippocampal CA1 region. J Neurophysiol 100(2):1041–1052. doi:10.1152/jn.90226.2008
Mechoulam R, Spatz M, Shohami E (2002) Endocannabinoids and neuroprotection. Sci STKE 2002 (129):RE5
Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA (2002) Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci 22(22):9771–9775
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B (2003) CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302(5642):84–88
Fowler CJ (2003) Plant-derived, synthetic and endogenous cannabinoids as neuroprotective agents. Non-psychoactive cannabinoids, ‘entourage’ compounds and inhibitors of N-acyl ethanolamine breakdown as therapeutic strategies to avoid pyschotropic effects. Brain Res Brain Res Rev 41(1):26–43
Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E (2001) An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413(6855):527–531
Degn M, Lambertsen KL, Petersen G, Meldgaard M, Artmann A, Clausen BH, Hansen SH, Finsen B, Hansen HS, Lund TM (2007) Changes in brain levels of N-acylethanolamines and 2-arachidonoylglycerol in focal cerebral ischemia in mice. J Neurochem 103(5):1907–1916. doi:10.1111/j.1471-4159.2007.04892.x
Chen X, Zhang J, Chen C (2011) Endocannabinoid 2-arachidonoylglycerol protects neurons against beta-amyloid insults. Neuroscience 178:159–168. doi:10.1016/j.neuroscience.2011.01.024
Melis M, Pillolla G, Bisogno T, Minassi A, Petrosino S, Perra S, Muntoni AL, Lutz B, Gessa GL, Marsicano G, Di Marzo V, Pistis M (2006) Protective activation of the endocannabinoid system during ischemia in dopamine neurons. Neurobiol Dis 24(1):15–27
Sugiura T, Kondo S, Kishimoto S, Miyashita T, Nakane S, Kodaka T, Suhara Y, Takayama H, Waku K (2000) Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J Biol Chem 275(1):605–612
Gonsiorek W, Lunn C, Fan X, Narula S, Lundell D, Hipkin RW (2000) Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Mol Pharmacol 57(5):1045–1050
Sugiura T, Kishimoto S, Oka S, Gokoh M (2006) Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res 45(5):405–446. doi:10.1016/j.plipres.2006.03.003
Facchinetti F, Del Giudice E, Furegato S, Passarotto M, Leon A (2003) Cannabinoids ablate release of TNFalpha in rat microglial cells stimulated with lypopolysaccharide. Glia 41(2):161–168. doi:10.1002/glia.10177
Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N (2003) Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci 23(4):1398–1405
Stella N (2009) Endocannabinoid signaling in microglial cells. Neuropharmacology 56(Suppl 1):244–253. doi:10.1016/j.neuropharm.2008.07.037
Cabral GA, Griffin-Thomas L (2009) Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med 11:e3. doi:10.1017/S1462399409000957
Kreutz S, Koch M, Bottger C, Ghadban C, Korf HW, Dehghani F (2009) 2-Arachidonoylglycerol elicits neuroprotective effects on excitotoxically lesioned dentate gyrus granule cells via abnormal-cannabidiol-sensitive receptors on microglial cells. Glia 57(3):286–294. doi:10.1002/glia.20756
Ikemoto S (2010) Brain reward circuitry beyond the mesolimbic dopamine system: a neurobiological theory. Neurosci Biobehav Rev 35(2):129–150. doi:10.1016/j.neubiorev.2010.02.001
Berger C, Schmid PC, Schabitz W-R, Wolf M, Schwab S, Schmid HHO (2004) Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem 88(5):1159–1167. doi:10.1046/j.1471-4159.2003.02244.x
