
Abstract
Endocannabinoids are ubiquitous lipid signaling molecules that mimic some of the actions of phytocannabinoids such as delta-9-tetrahydrocannabinol. Endocannabinoids are a component of the endocannabinoid signaling system, which comprises the endocannabinoids, the enzymes that synthesize and degrade endocannabinoids, and cannabinoid receptors. Within the central nervous system (CNS), endocannabinoids serve as modulators of both long-term and short-term synaptic plasticity. This review will briefly review the signaling of cannabinoid-1 (CB1) and cannabinoid-2 (CB2) receptors and then explore some of the roles endocannabinoids play in mediating diverse forms of synaptic plasticity, with an emphasis on recent findings.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Anandamide
- 2-arachidonoyl glycerol
- Long-term potentiation
- Long-term depression
- Astrocyte
- Spike
- Timing-dependent plasticity
Endocannabinoids (eCBs) are small lipid signaling molecules, so named because they often engage the same receptors as the well-known phytocannabinoid, delta-9-tetrahydrocannabinol (THC) . Work over the past 20 years firmly establishes that eCBs participate in signaling in many parts of the body, and especially in the nervous system (Katona and Freund 2012 ). This review will consider an important functional role for eCBs—their role in modulating diverse forms of synaptic plasticity. However, first, the components of the eCB signaling system will be considered, with an emphasis on those components that are most relevant for synaptic plasticity, and then their regulation.
All well-characterized eCBs are arachidonic acid derivatives. One of their key features is that they exist as precursor lipids in the cell membrane and are liberated by the action of specific lipases under certain physiological or pathological conditions. The two most studied eCBs are anandamide (arachidonoyl ethanolamide) and 2-arachidonoyl glycerol (2-AG) . The precursors of anandamide are the N-arachidonoyl phosphatidyl ethanolamines (NAPEs). Anandamide can be produced from NAPEs by several different pathways (Ahn et al., 2008 ). The precursor of 2-AG is chiefly phosphatidyl bisphosphate (PIP2). 2-AG is primarily produced from PIP2 by the sequential action of a phospholipase C (PLC) and one of two diacyl glycerol lipases (Tanimura et al. 2010 ). The completely different routes of anandamide and 2-AG synthesis suggest that they are produced under different physiological conditions. In general, this is what has been found (Hohmann et al. 2005 ; Liu et al. 2008 ; Puente et al. 2011 ).
Similar to eCB synthesis, eCB degradation largely occurs via different pathways. Most anandamide is degraded by fatty acid amino hydrolase (FAAH) (McKinney and Cravatt 2005 ; Ahn et al. 2009). In contrast, 2-AG can be degraded by several serine hydrolases (monoacyl glycerol lipase, alpha beta hydrolase domain-containing 6, alpha beta hydrolase domain-containing 12, and FAAH) (Blankman et al. 2007 ). Thus, anandamide and 2-AG breakdown will be differentially regulated and inhibition of the respective pathways can be a useful tool to identify the eCB involved in a specific form of eCB-mediated synaptic plasticity.
For the purposes of this review, we will primarily consider the cannabinoid-1 (CB1) and cannabinoid-2 (CB2) receptors. (It is important to note that eCBs can interact with a wide variety of receptors and other molecules including other G protein-coupled receptors (GPCRs), transcription factors, and ion channels (Zygmunt et al. 1999; Fu et al. 2003 ; Oz 2006 ; McHugh et al. 2010 ). However, with a few notable exceptions (Melis et al. 2008 ; Mazzola et al. 2009; Chavez et al. 2010 ), these do not yet have an established role in eCB-mediated synaptic plasticity). The CB1 and CB2 receptors were both cloned a little more than 20 years ago. CB1 receptors are highly expressed in the central nervous system (CNS) but are present throughout the body and play roles in processes as diverse as metabolism, reproduction, and immune regulation (Nagarkatti et al. 2009 ; Talwar and Potluri 2011 ; Ward and Raffa 2011 ). CB2 receptors are less highly expressed, which has made identification of the cell types expressing them somewhat more problematic (Atwood and Mackie 2010 ). It is well accepted that CB2 receptors are expressed in several types of immune cells, particularly cells of macrophage lineage, including microglia. The extent of their expression in neurons and other glia is more contentious (Atwood and Mackie 2010 ). A striking feature of CB2 receptors is their high inducibility. For example, in the experimental allergic encephalitis (EAE) model of multiple sclerosis, CB2 mRNA levels can increase more than 100 fold (Maresz et al. 2005 ). The low level of CB2 receptor expression under basal conditions and the lack of suitably sensitive antibodies has, at the level of anatomy, led to much confusion (reviewed in Atwood and Mackie 2010 ). Thus, the most conclusive evidence for a role of CB2 in the CNS outside of microglia comes from functional and molecular studies (e.g., Xi et al. 2011 ; den Boon et al. 2012 ; Zarruk et al. 2012) . However, these studies lack anatomical precision, and often it is hard to conclusively determine which cell type(s) are involved. Conclusive resolution of these issues will require cell type-specific deletion of CB2 receptors.
