
Abstract
MicroRNAs are endogenous non-coding small RNAs that have been described as highly conserved regulators of gene expression. They are involved in cancer and in the regulation of neural development and stem cell function. Recent studies suggest that a small subpopulation of cancer stem cells (CSCs) has the capacity to repopulate solid tumours such as glioblastoma (GBM), drive malignant progression and mediate radio- and chemoresistance. GBM-derived CSCs share the fundamental stem cell properties of self-renewal and multipotency with neural stem cells (NSCs) and may be regulated by miRNAs. In this review, we will summarize the current knowledge regarding the role of miRNAs in GBM development with a focus on the regulation of GBM-CSCs. We propose a list of miRNAs that could serve as molecular classifiers for GBMs and/or as promising therapeutic targets for such brain tumours.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is the most malignant and aggressive brain tumour, with a mean survival time of 9–12 months. The term “glioblastoma” is synonymous to “grade IV astrocytoma” using the World Health Organization (WHO) classification and grading system. Glioblastoma is defined by an uncontrolled cellular proliferation, diffuse infiltration, propensity for necrosis, robust angiogenesis, intense resistance to apoptosis, and rampant genomic instability [1]. Primary glioblastoma arises as a de novo process in the absence of a pre-existing low-grade lesion, whereas secondary glioblastoma develops progressively from grade II (astrocytoma) to grade III (anaplastic astrocytoma) and finally to grade IV (GBM) (Fig. 1) [2]. Lately, on the basis of mRNA expression profiles, GBMs have been also classified as proneural, proliferative, classical, and mesenchymal [3, 4]. The proneural signature predominates among low-grade gliomas and secondary GBMs and is characterised by markers associated with neurogenesis. Instead, the mesenchymal signature is more often related to primary GBMs. Although there is some disagreement regarding the correlation of the novel high-throughput signatures to patient outcome, the predominant view is that proneural tumours are associated to longer survival. However, the greatest response to aggressive therapy was found in the classical subtype.

MicroRNAs as regulators of the genes altered in primary and secondary glioblastoma multiforme. We have introduced in the classical scheme of GBM genetic alterations those miRs that may act as regulators. The so-called “onco-miRs” participate in the inhibition of tumour suppressor genes together with other well-known mechanisms, such as LOH, mutation or promoter methylation. The “tumour suppressor miRs” (TS-miRs) inhibit oncogenes under healthy conditions and would be inactivated in GBM, allowing for the pathological expression of the target oncogenes. Amp, amplification. Mut mutation, OE, over-expression
The traditional model of gliomagenesis predicts that gliomas arise stochastically from somatic mutations in terminally differentiated astrocytes that subsequently undergo a series of transformations to a less differentiated phenotype. A more recent perspective suggests instead that gliomas arise from adult neural stem cells (NSCs) or neural precursors that after transformation behave as cancer stem cells (CSCs) and maintain the tumour. CSCs are regulated by microRNAs (miRNAs), which are key players during normal mammalian development and become altered in multiple human cancers. In this review we will first introduce the CSC hypothesis and the classical signalling pathways that have been linked to primary and secondary glioblastoma. Next, we will summarize recent findings regarding a subset of miRNAs that control signalling pathways critical to glioblastoma biology and we will discuss their role in the context of neural/CSC-related programs. The expression profile of some of these microRNAs has allowed us to improve previous GBM classifications. We propose that miRNAs might be involved in the regulation of both the classical pathway of genetic alterations found in glioma and the self-renewal capacity of glioma CSCs.
The Cancer Stem Cell Hypothesis in Glioblastoma
In recent years, the traditional view of glioma progression has been challenged due to the identification of a distinct subpopulation of cancer cells with higher tumorigenic potential [5]. These cells retain the capacity to initiate and propagate tumours with very high efficiency and have been named generically as CSCs, due to their similarities with normal NSCs. CSCs show expression of stem cell markers (such as CD133, NESTIN, MUSASHI-1 and SOX2), they have capacity to differentiate into multiple lineages and most importantly, they have the ability to self-renew indefinitely [6, 7]. In the field of brain cancer, they are also referred to as brain tumour stem-like cells (BTSC) or brain tumour-initiating cells (BTICs). Glioma CSCs were among the first solid tumour CSCs described [8, 9]. They might arise from transformed NSCs/precursors or from other adult brain cell types that eventually de-differentiate and reacquire stem cell-like properties. Regardless of their putative origin, the CSC hypothesis proposes that aggressive gliomas are maintained from a reservoir of self-sustaining CSCs that self-renew and generate differentiated progeny. Thus, the concept of CSCs resembles the concept of normal stem cells, which are capable of generating the lineage-related cell types of a given tissue.