Amantea D, Spagnuolo P, Bari M, Fezza F, Mazzei C, Tassorelli C, Morrone LA, Corasaniti MT, Maccarrone M, Bagetta G (2007) Modulation of the endocannabinoid system by focal brain ischemia in the rat is involved in neuroprotection afforded by 17beta-estradiol. FEBS J 274(17):4464–4775. doi:10.1111/j.1742-4658.2007.05975.x
Iuvone T, Esposito G, De Filippis D, Bisogno T, Petrosino S, Scuderi C, Di Marzo V, Steardo L (2007) Cannabinoid CB1 receptor stimulation affords neuroprotection in MPTP-induced neurotoxicity by attenuating S100B up-regulation in vitro. J Mol Med 85(12):1379–1392. doi:10.1007/s00109-007-0233-y
Shouman B, Fontaine RH, Baud O, Schwendimann L, Keller M, Spedding M, Lelievre V, Gressens P (2006) Endocannabinoids potently protect the newborn brain against AMPA-kainate receptor-mediated excitotoxic damage. Br J Pharmacol 148(4):442–451. doi:10.1038/sj.bjp.0706755
Nucci C, Gasperi V, Tartaglione R, Cerulli A, Terrinoni A, Bari M, De Simone C, Agro AF, Morrone LA, Corasaniti MT, Bagetta G, Maccarrone M (2007) Involvement of the endocannabinoid system in retinal damage after high intraocular pressure-induced ischemia in rats. Invest Ophthalmol Vis Sci 48(7):2997–3004. doi:10.1167/iovs.06-1355
Karanian DA, Karim SL, Wood JT, Williams JS, Lin S, Makriyannis A, Bahr BA (2007) Endocannabinoid enhancement protects against kainic acid-induced seizures and associated brain damage. J Pharmacol Exp Ther 322(3):1059–1066. doi:10.1124/jpet.107.120147
Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Makriyannis A, Khanolkar A, Layward L, Fezza F, Bisogno T, Di Marzo V (2001) Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J 15(2):300–302. doi:10.1096/fj.00-0399fje
de Lago E, Fernandez-Ruiz J, Ortega-Gutierrez S, Cabranes A, Pryce G, Baker D, Lopez-Rodriguez M, Ramos JA (2006) UCM707, an inhibitor of the anandamide uptake, behaves as a symptom control agent in models of Huntington's disease and multiple sclerosis, but fails to delay/arrest the progression of different motor-related disorders. Eur Neuropsychopharmacol 16(1):7–18. doi:10.1016/j.euroneuro.2005.06.001
Wang Q, Peng Y, Chen S, Gou X, Hu B, Du J, Lu Y, Xiong L (2009) Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke 40(6):2157–2164. doi:10.1161/STROKEAHA.108.541490
Murillo-Rodriguez E, Sanchez-Alavez M, Navarro L, Martinez-Gonzalez D, Drucker-Colin R, Prospero-Garcia O (1998) Anandamide modulates sleep and memory in rats. Brain Res 812(1–2):270–274
Munguba H, Cabral A, Leao AH, Barbosa FF, Izidio GS, Ribeiro AM, Silva RH (2011) Pre-training anandamide infusion within the basolateral amygdala impairs plus-maze discriminative avoidance task in rats. Neurobiol Learn Mem 95(4):527–533. doi:10.1016/j.nlm.2011.03.006
Goonawardena AV, Sesay J, Sexton CA, Riedel G, Hampson RE (2011) Pharmacological elevation of anandamide impairs short-term memory by altering the neurophysiology in the hippocampus. Neuropharmacology 61(5–6):1016–1025. doi:10.1016/j.neuropharm.2011.07.003
Wise LE, Harloe JP, Lichtman AH (2009) Fatty acid amide hydrolase (FAAH) knockout mice exhibit enhanced acquisition of an aversive, but not of an appetitive, Barnes maze task. Neurobiol Learn Mem 92(4):597–601. doi:10.1016/j.nlm.2009.06.001
Varvel SA, Cravatt BF, Engram AE, Lichtman AH (2006) Fatty acid amide hydrolase (−/−) mice exhibit an increased sensitivity to the disruptive effects of anandamide or oleamide in a working memory water maze task. J Pharmacol Exp Ther 317(1):251–257