Both CB1 and CB2 receptors are GPCRs. They primarily couple to Gi/Go G proteins, thus their dominant signaling pathways include inhibition of adenylyl cyclase, activation of mitogen-activated protein (MAP) kinases, inhibition of some voltage-dependent calcium channels, and activation of G protein-gated inwardly rectifying potassium (GIRK) channels (Howlett et al. 2002 ). Nonetheless, it is important to appreciate that both receptors can couple to alternative pathways. For example, CB1 can stimulate adenylyl cyclase (Glass and Felder 1997 ; Felder et al. 1998 ) and both receptors can release calcium from intracellular stores (Sugiura et al. 1997; Lauckner et al. 2005 ; Shoemaker et al. 2005 ). In considering activation of CB1 and CB2 receptors by eCBs, two other properties of these ligands need to be considered. The first is efficacy. Efficacy is a measure of how completely a particular ligand can activate a receptor. There is good agreement that anandamide is a lower efficacy agonist than 2-AG (Mackie et al. 1993 ; Luk et al. 2004 ; Sugiura et al., 2006). The consequences of this depend on receptor number and the efficiency of the receptor’s coupling to downstream signaling pathways. In general, low receptor density and/or poor coupling to downstream effectors will cause a low-efficacy agonist to have a diminished cellular response relative to a high-efficacy agonist (e.g., Luk et al. 2004 ). Under these conditions the low-efficacy agonist is considered to be a partial agonist. Conversely, under conditions where receptor density is high or effector coupling is strong, low- and high-efficacy agonists may have indistinguishable cellular responses. The second important property is functional selectivity. Functional selectivity refers to the ability of different agonists to differentially activate distinct signaling pathways, despite both activating the receptor (Kenakin and Miller 2010 ). Both CB1 and CB2 ligands can show functional selectivity; however, the functional selectivity of commonly encountered CB2 agonists is particularly striking (Atwood et al. 2012a, b).
Most Gi/Go-coupled GPCRs also modulate ion channels. CB1 receptors inhibit several voltage-dependent calcium channels, particularly N (Cav2.2) and P/Q (Cav2.1) channels (Mackie and Hille 1992 ; Mackie et al. 1995; Twitchell et al. 1997 ). In addition, CB1 receptors activate GIRK channels (Mackie et al. 1995 ). CB2 receptors likely modulate the same types of ion channels, although the strong functional selectivity of different CB2 ligands means that only some ligands can do this. For example, 2-AG potently inhibits calcium channels, whereas anandamide does not (Atwood et al. 2012a, b ).
The property of cannabinoid receptors to activate GIRK channels and inhibit calcium channels suggests that they will likely dampen neuronal excitability and inhibit synaptic transmission. A large number of studies support this contention (Roth 1978 ; Shen et al. 1996 ; Levenes et al. 1998 ; Misner and Sullivan 1999 ; Vaughan et al. 1999 ; Hajos et al. 2000 ; Takahashi and Linden 2000 ). Inhibition of synaptic transmission by CB1 receptors is an example where ligand efficacy is important. For example, THC, a low-efficacy CB1 agonist, has little effect on synaptic transmission in some model systems and can actually antagonize inhibition of synaptic transmission by 2-AG (Shen and Thayer 1999 ; Straiker and Mackie 2005 ). However, whether THC inhibits synaptic transmission depends on many factors, including the frequency of stimulation (Roloff and Thayer 2009 ; Hoffman and Lupica 2012 ).
The concept that eCBs can inhibit synaptic transmission, coupled with the observation that eCBs are often produced under conditions encountered during vigorous synaptic transmission, gave rise to a series of studies to determine if eCBs produced in this way could modulate synaptic transmission. Indeed, eCBs generated during intense neuronal activity can modulate synaptic transmission in a surprisingly diverse number of ways (Kano et al. 2009 ; Castillo et al. 2012 ).
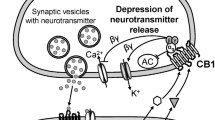
The first type of synaptic plasticity demonstrated to be mediated by eCBs was depolarization-induced suppression of inhibition (DSI) (Ohno-Shosaku et al. 2001 ; Wilson and Nicoll 2001 ). DSI is a phenomenon where intense depolarization (e.g., repeated action potentials or a 1–5 s step depolarization to 0 mV) of a postsynaptic neuron leads to a transient (tens of seconds) suppression of inhibitory transmission onto that neuron (Llano et al. 1991 ; Pitler and Alger 1992 ; Pitler and Alger 1994). An analogous phenomenon involving excitatory transmission mission is called depolarization-induced suppression of excitation (DSE) (e.g., Kreitzer and Regehr 2001 ). These phenomena have been extensively studied in both the hippocampus and cerebellum. Work from a number of investigators has arrived at the following canonical mechanism (however, note there is not complete agreement on these steps (Kano et al. 2009 )): depolarization of the postsynaptic cell leads to an increase in intracellular calcium (can be entry through calcium channels and/or release from intracellular stores) that activates diacyl glycerol lipase (DAGL) alpha. DAGL then cleaves the acyl chain in the one position on diacyl glycerol (DAG), generating 2-AG. 2-AG then travels (possibly by diffusion or via an undefined carrier) to the presynaptic terminal, where it engages presynaptic CB1 receptors, inhibiting calcium channels (and possibly also inhibiting the vesicular release machinery) to suppress synaptic transmission. DSI (and DSE) are terminated as 2-AG is degraded, either by MGL (Pan et al. 2009 ; Straiker et al. 2009 ) and/or cyclooxygenase-2 (COX-2) (Kim and Alger 2004 ; Straiker et al. 2011 ). The participation of COX-2 in terminating DSE may have important therapeutic implications. For example, if COX-2 is increased (e.g., following ischemic injury), the duration of DSE will be shortened and glutamate release increased, which may exacerbate excitotoxicity. In addition, COX-2 metabolites of 2-AG (e.g., prostaglandin E2 glycerol) can enhance excitatory synaptic transmission and long-term potentiation in the hippocampus (Sang et al. 2006 ; Yang et al. 2008 ).