The CSC hypothesis states that tumour cell populations have a hierarchical developmental structure. According to this view, the initiation and progression of the tumour (including GBM) depends on the presence of a rare fraction of CSCs. However, the CSC hypothesis has been challenged by recent findings that propose a different scenario, in which most GBM cells can behave as BTICs with different degrees of stemness, and therefore many cells (not a minority) have the capacity to drive glioblastoma progression [10]. An open question is whether the number of BTICs/CSCs reflects cancer progression, with more tumour cells acquiring the properties of CSCs in advanced GBM stages. Either way, subpopulations of cells that satisfy the functional characteristics of stem cells are highly resistant to current GBM therapies [11], underscoring the importance to elucidate the molecular mechanisms regulating CSC self-renewal and differentiation in order to develop potential therapeutic approaches for aggressive gliomas.
One such approach is to force the cells to differentiate, manipulating the signalling pathways that regulate NSCs during development and through adulthood. Amongst the pathways controlling embryonic and adult brain stem cells, the TGF-β/BMP pathway, the canonical WNT pathway, the NOTCH or the HEDGEHOG pathways have emerged as critical regulators of NSCs. For some of these pathways, pro-differentiative actions in the regulation of glioblastoma CSCs have already been reported. This is the case of the BMP pathway. BMP ligands belong to the TGF-β superfamily of cytokines that signal through tetrameric complexes formed by type II and type I receptors. The ligand–receptor interaction can trigger several signalling cascades, including the canonical pathway, in which activated type I receptors phosphorylate the DNA binding proteins SMADs, which regulate the expression of target genes. It has been proven that GBM-CSCs express type I and II BMP receptors and can respond to BMP ligands through SMAD-dependent signalling [12]. Following BMP4 treatment, CD133+ glioblastoma cells differentiate and this abrogates their stem cell phenotype, decreases the size and invasive capacity of the tumours in xenograft models of GBM and prolongs the overall survival of the treated animals [12]. These results suggest that it is possible to take advantage of the knowledge on NSC regulation to manipulate, at least in part, GBM-CSC behaviour.
More recently, it has been reported that other signalling pathways regulating self-renewal of NSCs, such as the HEDGEHOG or NOTCH pathways, are also activated in gliomas and contribute to GBM-CSC self-renewal [13, 14]. In the SONIC HEDGEHOG (SHH) pathway, the ligand activates a signal-transduction cascade that involves the action of the membrane proteins PATCHED1 and SMOOTHENED, and the activity of the GLI1 transcription factor. SHH-GLI1 signalling regulates the expression of stemness genes, the self-renewal of CD133+ GBM-CSCs, and is required for sustained glioma growth and survival [13]. Interestingly, interference of SHH-GLI1 signalling inhibits human glioma xenograft growth, indicating that down-regulation of the pathway may be of therapeutic value [13]. Regarding the NOTCH pathway, it has been shown that most human gliomas of different grades express moderate to high levels of NOTCH receptors (NOTCH 1–4), ligands (i.e., DELTA-like and JAGGED proteins) and downstream target genes. The NOTCH receptors are cleaved and release the intracellular domain of NOTCH (NICD), which translocates to the nucleus, associates with the DNA-binding protein CSL that is converted from a transcriptional repressor to an activator. In glioma cell lines, over-expression of active NICD promotes the acquisition of a CSC-like identity. Thus, NOTCH signalling plays a role in CSCs, and it may be possible to target these tumour-initiating cells by inhibiting the NOTCH pathway (reviewed by Stockhausen et al. [14]). However, as we will show in the next section, genetic mutations in stem cell pathways are very rarely found in gliomas, although changes in the expression of their components are commonly associated with high grade tumours (reviewed by Li et al. [15]), suggesting that epigenetic changes of developmental pathways are key drivers of GBM growth.
Classical Signalling Pathways Altered in Glioblastoma
After 20 years of research in gliomas, their main genetic alterations have been identified, helping to decipher the heterogeneity of these tumours. For example, distinctions between the genetic lesions found in primary and secondary GBMs have been made [1, 16] (Fig. 1). Primary glioblastomas exhibit frequent EGFR amplification, CDK4 amplification, MDM2 or MDM4 amplification, RB1 mutation/homozygous deletion, p16 INK4A and p14 ARF homozygous deletion, monosomy of chromosome 10 and PTEN mutations [16]. TP53 mutation is found in less than 30% of primary glioblastomas. In contrast, secondary glioblastomas arise from a lower-grade precursor lesion and carry TP53 mutations in more than two thirds of the cases [16, 17]. Also, allelic losses on 19q and 13q, and over-expression of PDGFRA are more common in secondary glioblastomas whereas amplification of EGFR or MDM2, PTEN mutation as well as homozygous p16 INK4A or p14 ARF deletions are all rare in secondary glioblastomas [16]. Therefore, malignant glioma cells frequently have increased EGFR and/or PDGFR tyrosine kinase signalling, either as a result of amplification of the gene, expression of a constitutively active variant or autocrine loops (in the case of the PDGF ligand).