Mazzola C, Medalie J, Scherma M, Panlilio LV, Solinas M, Tanda G, Drago F, Cadet JL, Goldberg SR, Yasar S (2009) Fatty acid amide hydrolase (FAAH) inhibition enhances memory acquisition through activation of PPAR-alpha nuclear receptors. Learn Mem 16(5):332–337. doi:10.1101/lm.1145209
Busquets-Garcia A, Puighermanal E, Pastor A, de la Torre R, Maldonado R, Ozaita A (2011) Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol Psychiatry 70(5):479–486. doi:10.1016/j.biopsych.2011.04.022
Manwell LA, Satvat E, Lang ST, Allen CP, Leri F, Parker LA (2009) FAAH inhibitor, URB-597, promotes extinction and CB(1) antagonist, SR141716, inhibits extinction of conditioned aversion produced by naloxone-precipitated morphine withdrawal, but not extinction of conditioned preference produced by morphine in rats. Pharmacol Biochem Behav 94(1):154–162. doi:10.1016/j.pbb.2009.08.002
Vigano D, Guidali C, Petrosino S, Realini N, Rubino T, Di Marzo V, Parolaro D (2009) Involvement of the endocannabinoid system in phencyclidine-induced cognitive deficits modelling schizophrenia. Int J Neuropsychopharmacol 12(5):599–614. doi:10.1017/S1461145708009371
Yoshida T, Uchigashima M, Yamasaki M, Katona I, Yamazaki M, Sakimura K, Kano M, Yoshioka M, Watanabe M (2011) Unique inhibitory synapse with particularly rich endocannabinoid signaling machinery on pyramidal neurons in basal amygdaloid nucleus. Proc Natl Acad Sci USA 108(7):3059–3064. doi:10.1073/pnas.1012875108
Cuellar JN, Isokawa M (2011) Ghrelin-induced activation of cAMP signal transduction and its negative regulation by endocannabinoids in the hippocampus. Neuropharmacology 60(6):842–851. doi:10.1016/j.neuropharm.2010.12.024
Iversen L, Chapman V (2002) Cannabinoids: a real prospect for pain relief? Curr Opin Pharmacol 2(1):50–55
Campbell FA, Tramer MR, Carroll D, Reynolds DJ, Moore RA, McQuay HJ (2001) Are cannabinoids an effective and safe treatment option in the management of pain? A qualitative systematic review. BMJ 323(7303):13–16
Pertwee RG (2001) Cannabinoid receptors and pain. Prog Neurobiol 63(5):569–611
Starowicz K, Makuch W, Osikowicz M, Piscitelli F, Petrosino S, Di Marzo V, Przewlocka B (2012) Spinal anandamide produces analgesia in neuropathic rats: possible CB(1)- and TRPV1-mediated mechanisms. Neuropharmacology 62(4):1746–1755. doi:10.1016/j.neuropharm.2011.11.021
Guindon J, Desroches J, Beaulieu P (2007) The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br J Pharmacol 150(6):693–701. doi:10.1038/sj.bjp.0706990
Olango WM, Roche M, Ford GK, Harhen B, Finn DP (2011) The endocannabinoid system in the rat dorsolateral periaqueductal grey mediates fear-conditioned analgesia and controls fear expression in the presence of nocicpetive tone. Br J Pharmacol. doi:10.1111/j.1476-5381.2011.01478.x
Liao HT, Lee HJ, Ho YC, Chiou LC (2011) Capsaicin in the periaqueductal gray induces analgesia via metabotropic glutamate receptor-mediated endocannabinoid retrograde disinhibition. Br J Pharmacol 163(2):330–345. doi:10.1111/j.1476-5381.2011.01214.x
Spradley JM, Guindon J, Hohmann AG (2010) Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacol Res Commun 62(3):249–258. doi:10.1016/j.phrs.2010.03.007
Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435(7045):1108–1112
Mitrirattanakul S, Ramakul N, Guerrero AV, Matsuka Y, Ono T, Iwase H, Mackie K, Faull KF, Spigelman I (2006) Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain 126(1–3):102–114
Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH (2009) Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther 330(3):902–910. doi:10.1124/jpet.109.155465