A second form of transient modulation of synaptic transmission by eCBs is metabotropic suppression of inhibition (MSI) or excitation (MSE) (Maejima et al. 2001 ; Varma et al. 2001 ; Kim et al. 2002 ). This is a functionally distinct pathway from DSI/DSE. In MSI/MSE, activation of a postsynaptic Gq/11-linked GPCR stimulates PLCbeta, leading to the production of DAG. DAGL then cleaves the DAG to 2-AG, which then traverses the synapse to activate presynaptic CB1 receptors, inhibiting synaptic transmission (Kano et al. 2009 ). In theory, any appropriately positioned, postsynaptic Gq/11-linked GPCR should be able to elicit MSI/MSE. However, the most commonly encountered receptors mediating MSI/MSE are the group I metabotropic glutamate receptors (i.e., mGluR1 and mGluR5) and the M1 and M3 muscarinic receptors.
Although DSI/DSE and MSI/MSE can occur independently of one another, they can also synergize. In this situation, a brief depolarization, combined with modest activation of the Gq/11-linked receptor, increases intracellular calcium. This increased intracellular calcium stimulates the activity of PLCbeta, leading to higher levels of DAG production (and possibly greater DAGL activity), which increases 2-AG production (Hashimotodani et al. 2005 ). In this way eCBs can serve as a coincidence detector between depolarization and activation of metabotropic receptors.
The preceding discussion has focused on CB1 receptor-mediated forms of short-term synaptic plasticity. In these cases, CB1 involvement has been firmly established by antagonism of the plasticity with a variety of CB1 receptor antagonists or the absence of the plasticity in CB1 receptor knockout mice. Thus, under normal conditions (i.e., an acute brain slice or cultured neurons), CB2 receptors have not been observed to participate in these short-term forms of synaptic plasticity in the brain regions studied. However, these experiments left open the question if CB2 receptors can participate in short-term forms of synaptic plasticity. We addressed this question by transfecting CB2 receptors into hippocampal neurons cultured from CB1 receptor knockout mice. Expression of CB2 receptors into these neurons recovered 2-AG-mediated inhibition of synaptic transmission as well as DSE (Atwood et al. 2012a, b ). Thus, CB2 appears capable of supporting short-term forms of eCB-mediated synaptic plasticity, if it is appropriately expressed in neurons.
Apart from this example, there is additional evidence that CB2 receptors can influence synaptic transmission or neuronal excitability. Activation of CB2 receptors in layer 2/3 of the rodent prefrontal cortex increased activity of calcium-activated chloride currents, reducing spontaneous activity (den Boon et al. 2012 ). In addition, activation of CB2 receptors decreased action potential (but not action potential-independent γ-aminobutyric acid (GABA) release in rat medial entorhinal cortex (Morgan et al. 2009 )).
The above has focused on short-term synaptic plasticity. However, shortly after the description of the depolarization and metabotropic receptor forms of eCB-mediated synaptic plasticity discussed above, long-term depression (LTD) mediated by eCBs (eLTD) was described (Gerdeman et al. 2002 ; Robbe et al. 2002 ). This has been thoroughly studied in both excitatory (e.g., Gerdeman et al. 2002 ; Robbe et al. 2002 ; Peterfi et al. 2012 ) and inhibitory connections (e.g., Chevaleyre and Castillo 2003 ). eLTD occurs at some CB1-expressing synapses following prolonged low-frequency stimulation (e.g., 1 Hz, 10 min (Robbe et al. 2002)). It often appears to require prolonged activation of postsynaptic group I mGluR receptors, leading to continued synthesis of eCBs (likely, 2-AG), sustained activation of CB1 receptors, and persistent inhibition of neurotransmitter release (possibly mediated by RIM1alpha (Chevaleyre et al. 2007)). Like other forms of LTD, eLTD synaptic depression persists after the cessation of the inducing stimulus (in this case, CB1 production). Other forms of eLTD that vary from this canonical pathway have been reported in hippocampus (CA1) from young (< P10) rats (Yasuda et al. 2008 ) and in cultured autaptic hippocampal neurons (Kellogg et al. 2009 ). The former form of eLTD is notable for likely involving activation of potassium channels (Yasuda et al. 2008); whereas the latter form involves CB1 receptors signaling via Gi/o-independent G proteins (Kellogg et al. 2009 ).