Interestingly, the signalling pathways downstream of these tyrosine kinase receptors are involved in the regulation of adult NSCs, suggesting they may also operate in the regulation of GBM-CSCs. For instance, in cell culture studies, proliferation and neurosphere formation of glioma CSCs was dependent on EGFR [10, 18]. In the mouse model, EGFR expression has been proposed as a marker of activated NSCs [19, 20]. It has also been shown that adult mouse NSCs depend on EGF signalling for self-renewal and that EGFR activity, in the absence of PTEN expression, can transform murine NSCs [21]. Accordingly, glioma CSCs are responsive to anti-EGFR drugs but PTEN expression and AKT inhibition seems to be necessary for such effect [22]. In addition, the activity of adult NSCs is also regulated by PDGFRA [23]. Excessive PDGF activation in stem cells is sufficient to initiate tumour formation and can transform INK4a/ARF-deficient astrocytes [24].
Apart from the alteration in tyrosine kinase receptors, most GBMs bear mutations in genes encoding components of one of three pathways, p53, RB1 and PI3K/PTEN/AKT [16]. These pathways participate in the regulation of NSCs. For instance, p53 regulates adult NSC behaviour [25–27] and accumulation of mutant p53 in mouse leads to the expansion of mutant p53-expressing OLIG2(+) transit-amplifying progenitor-like cells and initiates glioma formation [28]. Recently, murine modelling studies, together with confirmatory transcriptomic/promoter studies in human primary GBM, have validated an important cooperative action between p53 and PTEN in the regulation of normal and malignant stem cell differentiation, self-renewal and tumorigenic potential, which was unexpected from the previous genomic analysis on primary and secondary GBMs. This cooperative action seems to be mediated by increased MYC activity [29].
The stem cell regulator BMI-1 controls proliferation and RB1 signalling through the repression of p16 INK4A [30]. Cyclin/cyclin-dependent kinase (Cyclin/CDK) complexes phosphorylate RB, inhibit its activity and allow cell cycle progression. In the absence of BMI-1, the CDK inhibitor p16 INK4A is up-regulated, therefore blocking the cell cycle and reducing the rate of NSC proliferation. Thus, BMI-1 plays an important role in sustaining the replication-competent state of normal NSCs. Interestingly, p16 INK4A is also linked to senescence, and deletion of p16 INK4A can partially oppose the age-related decline in the number and self-renewal potential of neural progenitors [31].
Another cyclin-dependent kinase inhibitor that regulates the behaviour of NSCs is p21cip1/waf. It has been reported that the relative quiescence of adult NSCs in vivo depends on p21cip1/waf, which is necessary for the maintenance of these cells throughout life [32]. Interestingly, MUSASHI-1, a marker of CSCs that is enriched in NSCs and in many brain tumours including gliomas, represses p21cip1/waf during active NSC proliferation. MUSASHI-1 exerts a pro-proliferative effect that is mediated through direct binding of the protein to the p21cip1/waf 3′UTR, which results in translational inhibition and reduced p21cip1/waf protein. In addition, it has been recently shown that MUSASHI-1 converts to an activator during the early phases of NSC differentiation, possibly inducing p21cip1/waf expression and cell cycle exit [33]. Thus, deciphering MUSASHI-mediated mRNA translational regulation in CSCs may be relevant to understand and control pathological stem cell proliferation.
We speculate that a subset of pathways may be shared by NSCs and CSCs, in particular those required for continuous self-renewal/proliferation of the cells. As we will discuss in this review, some of these pathways are targets of multiple microRNAs that become deregulated in GBM.
MicroRNAs in Glioblastoma
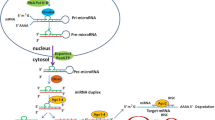
Several studies have shown that miRNA expression profiles are altered in tumours, including GBM. MicroRNAs are endogenous, single-stranded RNA molecules consisting of approximately 22 non-coding nucleotides that regulate target genes [34, 35]. There are approximately 500–1,000 different mammalian miRNA genes, most of which are transcribed by RNA polymerase II as long primary transcripts (pri-miRNAs) that form a stem–loop structure [34, 35]. In the nucleus, pri-miRNAs are processed into 70–100 nucleotide-long hairpin pre-miRNAs by the RNAse III Drosha. These pre-miRNAs are then exported into the cytoplasm by Exportin-5 and are further processed by another RNAse III, Dicer. The resultant RNA duplexes contain the mature miRNA and the passenger miRNA strand. The mature miRNA is incorporated into the RNA-induced silencing complex (RISC) and is directed to specific (complementary) binding sites in the 3′-untranslated region (UTR) of mRNA transcripts. This miRNA–mRNA interaction blocks translation and only very rarely, guides endonucleolytic cleavage of the mRNA [36]. In mammals, microRNAs can guide recruitment of deadenylases that remove the poly(A) tail form the mRNA leading to exonucleolytic cleavage. It is believed that the expression of ~30% of all human genes may be regulated by miRNAs. It is also thought that a single mRNA can be regulated by several miRNAs, and that one miRNA can recognize several targets, highlighting the great modulatory potential of these small non-coding RNA molecules.