Khasabova IA, Khasabov SG, Harding-Rose C, Coicou LG, Seybold BA, Lindberg AE, Steevens CD, Simone DA, Seybold VS (2008) A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. J Neurosci 28(44):11141–11152. doi:10.1523/JNEUROSCI.2847-08.2008
Khasabova IA, Chandiramani A, Harding-Rose C, Simone DA, Seybold VS (2011) Increasing 2-arachidonoyl glycerol signaling in the periphery attenuates mechanical hyperalgesia in a model of bone cancer pain. Pharmacol Res 64(1):60–67. doi:10.1016/j.phrs.2011.03.007
Petrosino S, Palazzo E, de Novellis V, Bisogno T, Rossi F, Maione S, Di Marzo V (2007) Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology 52(2):415–422. doi:10.1016/j.neuropharm.2006.08.011
Maione S, Bisogno T, de Novellis V, Palazzo E, Cristino L, Valenti M, Petrosino S, Guglielmotti V, Rossi F, Di Marzo V (2006) Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J Pharmacol Exp Ther 316(3):969–982. doi:10.1124/jpet.105.093286
Hall W, Solowij N (1998) Adverse effects of cannabis. Lancet 352(9140):1611–1616. doi:10.1016/S0140-6736(98)05021-1
Marco EM, Perez-Alvarez L, Borcel E, Rubio M, Guaza C, Ambrosio E, File SE, Viveros MP (2004) Involvement of 5-HT1A receptors in behavioural effects of the cannabinoid receptor agonist CP 55,940 in male rats. Behav Pharmacol 15(1):21–27
Viveros MP, Marco EM, File SE (2005) Endocannabinoid system and stress and anxiety responses. Pharmacol Biochem Behav 81(2):331–342. doi:10.1016/j.pbb.2005.01.029
Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D (2006) Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev 12(1):21–38
Moreira FA, Kaiser N, Monory K, Lutz B (2008) Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology 54(1):141–150. doi:10.1016/j.neuropharm.2007.07.005
Rossi S, De Chiara V, Musella A, Sacchetti L, Cantarella C, Castelli M, Cavasinni F, Motta C, Studer V, Bernardi G, Cravatt BF, Maccarrone M, Usiello A, Centonze D (2010) Preservation of striatal cannabinoid CB1 receptor function correlates with the antianxiety effects of fatty acid amide hydrolase inhibition. Mol Pharmacol 78(2):260–268. doi:10.1124/mol.110.064196
Cippitelli A, Astarita G, Duranti A, Caprioli G, Ubaldi M, Stopponi S, Kallupi M, Sagratini G, Rodriguez de Fonseca F, Piomelli D, Ciccocioppo R (2011) Endocannabinoid regulation of acute and protracted nicotine withdrawal: effect of FAAH inhibition. PLoS One 6(11):e28142. doi:10.1371/journal.pone.0028142
Bortolato M, Campolongo P, Mangieri RA, Scattoni ML, Frau R, Trezza V, La Rana G, Russo R, Calignano A, Gessa GL, Cuomo V, Piomelli D (2006) Anxiolytic-like properties of the anandamide transport inhibitor AM404. Neuropsychopharmacology 31(12):2652–2659
Patel S, Hillard CJ (2006) Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: further evidence for an anxiolytic role for endogenous cannabinoid signaling. J Pharmacol Exp Ther 318(1):304–311. doi:10.1124/jpet.106.101287
Hillard CJ, Jarrahian A (2003) Cellular accumulation of anandamide: consensus and controversy. Br J Pharmacol 140(5):802–808
Ronesi J, Gerdeman GL, Lovinger DM (2004) Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci 24(7):1673–1679
Roohbakhsh A, Keshavarz S, Hasanein P, Rezvani ME, Moghaddam AH (2009) Role of endocannabinoid system in the ventral hippocampus of rats in the modulation of anxiety-like behaviours. Basic Clin Pharmacol Toxicol 105(5):333–338. doi:10.1111/j.1742-7843.2009.00449.x