The above paradigm for eLTP generally involves the direct action of a Gq/11-linked GPCR, followed by 2-AG production, prolonged stimulation of CB1 receptors, and inhibition of neurotransmission that exceeds the duration of eCB production. A related form of long-term synaptic depression has been demonstrated following the activation of two (membrane) steroid hormone receptors, the glucocorticoid receptor and the α isoform of the estrogen receptor. In the case of the glucocorticoid receptor, activation of this receptor in hypothalamic parvocellular neurons leads to a long-lasting inhibition of glutamate release (Di et al. 2003; Evanson et al. 2010 ; Tasker and Herman 2011 ). This leads to inhibition of corticotropin-releasing hormone (CRH)-secreting neurons and suppression of the hypothalamic–pituitary–adrenal (HPA) axis. In the case of estrogen receptor-mediated eLTD, activation of a membrane-associated α form of the estrogen receptor leads to long-term inhibition of CB1-expressing inhibitory synapses onto CA1 pyramidal neurons. Notable aspects of this latter form of eLTD is that it (1) only occurs in female rats, (2) requires mGluR1 signaling, and (3) appears to involve anandamide , and not 2-AG (Huang and Woolley 2012 ). The glucocorticoid receptor-mediated form of eLTD requires a G protein (as it is blocked by inclusion of GDPβS in the recording pipette), but whether this is a metabotropic glutamate receptor has not been tested. With the identification of these two steroid hormone receptor–mediated forms of eLTD, it is interesting to speculate that similar forms of LTD may be evoked by mineralocorticoid or androgen receptors stimulating GPCR activation.
Another form of activity-dependent modulation of neuronal excitability is slow self-inhibition (SSI). This form of eCB-mediated modulation of neuronal excitability has been reported in neocortical low-threshold spiking interneurons (Bacci et al. 2004 ), in a population of cerebellar basket cells (Kreitzer et al. 2002 ), and in a fraction of cortical pyramidal neurons (Marinelli et al. 2009 ). The likely signaling pathway for this phenomenon is that repeated depolarization of the neuron increases intracellular calcium, which activates DAGL and increases 2-AG production. 2-AG then activates a potassium conductance (likely GIRK channels) (Marinelli et al. 2008 ). In contrast to the forms of synaptic plasticity discussed earlier, SSI involves cell autonomous 2-AG signaling.
The above forms of eCB-mediated modulation of synaptic transmission and neuronal excitability have considered exclusively the domain of inter- or intra-neuronal signaling, with no involvement of glial cells. There are two major ways that glial cells may influence neuronal excitability and synaptic transmission in an eCB-dependent fashion. One is that glial cells, particularly astrocytes and microglial cells, can produce prodigious amounts of eCBs (Walter et al. 2002 ; Walter et al. 2003; Stella 2004). The other is that the glial cells may be expressing the cannabinoid receptors and influencing synaptic plasticity in a paracrine fashion. Considerable evidence has emerged over the past 5 years that glial cells, particularly astrocytes, participate as active CB1-expressing partners in some forms of eCB-mediated synaptic plasticity. This involvement was surprising to some in the field, as immunocytochemical studies showed high levels of CB1 expression in some GABAergic neurons and intermediate levels in a subset of excitatory synapses. CB1 expression in astrocytes, when noted. for example (Rodriguez et al., 2001 ), was only a small fraction of the levels observed in neuronal elements. However, density of CB1 receptor expression does not necessarily correlate with “importance,” as has been amply shown in prior studies (Azad et al. 2003 ; Marsicano et al. 2003; Domenici et al. 2006 ; Monory and Lutz 2009 ).
Several anatomical features of astrocytes are important when considering their role as potential mediators and modulators of eCB action (Ventura and Harris 1999 ). The first is that most central synapses are embedded in glial endfeet. This means that glial membranes are never far from the source of eCB production (primarily dendrites). The second is that the ramifications of a single astrocyte can extend over a considerable range, thus potentially transducing a local signal into one covering several hundred cubic microns. The third is that gap junction coupling between astrocytes will further increase the potential distance a signal can be transmitted. Thus, if CB1 stimulation increases the concentration of a diffusible messenger (e.g., calcium) in one astrocyte , that messenger may affect a number of neighboring astrocytes, potentially influencing a volume of several thousand cubic microns.
That eCBs released from neurons can signal via astrocytic CB1 receptors was first demonstrated about 5 years ago (Navarrete and Araque 2008 ). In these experiments, the investigators found that by depolarizing one neuron, eCBs were produced that activated neighboring astrocytic CB1 receptors. These CB1 receptors increased astrocytic intracellular calcium (interestingly, in a non-Gi/o-mediated fashion; this is a common feature of all forms of synaptic plasticity mediated by astrocytic CB1 receptors and deserves additional study), causing release of glutamate from the astrocyte , which activated (presynaptic) mGluR1 receptors to increase glutamate release and synaptic efficacy (Navarrete and Araque 2010 ). Therefore, in classic DSE, glutamatergic transmission onto the depolarized cell is inhibited by activation of presynaptic CB1 receptors, but when astrocytic CB1 receptors are activated, glutamatergic transmission is enhanced. Thus, eCB production could either stimulate or inhibit glutamatergic transmission, leading to the question of what happens in vivo. These investigators found that whether eCB production enhanced or suppressed glutamatergic transmission had strong spatial dependency. If glutamatergic terminals were close to the site (< 40 microns) (using the location of the neuron’s soma) of eCB production, then inhibition dominated, whereas at more distant sites (between 60 and 100 microns), enhancement was seen (Navarrete and Araque 2008 ). Interestingly, if presynaptic CB1 inhibition was prevented (by treatment of the slice with pertussis toxin), then activation of astrocytic CB1 led to more pronounced synaptic enhancement (Navarrete and Araque 2008 ), suggesting that under basal conditions, production of eCBs in this system (CA1) leads to a suppression of proximal glutamatergic transmission and an enhancement of more distal glutamatergic transmission. This provides an additional mechanism for a network to “tune” synaptic strength, where neuronal activation strong enough to produce eCBs will decrease subsequent glutamatergic input onto those neurons, yet will strengthen glutamatergic inputs onto more distant, presumably less-stimulated, neurons.