Many mammalian miRNAs are tissue-specific and are expressed in a temporally regulated manner during development. In recent years, miRNAs are emerging as important regulators of key developmental processes such as cell differentiation, proliferation and apoptosis. These processes are altered in tumours. Accordingly, it has been shown that microRNAs are highly deregulated in a wide variety of cancers, including brain tumours such as GBM, and their abnormal expression has been linked to tumour initiation and progression [37]. Some miRNAs are down-regulated in tumours (acting as tumour-suppressor genes) whereas others are over-expressed (acting as oncogenes or oncomiRs) [38]. In the following sections, we will summarise the information available on reported miRNA signatures in GBM and on the role miRs may have in the regulation of GBM-related signalling pathways and in the regulation of GBM-CSCs.
MicroRNA Expression Profiles in Glioblastoma
Provided microRNA deregulation appears to be a hallmark of cancer, several studies have already investigated whether miRNAs are differentially expressed in glioblastoma versus normal brain tissue, in low grade versus high grade gliomas or in anaplastic astrocytomas versus GBM. Although Northern blot, direct cloning, microarrays and RT-qPCR are all methods of choice to study miRNA expression patterns, most of the studies in gliomas have been microarray-based and are summarized in Table 1 (up-regulated miR genes in GBM) and Table 2 (down-regulated miR genes). It is interesting to note that approximately 30% of these microRNAs have been consistently found as deregulated in two or more independent studies (i.e., miR-9/9*, miR-21, miR-124, miR-128, miR-137), whereas others have been only detected in a single study and will therefore require further validation. These emerging profiles may be useful in determining specific miRNA signatures that might help to classify brain tumours and to define some miRNA subsets as biomarkers of prognosis and therapeutic outcome (reviewed in [39, 40]). In addition, miRNA signatures may be also useful to understand the origin of the tumour itself. In this regard, recent microRNA expression profiles from the Cancer Genome Atlas (TCGA) have allowed for the identification of five glioblastoma subgroups that refine previous classifications and that markedly correspond to the expression profiles of neural precursors at different stages of differentiation. According to this novel classification, GBM subgroups correlate with the signatures of multipotent/radial glia precursors, oligondendroglial precursors, neuronal precursors, neuroepithelial/neural crest precursors or astrocyte precursors. This finding points to different cells of origin for brain tumours and uncovers a great heterogeneity within previous classifications, which were solely based on genetic alterations or mRNA profiles, highlighting the enormous potential of analysing and understanding miRNA expression and function in GBM [41].
MicroRNA Regulation of Classical Glioblastoma Genetic Pathways
Although the distinct miRNA expression profiles observed in glioma underscore the importance of miRNAs in glioblastoma pathogenesis, functional studies addressing the role of miRNAs in GBMs are scarce. From all the deregulated miRNAs shown in Tables 1 and 2, only a few have been partially characterized from a functional perspective. Functional analysis is often performed in vitro by manipulating microRNA levels, using synthetic microRNA precursors or modified oligonucleotides that antagonize miRNAs (antagomiRs). However, very little information is available regarding their role in vivo. Some GBM-related miRNAs can be placed in the scheme of genetic alterations involved in gliomagenesis according to their predicted and/or validated targets (Fig. 1). This underscores the many processes in which miRNAs could be participating and the increasing complexity of the gliomagenesis network.
Among the miRNAs up-regulated in brain tumours, miR-21 has emerged as one of the most consistently highly expressed microRNA in malignant glioma tissue versus normal tissue, and Kaplan-Meier survival analysis have revealed that high expression of this microRNA is significantly associated with poor patient survival [42]. According to the novel miR-based classification of GBM provided by Kim et al. [41], miR-21 over-expression would preferentially correlate to GBMs with astrocytic signature. Most groups suggest that miR-21 acts as an oncogene that regulates multiple malignancy parameters in GBM. Inhibition of miR-21 increases apoptosis, decreases growth and invasion and may decrease chemoresistance. Suppression of cell death or apoptosis is one of the key roles of miR-21. It has been shown that inhibition of miR-21 (by locked nucleic acid (LNA) or 2-O-Me-miR-21 antagomirs) in glioma cell lines leads to an increase in Caspase-3 and Caspase-9 dependent apoptosis [43, 44]. Chen et al. [45] found that inhibition of miR-21 in a number of GBM cell lines increases endogenous levels of programmed cell-death 4 (PDCD4) and so increases PDCD4-dependent apoptosis. Silencing of miR-21 not only inhibits apoptosis but also stops cell cycle progression in GBM cell lines, by decreasing EGFR signalling, a crucial pathway in gliomagenesis [46], possibly through STAT3 inhibition [47]. It is interesting to note that PTEN, a major tumour suppressor in GBM, is a miR-21 target. However, although down-regulation of miR-21 leads to increased PTEN expression, the GBM suppressor effect of blocking miR-21 was found in both PTEN mutant and wild-type GBM cells, and is most likely independent of PTEN regulation [46, 47]. Thus, miR-21 may serve as a novel therapeutic target for glioblastoma independent of PTEN status. By performing pathway analysis of computationally predicted miR-21 targets, Papagiannakopoulos et al. [48] identified that some miR-21 regulated genes participated in three key tumour suppressor pathways involved in glioblastoma: p53, TGF-β and mitochondrial apoptosis. MiR-21 also contributes to the invasiveness of glioma cells by targeting inhibitors of the matrix metalloproteinases TIMP3 and RECK [49] or by disrupting the negative feedback circuit of RAS/MAPK signalling mediated by SPRY2 [50].