Haller J, Barna I, Barsvari B, Gyimesi Pelczer K, Yasar S, Panlilio LV, Goldberg S (2009) Interactions between environmental aversiveness and the anxiolytic effects of enhanced cannabinoid signaling by FAAH inhibition in rats. Psychopharmacology (Berl) 204(4):607–616. doi:10.1007/s00213-009-1494-7
Scherma M, Medalie J, Fratta W, Vadivel SK, Makriyannis A, Piomelli D, Mikics E, Haller J, Yasar S, Tanda G, Goldberg SR (2008) The endogenous cannabinoid anandamide has effects on motivation and anxiety that are revealed by fatty acid amide hydrolase (FAAH) inhibition. Neuropharmacology 54(1):129–140. doi:10.1016/j.neuropharm.2007.08.011
Rubino T, Realini N, Castiglioni C, Guidali C, Vigano D, Marras E, Petrosino S, Perletti G, Maccarrone M, Di Marzo V, Parolaro D (2008) Role in anxiety behavior of the endocannabinoid system in the prefrontal cortex. Cereb Cortex 18(6):1292–1301. doi:10.1093/cercor/bhm161
Campos AC, Ferreira FR, Guimaraes FS, Lemos JI (2010) Facilitation of endocannabinoid effects in the ventral hippocampus modulates anxiety-like behaviors depending on previous stress experience. Neuroscience 167(2):238–246. doi:10.1016/j.neuroscience.2010.01.062
Lisboa SF, Guimaraes FS (2012) Differential role of CB1 and TRPV1 receptors on anandamide modulation of defensive responses induced by nitric oxide in the dorsolateral periaqueductal gray. Neuropharmacology 62(8):2455–2462. doi:10.1016/j.neuropharm.2012.02.008
Umathe SN, Manna SS, Jain NS (2012) Endocannabinoid analogues exacerbate marble-burying behavior in mice via TRPV1 receptor. Neuropharmacology 62(5–6):2024–2033. doi:10.1016/j.neuropharm.2011.12.030
Casarotto PC, Terzian AL, Aguiar DC, Zangrossi H, Guimaraes FS, Wotjak CT, Moreira FA (2012) Opposing roles for cannabinoid receptor type-1 (CB(1)) and transient receptor potential vanilloid type-1 channel (TRPV1) on the modulation of panic-like responses in rats. Neuropsychopharmacology 37(2):478–486. doi:10.1038/npp.2011.207
Sumislawski JJ, Ramikie TS, Patel S (2011) Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: a potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology 36(13):2750–2761. doi:10.1038/npp.2011.166
Sciolino NR, Zhou W, Hohmann AG (2011) Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol Res 64(3):226–234. doi:10.1016/j.phrs.2011.04.010
Kinsey SG, O'Neal ST, Long JZ, Cravatt BF, Lichtman AH (2011) Inhibition of endocannabinoid catabolic enzymes elicits anxiolytic-like effects in the marble burying assay. Pharmacol Biochem Behav 98(1):21–27. doi:10.1016/j.pbb.2010.12.002
Justinova Z, Solinas M, Tanda G, Redhi GH, Goldberg SR (2005) The endogenous cannabinoid anandamide and its synthetic analog R(+)-methanandamide are intravenously self-administered by squirrel monkeys. J Neurosci 25(23):5645–5650. doi:10.1523/JNEUROSCI.0951-05.2005
Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, King AR, Redhi GH, Yasar S, Piomelli D, Goldberg SR (2008) Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiatry 64(11):930–937. doi:10.1016/j.biopsych.2008.08.008
Justinova Z, Yasar S, Redhi GH, Goldberg SR (2011) The endogenous cannabinoid 2-arachidonoylglycerol is intravenously self-administered by squirrel monkeys. J Neurosci 31(19):7043–7048. doi:10.1523/JNEUROSCI.6058-10.2011
Kauer JA, Malenka RC (2007) Synaptic plasticity and addiction. Nat Rev Neurosci 8(11):844–858
Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. PNAS 102(51):18620–18625
Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Q-s Liu, Lichtman AH, Cravatt BF (2010) Chronic monoacylglycerol lipase bloc\kade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13(9):1113–1119. doi:10.1038/nn.2616
Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF (2009) Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci 106(48):20270–20275. doi:10.1073/pnas.0909411106
Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, Justinova Z, Mikics E, Haller J, Medalie J, Stroik J, Barnes C, Yasar S, Tanda G, Piomelli D, Fratta W, Goldberg SR (2008) Inhibition of anandamide hydrolysis by cyclohexyl carbamic acid 3′-carbamoyl-3-yl ester (URB597) reverses abuse-related behavioral and neurochemical effects of nicotine in rats. J Pharmacol Exp Ther 327(2):482–490. doi:10.1124/jpet.108.142224
Melis M, Pillolla G, Luchicchi A, Muntoni AL, Yasar S, Goldberg SR, Pistis M (2008) Endogenous fatty acid ethanolamides suppress nicotine-induced activation of mesolimbic dopamine neurons through nuclear receptors. J Neurosci 28(51):13985–13994
Luchicchi A, Lecca S, Carta S, Pillolla G, Muntoni AL, Yasar S, Goldberg SR, Pistis M (2010) Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: involvement of PPAR-alpha nuclear receptors. Addict Biol 15(3):277–288. doi:10.1111/j.1369-1600.2010.00222.x
Scherma M, Justinova Z, Zanettini C, Panlilio LV, Mascia P, Fadda P, Fratta W, Makriyannis A, Vadivel SK, Gamaleddin I, Le Foll B, Goldberg SR (2011) The anandamide transport inhibitor AM404 reduces the rewarding effects of nicotine and nicotine-induced dopamine elevations in the nucleus accumbens shell in rats. Br J Pharmacol. doi:10.1111/j.1476-5381.2011.01467.x
Gamaleddin I, Guranda M, Goldberg SR, Le Foll B (2011) The selective anandamide transport inhibitor VDM11 attenuates reinstatement of nicotine seeking behaviour, but does not affect nicotine intake. Br J Pharmacol 164(6):1652–1660. doi:10.1111/j.1476-5381.2011.01440.x
Merritt LL, Martin BR, Walters C, Lichtman AH, Damaj MI (2008) The endogenous cannabinoid system modulates nicotine reward and dependence. J Pharmacol Exp Ther 326(2):483–492. doi:10.1124/jpet.108.138321
Gonzalez S, Cascio MG, Fernandez-Ruiz J, Fezza F, Di Marzo V, Ramos JA (2002) Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res 954(1):73–81
Adamczyk P, McCreary AC, Przegalinski E, Mierzejewski P, Bienkowski P, Filip M (2009) The effects of fatty acid amide hydrolase inhibitors on maintenance of cocaine and food self-administration and on reinstatement of cocaine-seeking and food-taking behavior in rats. J Physiol Pharmacol 60(3):119–125
Caille S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH (2007) Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci 27(14):3695–3702. doi:10.1523/jneurosci.4403-06.2007
Centonze D, Battista N, Rossi S, Mercuri NB, Finazzi-Agro A, Bernardi G, Calabresi P, Maccarrone M (2004) A critical interaction between dopamine D2 receptors and endocannabinoids mediates the effects of cocaine on striatal gabaergic Transmission. Neuropsychopharmacology 29(8):1488–1497
Perra S, Pillolla G, Melis M, Muntoni AL, Gessa GL, Pistis M (2005) Involvement of the endogenous cannabinoid system in the effects of alcohol in the mesolimbic reward circuit: electrophysiological evidence in vivo. Psychopharmacology (Berl) 183(3):368–377