A second form of astrocyte-mediated cannabinoid synaptic plasticity has been described more recently (Han et al. 2012 ). In this study, it was shown that activation of astrocyte CB1 receptors by exogenous cannabinoids (systemically administered) led to LTD of CA3 to CA1 glutamatergic synapses. In addition to requiring astrocyte CB1 receptors, this cannabinoid-dependent form of LTD (CB-LTD) also required activation of N-methyl d-aspartic acid (NMDA) receptors and was mediated by the loss of cell surface α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors. Interestingly, the authors of this study were able to show a strong correlation between CB-LTD and THC-induced impairment of spatial working memory (Han et al. 2012 ), suggesting that the deleterious effects of THC on spatial working memory may be due to astrocyte-mediated synaptic depression at CA3 > CA1 synapses.
The observation that CB1 receptors on astrocytes can mediate opposing forms of synaptic plasticity (that is, LTD or long-term potentiation ) deserves further consideration. In both cases the common mediator appears to be glutamate release from astrocytes. Drawing from the extensive literature of glutamate-mediating diverse forms of synaptic plasticity (Citri and Malenka 2008 ), it is conceivable that the discrepancy in the two studies discussed earlier may be due to the different time courses of synaptic stimulation (that is, brief and punctate in the first study and prolonged with the second study). The first form of astrocytic CB1 stimulation may lead to brief, high local levels of glutamate , whereas the second will lead to more diffuse and prolonged elevations of extracellular glutamate. (This possibility is strengthened by the observation that blockade of extracellular glutamate uptake by threo-beta-benzyloxyaspartate (TBOA) induces a mechanistically similar form of LTD (Han et al. 2012 ).) There may also be experimental differences that explain the discrepancy. In the first study, experiments were conducted in slices prepared from hippocampus, which, while effectively preserving laminar neuronal connectivity, may disrupt the extensive network of interconnected astrocytes . In addition, the first study used minimal stimulation to investigate a small number of synaptic connections. The second study examined field potentials in anesthetized animals, so astrocyte connectivity would be maintained. (However, most of the experiments were conducted in anesthetized animals, which brings in the potential complicating factors of anesthesia.) Despite some uncertainties in interpretation, the above experiments establish that astrocytes can participate in cannabinoid-mediated synaptic plasticity and in cannabinoid-mediated behaviors. It is probable that future studies will more completely establish the precise mechanisms involved, which are likely varied depending on the form of stimulation and the precise behaviors involved.
A very recent study expanded the role of astrocytes in eCB-mediated synaptic plasticity to LTD in spike-timing-dependent plasticity (STDP) . STDP is a phenomenon where repeated pairing of presynaptic stimulation with postsynaptic depolarization leads to persistent changes in synaptic strength (Feldman 2012). A key feature of most forms of STDP is that the order of stimulation is critically important, where the sign of plasticity (LTP vs. LTD) depends on whether presynaptic or postsynaptic stimulation occurs first. Some forms of STDP have been shown to involve CB1 receptors and eCBs (e.g., Sjostrom et al. 2003 ; Tzounopoulos et al. 2007 ; Fino et al. 2010 ); however, a potential role for astrocytic CB1 receptors was not investigated. A recent study examining STDP in the neocortex during the development of somatosensory barrel cortex found that STDP producing LTD at the glutamatergic synapse from layer IV to layer II/III required astrocytic CB1 receptors (Min and Nevian 2012 ). This was established by using an LTD-producing STDP protocol where the postsynaptic neuron was depolarized 25 ms before afferent fibers were stimulated. The LTD produced under these circumstances was NMDA receptor dependent, was occluded by clamping astrocyte calcium levels, and was mimicked by inducing astrocytic calcium spikes together with afferent stimulation. Thus, the model in this case appears to be that prolonged production of eCBs during the STDP protocol coupled with glutamate release leads to activation of presynaptic NMDA receptors and persistent enhancement of glutamate release. It is important to note that this model requires compartmentalization of the astrocyte glutamate release (to the presynapse) and presynaptic glutamate release during the induction stage (simply increasing astrocyte calcium levels was insufficient to cause this form of LTD). However, the tight investiture of excitatory synapses by astrocyte processes and highly active glutamate uptake by astrocytes provides the necessary anatomical and functional substrates for compartmentalization of extracellular glutamate. It is interesting to note the similarities between this form to STDP LTD and CB LTD (Han et al. 2012 ) discussed previously.