Altogether, these data support an important role for miR-21 in GBM pathogenesis and some groups have already considered that targeting miR-21 may be a valid approach for enhancing the chemotherapeutic effects in glioblastoma treatment. Corsten et al. [51] revealed that microRNA-21 knockdown disrupts glioma growth in an in vivo glioma model and displays synergistic cytotoxicity with S-TRAIL in human gliomas. Ren et al. [52, 53] found an increase of the chemotherapeutic activity of 5-fluorouracil (5-FU) by combining this compound with antisense-miR-21 oligonucleotides on in vitro studies with the GBM cell line U251. Both the apoptosis and the migration ability of the cells were decreased. In a similar study, the same group has reported that IC50 values are dramatically decreased in cells treated with a miR-21 inhibitor combined with taxol, relative to cells treated with the cytotoxic drug alone.
Temozolamide (TMZ) is an alkylating agent commonly used as a first line treatment for GBM patients. TMZ treated cells undergo apoptosis by an increase in Caspase-3 activity and in the BAX/BCL-2 ratio. Acquired chemoresistance is a severe limitation to this therapy with more than 90% of recurrent gliomas showing no response to a second cycle of chemotherapy. In a recent study, Shi et al. [54] discovered that miR-21 over-expression protects the human GBM cell line U87 from TMZ by decreasing both BAX/BCL-2 ratio and Caspase-3 activity, which points to a possible role of miR-21 in the clinical resistance to this drug. In a similar way, suppression of miR-21 in the U373 GBM cell line increases the cytotoxicity of another chemotherapeutic agent, VM-26 [55], perhaps through the regulation of a new miR-21 target, LRRFIP1, an inhibitor of NF-kB signalling.
MiR-221/222 are two other miRNAs over-expressed in GBM with oncogenic characteristics. MiR-221 is over-expressed only in high grade astrocytomas (WHO grade III and IV) [56]. The function of miR-221 and miR-222 has been explored together since their expression is co-regulated and they have the same target specificity. MiR-221/222 have been found to repress the expression of the cell cycle regulator p27kip1 [57–59]. This protein is an inhibitor of cyclin-dependent protein kinases (CDK) and triggers cell cycle arrest in the G1 phase [60]. le Sage et al. [60] found that U87 glioma cells require high activity of these two miRNAs to maintain low levels of p27kip1 and to remain in a proliferative state. Zhang et al. [61] performed a bioinformatic analysis and found that 16 of the miR-221/22 target genes present a direct or an indirect interaction with AKT and may co-ordinately regulate the AKT pathway. Moreover, the authors found that miR-221/222 over-expression increases glioma cell proliferation and invasion in vitro and induces glioma growth in a subcutaneous xenograft mouse model. Importantly, the effects correlate with increased phosphorylation of AKT and therefore with the activation of AKT signalling [61].
Huse et al. [62] found that miR-26a is a regulator of PTEN expression. The phosphatase PTEN is a molecular antagonist of the AKT pathway that has a central role in glioma biology. Mutations in the PTEN gene have been found in 40% and 10% of primary and secondary GBM, respectively. MiR-26 is frequently amplified and over-expressed in GBM samples and this amplification correlates with monoallelic PTEN loss and decreased patient survival. Additionally, this group has demonstrated that PTEN repression by miR-26a increases de novo tumour formation in a mouse model of high grade glioma, demonstrating that this miR enhances gliomagenesis in vivo [63]. According to the miR-based classification of GBM, miR-26a over-expression would correlate to GBMs with oligodendroglial signature [41].
On the other hand miR-7, miR-128, miR-124, miR-137 and miR-181 are down-regulated in GBM. Kefas et al. [64] identified miR-7 down-regulation in GBM tissue compared to the surrounding brain. MiR-7 is a potential tumour suppressor in GBM because it targets the signalling pathway activated by EGFR, a receptor over-expressed in 60% of primary GBM patients [16]. Since miR-7 directly represses EGFR by binding to its 3′UTR in GBM cells, it is possible that increased EGFR levels/AKT signalling in some patients may be related to decreased miR-7 expression. The AKT pathway can also be activated independently of the EGF receptor, one example is through signalling downstream of IRS-1 and IRS-2. Interestingly, IRS-2 is also a direct target of miR-7 and over-expression of miR-7 reduces viability and invasiveness of GBM cells [64].