Serrano A, Rivera P, Pavon FJ, Decara J, Suarez J, Rodriguez de Fonseca F, Parsons LH (2011) Differential effects of single versus repeated alcohol withdrawal on the expression of endocannabinoid system-related genes in the rat amygdala. Alcohol Clin Exp Res. doi:10.1111/j.1530-0277.2011.01686.x
Blednov YA, Harris RA (2009) Deletion of vanilloid receptor (TRPV1) in mice alters behavioral effects of ethanol. Neuropharmacology 56(4):814–820
Adamczyk P, Miszkiel J, McCreary AC, Filip M, Papp M, Przegalinski E (2012) The effects of cannabinoid CB1, CB2 and vanilloid TRPV1 receptor antagonists on cocaine addictive behavior in rats. Brain Res 1444:45–54. doi:10.1016/j.brainres.2012.01.030
Gutierrez-Lopez MD, Llopis N, Feng S, Barrett DA, O'Shea E, Colado MI (2010) Involvement of 2-arachidonoyl glycerol in the increased consumption of and preference for ethanol of mice treated with neurotoxic doses of methamphetamine. Br J Pharmacol 160(3):772–783. doi:10.1111/j.1476-5381.2010.00720.x
Ramesh D, Ross GR, Schlosburg JE, Owens RA, Abdullah RA, Kinsey SG, Long JZ, Nomura DK, Sim-Selley LJ, Cravatt BF, Akbarali HI, Lichtman AH (2011) Blockade of endocannabinoid hydrolytic enzymes attenuates precipitated opioid withdrawal symptoms in mice. J Pharmacol Exp Ther 339(1):173–185. doi:10.1124/jpet.111.181370
Vigano D, Grazia Cascio M, Rubino T, Fezza F, Vaccani A, Di Marzo V, Parolaro D (2003) Chronic morphine modulates the contents of the endocannabinoid, 2-arachidonoyl glycerol, in rat brain. Neuropsychopharmacology 28(6):1160–1167. doi:10.1038/sj.npp. 1300117
Malinen H, Lehtonen M, Hyytia P (2009) Modulation of brain endocannabinoid levels by voluntary alcohol consumption in alcohol-preferring AA rats. Alcohol Clin Exp Res 33(10):1711–1720. doi:10.1111/j.1530-0277.2009.01008.x
Vigano D, Valenti M, Grazia Cascio M, Di Marzo V, Parolaro D, Rubino T (2004) Changes in endocannabinoid levels in a rat model of behavioural sensitization to morphine. Eur J Neurosci 20(7):1849–1857
Dimarzo V, Maccarrone M (2008) FAAH and anandamide: is 2-AG really the odd one out? Trends Pharmacol Sci 29(5):229–233. doi:10.1016/j.tips.2008.03.001
Sun Y, Alexander SP, Garle MJ, Gibson CL, Hewitt K, Murphy SP, Kendall DA, Bennett AJ (2007) Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br J Pharmacol 152(5):734–743
Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D (2003) Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 425(6953):90–93
LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D (2006) Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther 319(3):1051–1061
Ross RA (2003) Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol 140(5):790–801. doi:10.1038/sj.bjp.0705467
Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agrò A, Cravatt BF, Centonze D (2008) Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci 11(2):152–159. doi:10.1038/nn2042
Acknowledgments
This work was sponsored by a grant from the Italian Ministry of University to Marco Pistis (PRIN 2009: 200928EEX4). We wish to thank the Regione Autonoma della Sardegna, Assessorato alla Programmazione for the support given to Antonio Luchicchi (Bursaries for Young Researchers, Legge Regionale 7/2007).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Luchicchi, A., Pistis, M. Anandamide and 2-arachidonoylglycerol: Pharmacological Properties, Functional Features, and Emerging Specificities of the Two Major Endocannabinoids. Mol Neurobiol 46, 374–392 (2012). https://doi.org/10.1007/s12035-012-8299-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-012-8299-0