The above brief overview of eCB-mediated synaptic plasticity highlights the rich repertoire of eCB signaling in the brain. Despite the many forms of eCB-mediated synaptic plasticity, several themes emerge: eCB plasticity is widespread and is involved in many different circuits, from the spinal cord to the cortex. Because CB1-mediated synaptic plasticity occurs on both inhibitory and excitatory terminals, activation of these receptors and the various forms of synaptic plasticity that follow will be very state dependent and can be expected to have quite unpredictable effects at the network level, generally requiring experimentation to validate. The density of CB1 receptors correlates poorly to their functional importance. An example of this is CB1 receptors on astrocytes. While these receptors are very sparsely seen in immunocytochemical studies, synaptic plasticity elicited by these receptors appears to exert significant effects at the network and behavioral levels (Navarrete and Araque 2010 ; Han et al. 2012 ; Min and Nevian 2012 ). While in many cases CB1-mediated synaptic plasticity is elicited by 2-AG, anandamide also frequently participates. The difficulties in demonstrating anandamide involvement primarily arise because of the diverse synthetic pathways for this eCB and the lack of specific inhibitors for its synthesis. Thus, more indirect approaches, such as inhibiting anandamide degradation by FAAH, must be used (keeping in mind the caveat that inhibition of anandamide degradation also affects other FAAH metabolites such as N-arachidonoyl glycerol and the acyl amides. In conclusion, in the 12 years since eCB-mediated synaptic plasticity was first demonstrated, it has been shown to involve an unexpectedly large number of types of synaptic plasticity as well as utilize numerous intracellular signaling pathways. It is likely that additional forms of eCB -mediated synaptic plasticity remain to be elucidated and our understanding of their roles in behavior will increase over the coming years.
References
Ahn K, McKinney MK, Cravatt BF (2008) Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chemi Rev 108(5):1687–1707
Ahn K, Johnson DS, Cravatt BF (2009) Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin Drug Discov 4(7):763–784
Atwood BK, Mackie K (2010) CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol 160(3):467–479
Atwood BK, Straiker A, Mackie K (2012a) CB(2) cannabinoid receptors inhibit synaptic transmission when expressed in cultured autaptic neurons. Neuropharmacology 63(4):514–523
Atwood BK, Wager-Miller J, Haskins C, Straiker A, Mackie K (2012b) Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB(2) ligands. Mol Pharmacol 81(2):250–263
Azad SC, Eder M, Marsicano G, Lutz B, Zieglgansberger W, Rammes G (2003) Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem 10(2):116–128
Bacci A, Huguenard JR, Prince DA (2004) Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature 431(7006):312–316
Blankman JL, Simon GM, Cravatt BF (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chemi Biol 14(12):1347–1356
Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y (2012) Endocannabinoid signaling and synaptic function. Neuron 76(1):70–81
Chavez AE, Chiu CQ, Castillo PE (2010) Trpv1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci 13(12):1511–1518
Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38(3):461–472
Chevaleyre V, Heifets BD, Kaeser PS, Sudhof TC, Castillo PE (2007) Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron 54(5):801–812
Citri A, Malenka RC (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacol 33(1):18–41
den Boon FS, Chameau P, Schaafsma-Zhao Q, van Aken W, Bari M, Oddi S, Kruse CG, Maccarrone M, Wadman WJ, Werkman TR (2012) Excitability of prefrontal cortical pyramidal neurons is modulated by activation of intracellular type-2 cannabinoid receptors. Proc Natl Acad Sci U S A 109(9):3534–3539.
Di S, Malcher-Lopes R, Halmos KC, Tasker JG (2003) Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci 23(12):4850–4857
Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, Zieglgansberger W, Lutz B, Rammes G (2006) Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci 26(21):5794–5799
Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP (2010) Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology 151(10):4811–4819
Felder CC, Joyce KE, Briley EM, Glass M, Mackie KP, Fahey KJ, Cullinan GJ, Hunden DC, Johnson DW, Chaney MO, Koppel GA, Brownstein M (1998) Ly320135, a novel cannabinoid CB1 receptor antagonist, unmasks coupling of the CB1 receptor to stimulation of camp accumulation. J Pharmacol Exp Ther 284(1):291–297
Feldman DE (2012) The spike-timing dependence of plasticity. Neuron 75(4):556–571
Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L (2010) Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol 588(Pt 16):3045–3062
Fu J, Gaetani S, Oveisi F, Verme JLo, Serrano A, Rodriguez De Fonseca F, Rosengarth A, Luecke H, Giacomo BDi, Tarzia G, Piomelli D (2003) Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 425(6953):90–93
Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5(5):446–451
Glass M, Felder CC (1997) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments camp accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17(14):5327–5333
Hajos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF (2000) Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci 12(9):3239–3249
Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, Koehl M, Abrous DN, Mendizabal-Zubiaga J, Grandes P, Liu Q, Bai G, Wang W, Xiong L, Ren W et al (2012) Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 148(5):1039–1050
Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, moto KE, Maejima T, Araishi K, Shin HS, Kano M (2005) Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron 45(2):257–268
Hoffman AF, Lupica CR (2012) Synaptic targets of Δ9-tetrahydrocannabinol in the central nervous system. Cold Spring Harb Perspect Med. doi:10.1101/cshperspect.a012237
Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435(7045):1108–1112
Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG (2002) International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 54(2):161–202
Huang GZ, Woolley CS (2012) Estradiol acutely suppresses inhibition in the hippocampus through a sex-specific endocannabinoid and mGluR-dependent mechanism. Neuron 74(5):801–808
Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89(1):309–380
Katona I, Freund TF (2012) Multiple functions of endocannabinoid signaling in the brain. Ann Rev Neurosci 35:529–558
Kellogg R, Mackie K, Straiker A (2009) Cannabinoid CB1 receptor-dependent long-term depression in autaptic excitatory neurons. J Neurophysiol 102(2):1160–1171
Kenakin T, Miller LJ (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62(2):265–304
Kim J, Alger BE (2004) Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci 7(7):697–698
Kim J, Isokawa M, Ledent C, Alger BE (2002) Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci 22(23):10182–10191
Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29(3):717–727
Kreitzer AC, Carter AG, Regehr WG (2002) Inhibition of interneuron firing extends the spread of endocannabinoid signaling in the cerebellum. Neuron 34(5):787–796
Lauckner JE, Hille B, Mackie K (2005) The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A 102(52):19144–19149
Levenes C, Daniel H, Soubrie P, Crepel F (1998) Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. J Physiol 510(3):867–879
Liu J, Wang L, Harvey-White J, Huang BX, Kim HY, Luquet S, Palmiter RD, Krystal G, Rai R, Mahadevan A, Razdan RK, Kunos G (2008) Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 54(1):1–7
Llano I, Leresche N, Marty A (1991) Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron 6(4):565–574
Luk T, Jin W, Zvonok A, Lu D, Lin XZ, Chavkin C, Makriyannis A, Mackie K (2004) Identification of a potent and highly efficacious, yet slowly desensitizing CB1 cannabinoid receptor agonist. Br J Pharmacol 142(3):495–500
Mackie K, Devane WA, Hille B (1993) Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol Pharmacol 44(3):498–503
Mackie K, Hille B (1992) Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A 89(9):3825–3829
Mackie KY, Westenbroek Lai R, Mitchell R (1995) Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci 15(10):6552–6561
Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31(3):463–475
Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN (2005) Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem 95(2):437–445
Marinelli S, Pacioni S, Bisogno T, Marzo VDi, Prince DA, Huguenard JR, Bacci A (2008) The endocannabinoid 2-arachidonoylglycerol is responsible for the slow self-inhibition in neocortical interneurons. J Neurosci 28(50):13532–13541
Marinelli S, Pacioni S, Cannich A, Marsicano G, Bacci A (2009) Self-modulation of neocortical pyramidal neurons by endocannabinoids. Nat Neurosci 12(12):1488–1490
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, Mvan der S, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Marzo VDi et al (2003) CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302(5642):84–88
Mazzola C, Medalie J, Scherma M, Panlilio LV, Solinas M, Tanda G, Drago F, Cadet JL, Goldberg SR, Yasar S (2009) Fatty acid amide hydrolase (FAAH) inhibition enhances memory acquisition through activation of PPAR-alpha nuclear receptors. Learn Mem 16(5):332–337
McHugh D, Hu SS, Rimmerman N, Juknat A, Vogel Z, Walker JM, Bradshaw HB (2010) N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci 11:44
McKinney MK, Cravatt BF (2005) Structure and function of fatty acid amide hydrolase. Ann Rev Biochem 74:411–432
Melis M, Pillolla G, Luchicchi A, Muntoni AL, Yasar S, Goldberg SR, Pistis M (2008) Endogenous fatty acid ethanolamides suppress nicotine-induced activation of mesolimbic dopamine neurons through nuclear receptors. J Neurosci 28(51):13985–13994
Min R, Nevian T (2012) Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat Neurosci 15(5):746–53
Misner DL, Sullivan JM (1999) Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci 19(16):6795–6805
Monory K, Lutz B (2009) Genetic models of the endocannabinoid system. Curr Top Behav Neurosci 1:111–139
Morgan NH, Stanford IM, Woodhall GL (2009) Functional CB2 type cannabinoid receptors at CNS synapses. Neuropharmacology 57(4):356–368
Nagarkatti P, Pandey R, Rieder SA, Hegde VL, Nagarkatti M (2009) Cannabinoids as novel anti-inflammatory drugs. Future Med Chem 1(7):1333–1349
Navarrete M, Araque A (2008) Endocannabinoids mediate neuron-astrocyte communication. Neuron 57(6):883–893
Navarrete M, Araque A (2010) Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68(1):113–126
Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29(3):729–738
Oz M (2006) Receptor-independent effects of endocannabinoids on ion channels. Curr Pharm Design 12(2):227–239
Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS (2009) Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther 331(2):591–597
Peterfi Z, Urban GM, Papp OI, Nemeth B, Monyer H, Szabo G, Erdelyi F, Mackie K, Freund TF, Hajos N, Katona I (2012) Endocannabinoid-mediated long-term depression of afferent excitatory synapses in hippocampal pyramidal cells and GABAergic interneurons. J Neurosci 32(41):14448–14463
Pitler TA, Alger BE (1992) Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12(10):4122–4132
Pitler TA, Alger BE (1994) Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron 13(6):1447–1455
Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ (2011) Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci 14(12):1542–1547
Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A 99(12):8384–8388
Rodriguez JJ, Mackie K, Pickel VM (2001) Ultrastructural localization of the CB1 cannabinoid receptor in mu-opioid receptor patches of the rat caudate putamen nucleus. J Neurosci 21(3):823–833
Roloff AM, Thayer SA (2009) Modulation of excitatory synaptic transmission by Delta 9-tetrahydrocannabinol switches from agonist to antagonist depending on firing rate. Mol Pharmacol 75(4):892–900
Roth SH (1978) Stereospecific presynaptic inhibitory effect of delta9-tetrahydrocannabinol on cholinergic transmission in the myenteric plexus of the guinea pig. Can J Physiol Pharmacol 56(6):968–975
Sang N, Zhang J, Chen C (2006) PGE2 glycerol ester, a COX-2 oxidative metabolite of 2-arachidonoyl glycerol, modulates inhibitory synaptic transmission in mouse hippocampal neurons. J Physiol 572(Pt 3):735–745
Shen M, Piser TM, Seybold VS, Thayer SA (1996) Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci 16(14):4322–4334
Shen M, Thayer SA (1999) Delta9-tetrahydrocannabinol acts as a partial agonist to modulate glutamatergic synaptic transmission between rat hippocampal neurons in culture. Mol Pharmacol 55(1):8–13
Shoemaker JL, Ruckle MB, Mayeux PR, Prather PL (2005) Agonist-directed trafficking of response by endocannabinoids acting at CB2 receptors. J Pharmacol Exp Ther 315(2):828–838
Sjostrom PJ, Turrigiano GG, Nelson SB (2003) Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39(4):641–654
Stella N (2004) Cannabinoid signaling in glial cells. Glia 48(4):267–277
Straiker A, Mackie K (2005) Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol 569(Pt 2):501–517
Straiker A, Hu SS, Long JZ, Arnold A, Wager-Miller J, Cravatt BF, Mackie K (2009) Monoacylglycerol lipase limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation in autaptic hippocampal neurons. Mol Pharmacol 76(6):1220–1227
Straiker A, Wager-Miller J, Hu SS, Blankman JL, Cravatt BF, Mackie K (2011) COX-2 and fatty acid amide hydrolase can regulate the time course of depolarization-induced suppression of excitation. Br J Pharmacol 164(6):1672–1683
Sugiura T, Kodaka T, Kondo S, Nakane S, Kondo H, Waku K, Ishima Y, Watanabe K, Yamamoto I (1997) Is the cannabinoid CB1 receptor a 2-arachidonoylglycerol receptor? Structural requirements for triggering a Ca2+ transient in NG108-15 cells. J Biochem 122(4):890–895
Sugiura T, Kishimoto S, Oka S, Gokoh M (2006) Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res 45(5):405–446
Takahashi KA, Linden DJ (2000) Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 83(3):1167–80
Talwar R, Potluri VK (2011) Cannabinoid 1 (CB1) receptor–pharmacology, role in pain and recent developments in emerging CB1 agonists. CNS Neurol Disord Drug Targets 10(5):536–544
Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M (2010) The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 65(3):320–327
Tasker JG, Herman JP (2011) Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress 14(4):398–406
Twitchell W, Brown S, Mackie K (1997) Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 78(1):43–50
Tzounopoulos T, Rubio ME, Keen JE, Trussell LO (2007) Coactivation of pre- and postsynaptic signaling mechanisms determines cell-specific spike-timing-dependent plasticity. Neuron 54(2):291–301
Varma N, Carlson GC, Ledent C, Alger BE (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21(24):188
Vaughan CW, McGregor IS, Christie MJ (1999) Cannabinoid receptor activation inhibits GABAergic neurotransmission in rostral ventromedial medulla neurons in vitro. Br J Pharmacol 127(4):935–940
Ventura R, Harris KM (1999) Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci 19(16):6897–6906
Walter L, Franklin A, Witting A, Moller T, Stella N (2002) Astrocytes in culture produce anandamide and other acylethanolamides. J Biol Chem 277(23):20869–20876
Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N (2003) Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci 23(4):1398–1405
Ward SJ, Raffa RB (2011) Rimonabant redux and strategies to improve the future outlook of CB1 receptor neutral-antagonist/inverse-agonist therapies. Obesity 19(7):1325–1334
Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410(6828):588–92
Xi ZX, Peng XQ, Li X, Song R, Zhang HY, Liu QR, Yang HJ, Bi GH, Li J, Gardner EL (2011) Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat Neurosci 14(9):1160–1166
Yang H, Zhang J, Andreasson K, Chen C (2008) COX-2 oxidative metabolism of endocannabinoids augments hippocampal synaptic plasticity. Mol Cell Neurosci 37(4):682–695
Yasuda H, Huang Y, Tsumoto T (2008) Regulation of excitability and plasticity by endocannabinoids and PKA in developing hippocampus. Proc Natl Acad Sci U S A 105(8):3106–3111
Zarruk JG, Fernandez-Lopez D, Garcia-Yebenes I, Garcia-Gutierrez MS, Vivancos J, Nombela F, Torres M, Burguete MC, Manzanares J, Lizasoain I, Moro MA (2012) Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 43(1):211–219
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Marzo VDi, Julius D, Hogestatt ED (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400(6743):452–457
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Mackie, K. (2013). Endocannabinoid-Mediated Synaptic Plasticity. In: Van Bockstaele, E. (eds) Endocannabinoid Regulation of Monoamines in Psychiatric and Neurological Disorders. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7940-6_2
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7940-6_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7939-0
Online ISBN: 978-1-4614-7940-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)