MiR-128 belongs to the class of brain specific miRNAs [65] and is down-regulated in glioma tissues [66–69] and, to a lesser extent, in lower grade gliomas [66]. Three independent groups have found that over-expression of miR-128 in glioma cells inhibits their proliferation in vitro by decreasing the S-phase population without inducing apoptosis [66, 68, 70]. However it has been recently demonstrated in other cellular systems that miR-128 down-regulates BAX and induces apoptosis [71]. An in silico analysis has revealed a conserved miR-128 target site in the 3′-UTR of the transcription factor E2F3a, which is essential for cell cycle progression [66]. In vitro studies have confirmed that miR-128 inhibits proliferation of brain cells by direct negative regulation of E2F3. Another direct target of miR-128 is the oncogene BMI-1 [68, 70], that functions in epigenetic silencing of certain gliomagenesis genes, such as the tumour suppressors TP53 and p16 INK4a [72]. A study in medulloblastoma reported that miR-128a targets BMI-1 and as a consequence the intracellular redox state of the tumour cells is altered and tumour cell growth is inhibited by promoting senescence [73].
The expression levels of miR-124 and miR-137 are also down-regulated in glioblastomas and/or anaplastic astrocytomas [67, 68]. Silber et al. [67] found that miR-124 and miR-137 promote G0/G1 cell cycle arrest in glioblastoma cell lines. The cyclin-dependent kinase 6 (CDK6) is an important regulator of the G1 to S phase transition, and both miRs directly target CDK6 and inhibit its expression.
On the other hand miR-181a/b down-regulation has a functional significance in GBM development [56, 69, 74]. Shi et al. [74] have demonstrated an association between diminished miR-181a and glioma grade, and Zhi et al. [42] have shown that miR-181b low expression is significantly associated with poor patient survival. Recently, Slaby et al. [75] have published that miR-181b and miR-181c can serve as predictive markers of response to TMZ since expression of these miRs in patients who respond to this therapy was significantly down-regulated in comparison to patients with progressive disease. Radiotherapy is also widely used in GBM patients. Transient over-expression of miR-181a in the U87 GBM cell line significantly sensitized the cells to radiation treatment concurrent with the down-regulation of the BCL-2 protein, suggesting that this miR can be used for enhancing the effect of radiation treatment on GBM patients [76].
Finally, microRNAs also regulate angiogenesis, which is a hallmark of glioma and is a major therapeutic target. A recent study used expression-profiling to examine the levels of microRNA in tumour associated endothelial cells. They found that miR-296 levels are up-regulated in cultured primary human microvascular endothelial cells exposed to pro-angiogenic factors, and also in primary tumour endothelial cells isolated from human brain tumours compared to normal brain endothelial cells. Interestingly these authors have shown that inhibition of miR-296 with antagomiRs reduces angiogenesis in tumour xenografts in vivo [77].
Altogether, these results underscore the relevance of miRNAs as multilevel regulators of gliomagenesis. They modulate proliferation (miR-7, miR-124, miR128, miR-137, miR-221), apoptosis (miR-7 and miR-21), angiogenesis (miR-296), tumour invasion (miR-7 and miR-21) and chemo and radiotherapy resistance (miR-21, miR-181a). Amongst the targets of several miRs up-regulated in GBM (putative oncomiRs), we encounter a fistful of genes that undergo loss of function in GBM, suggesting that these miRs further contribute to the repression of certain signalling pathways. For instance, the genes PTEN and TP53 are often mutated or epigenetically silenced in GBM and miR-21/miR-26, which are over-expressed in GBM, target the mRNA of these genes, reinforcing their functional silencing. On the contrary, amongst the targets of the miRs that are down-regulated in GBM (putative tumour suppressor miRs) we find targets that are amplified or undergo gain of function in GBM. This is the case of miR-7 and its target gene, EGFR.
In sum, the data so far reviewed evidence the impact that microRNAs may have in the regulation of the fundamental signalling pathways altered in glioblastoma. In addition, the recent microRNA expression profiles from the Cancer Genome Atlas underlines the necessity to revise the microarray-based data that consider GBMs as a unique group. For instance, some of the tumour suppressor miRs, like miR-7, miR-128 and miR-124, are over-represented in the neuronal precursor microRNA cluster defined by Kim et al. [41], suggesting that they could have a different action (even as onco-miRs) in this subtype of GBMs.
MicroRNA Regulation of Glioblastoma Stem Cell Proliferation and Differentiation
The parallelism pointed out between brain CSCs and NSCs raises the hypothesis that they may share common regulatory networks, which may be modulated by microRNAs. It has been reported that normal NSCs, and other stem cell types as well, express certain miR genes that are involved in the maintenance of the undifferentiated and self-renewing state of the cells (see below). It is conceivable that a subset of these microRNAs might be also expressed in GBM-CSCs, perhaps playing a similar role in the regulation of the stem-like properties of these tumour initiating cells. Thus, NSCs and CSCs possibly display partially overlapping miRNA profiles, with high levels of oncomiRs regulating self-renewal/proliferation of the cells. As shown in Fig. 2, this model would predict a decrease in oncomiR expression and an increase the expression of tumour suppressor microRNAs upon differentiation.

Regulation of GBM cancer stem cells and normal neural stem cells by miRNAs. Due to their common undifferentiated, multipotent status and their self-renewal property, it has been suggested that NSCs and CSCs share common regulatory networks. Therefore, we hypothesise that there must be a set of common miRs expressed by both cell types. Differentiation would be characterised by a particular miRNA signature that would reflect the down-regulation of Stem-miRs and Onco-miRs and the up-regulation of Pro-differentiation miRs and Tumour suppressor miRs
Chen et al. were the first to point out that stem cells have a less complex miRNA profile than mature somatic tissues and suggested that the degree of cellular differentiation can be characterized by a particular miRNA signature [78–80]. In mammals, several miRNAs such as miR-124, miR-125 and miR-137 are specifically enriched in the central nervous system (CNS), and changes in the pattern of miR expression during CNS development suggest that these microRNAs play a role in neural differentiation [81, 82]. The most significant changes in miRNA expression have been reported in the transition from neural stem/precursors to differentiated neurons.
Some microRNAs such as miR-124, miR-9, miR-125b and miR-22 are absent in undifferentiated cells but are highly up-regulated as differentiation proceeds. The role of these microRNAs has been partially elucidated in normal NSCs. More recently, Lavon et al. [83] have reported the results from a microarray assay comparing neural precursor cells with glioma samples, showing that 71 microRNAs in glioma had a distinct expression pattern relative to normal brain, and that those miRs were remarkably reminiscent of the expression pattern observed in embryonic stem cells and neural progenitor cells. About half of them were clustered in seven genomic regions: miR-17-92, miR106b-25, miR-106a-363, miR-183-96-182, miR-367-302, miR-371-373 and the large miRNA cluster in the Dlk1-Dio3 region [83]. Although the role of these miR clusters in NSCs/CSCs remains to be explored, it is interesting to note that members of these clusters are enriched in the oligoneural precursor GBM subclass defined by Kim et al. [41]. Of note, the oncogenic miR-17-92 is induced by MYC in many cancers and the MYC oncogene is specifically amplified in the oligoneural precursor GBM subclass [41], suggesting that miR-17-92 could function as a component of the MYC oncogenic program in GBM.
From a mechanistic point of view, miR-124 appears to ensure that progenitor genes like Laminin-1 and Integrin-1 are post-transcriptionally inhibited in mouse neurons [84]. MiR-124 also impacts the transition towards neuronal differentiation by directly targeting PTBP1, a protein involved in alternative splicing patterns related to neuronal development [85]. Moreover, miR-124 represses Sox-9 in adult NSCs from the mouse subventricular zone, and has the capacity to promote differentiation of dividing precursors into neurons [86]. Interestingly, miR-124 is one of the most abundant miRs in the adult brain, accounting for more than 25% of the total miR content of the brain [87]. As shown in Table 2, its down-regulation in high grade gliomas is remarkable, and also it has been frequently down-regulated in medulloblastoma [88] suggesting that over-expression of miR-124 could potentially cause the inhibition of CSC proliferation by promoting differentiation. In this regard, Silber et al. have found that miR-124 (and also miR-137) promote G0/G1 cell cycle arrest and induce neuronal-like differentiation of GBM-derived stem cells in the absence of growth factor signalling [67].
MiR-125b is other microRNA that promotes neuronal differentiation [89] and as many other miRs its expression levels are altered in gliomas but its function still remains obscure. Intriguingly, miR-125b has been reported as either up-regulated or unchanged in independent studies, suggesting it may function as an oncogene only in a subset of tumours. Accordingly, Kim et al. reported that miR-125b is over-expressed only in the multipotent precursor class of GBM [41]. Shi et al. have recently found that miR-125b is down-regulated in human U251 glioma stem cells. Its up-regulation in vitro leads to growth inhibition by decreased CDK6 and CDC25A cell cycle regulators [90]. However, one report seemed to contradict previous findings and reported that over-expression of miR-125b suppresses ATRA (all-trans retinoic acid)-induced apoptosis of human glioma cells and that low expression of miR-125b decreases proliferation and enhances the sensitivity of the cells to ATRA, by blocking the translation of the mRNA that codes for the cell apoptosis-related protein BCL-2 modifying factor (BMF) [91].
Gal et al. [92] demonstrated that transfection of GBM-CSCs with miR-451 inhibited their growth and their capacity to form clonal aggregates/spheres in vitro. But as with miR-125b, the in vitro results for miR-451 are controversial. Godlewski et al. [93] have shown that in glioblastoma patients, elevated miR-451 is associated with shorter survival. Abundant glucose allows relatively high miR-451 expression, promoting cell growth whereas low glucose levels decrease miR-451 expression, slowing proliferation but enhancing migration and survival [93, 94]. On the other hand, Nan et al. [95] have published that miR-451 plays a role as tumour suppressor in three human glioblastoma cell lines because increased expression of this miR inhibits cell growth, inducing G0/G1 phase arrest, increasing cell apoptosis and diminishing the invasive capacity through matrigel. Consequently, future studies are needed to elucidate the function of this microRNA in gliomagenesis. Provided miR-451 expression correlated with the neuromesenchymal GBM subgroup in the classification reported by Kim et al. [41], which was heterogeneous and contained a mixture of tumours from all previously reported mRNA-based GBM classes, it is possible that the functional role of miR-451 may be greatly dependent on the GBM subtype.
For some miRs, no apparent correlation is found between the expression in glioma samples and the role in stem cell self-renewal/differentiation. For instance, miR9/9* are over-expressed specifically in brain primary tumours as compared to primary tumours in other tissues and brain metastasis, and have been pointed out as valuable biomarkers [96]. Previous reports have found that over-expression of miR-9 promotes premature differentiation of neural progenitor cells. Amongst other genes, miR-9 targets TLX, a nuclear receptor that is involved in NSC self-renewal (reviewed by Fineberg et al. [97]). More recently, Kim et al. have found that miR-9 is enriched in the GBM subclass related to oligoneural precursors. In their hands, miR-9 down-regulates JAK kinases and inhibits the activation of STAT3, decreases the expression of mesenchymal/astrocytic markers, promotes the expression of neuronal markers and increases CD133+ GBM-CSC proliferation [41]. However, it remains to be assessed if miR-9 expression is enriched in the bulk of the tumour cells, which may be differentiated progeny of CSCs, but remains down-regulated in the CSC subpopulation.
Another interesting microRNA that regulates CSCs is miR-128, which represses the oncogene BMI-1, an important self-renewal factor for several types of stem cells. Indeed, it has been demonstrated that miR-128 inhibits glioma proliferation by targeting BMI-1 [68], providing the first link between a microRNA that acts specifically on a self-renewal factor and the regulation of glioma CSCs. Upon miR-128 over-expression, BMI-1 levels are reduced, and a significant decrease in the number and volume of glioma spheres is observed [68]. In mouse NSCs, the loss of Bmi-1 is associated with the up-regulation of p21 cip1/waf, raising the possibility that BMI-1 not only has a critical role in normal NSCs, but that it may also be key for glioma stem cell proliferation and self-renewal [98]. Recently, Cui et al. have found another miR-128 target, ARP5, a transcription suppressor that promotes stem cell renewal and inhibits the expression of known tumour suppressor genes involved in senescence and differentiation [70]. Finally, it is important to note that miR-128 over-expression also limited glioma xenograft growth in vivo [98].
Two microRNAs have been recently found to regulate the NOTCH pathway, which plays critical roles in glioma cell and stem cell survival and proliferation [14]. MiR-34a was described as tumour suppressor in glioma cells as it was found to be a direct target of p53 [99]. This miR is down-regulated in human glioma samples and its over-expression leads to the inhibition of cell proliferation, cell migration and cell invasion and also induces glioma stem cell differentiation. Mir-34a acts by targeting multiple oncogenes such as c-MET, CDK6, NOTCH-1 and NOTCH-2 [100, 101]. Forced NOTCH1/2 expression partially rescued the effects of miR-34a on cell death in glioma stem cells. On the other hand, Purow et al. have reported that miR-326 is also down-regulated in glioma samples. This neuronally expressed microRNA is up-regulated following NOTCH-1 knockdown and inhibited by NOTCH over-expression. In addition this miR inhibited the activity of NOTCH proteins, establishing a novel regulatory feedback loop in this important pathway in glioma [102].
Taken together, increasing data suggest that down- or up-regulation of certain miRNA species has great potential in the modulation of CSCs. However, most of the studies resumed here are based on whole tumour microarray data and on assays using glioma cell lines that represent a mixture of CSCs and their differentiated progeny. Considering that CSCs represent only a sub-population in the glioma tissue and in the cultured cell lines, it is possible that miRNAs with important CSC regulatory capacities are not being pulled out in microarray-based approaches. Another limitation of this approach is the heterogeneity of glioblastomas and the fact that CSC could be very different entities in the proneural and the mesenchymal subtype (e.g., [1, 2, 41]), and therefore they could be modulated by a distinct set of miRNAs. One alternative to circumvent these limitations would be to study the function in gliomas of those miRNAs that have been described to modulate CSC-related pathways in other systems (BMPs, SHH, NOTCH). Moreover, it would be interesting to perform comparative studies between CSCs and their differentiated progeny in the different GBM subtypes. In any case, the development of miRNA-based therapies that promote glioma CSC differentiation and/or limit glioma CSC self-renewal may be of great interest for diagnosis, prognosis and therapeutic purposes.
Conclusions
The discovery of miRNAs has given us a deeper insight into regulation of gene expression. Recent data demonstrate that deregulation of miRNA expression may be part of the basic process of cancer pathogenesis. In gliomas, some over-expressed miRNAs behave as potent oncogenes that down-regulate multiple targets. The emergence of miRNAs as important cancer related genes is likely to have a large impact on therapies designed to block tumour progression. In the future, techniques to over-express miRNAs that function as tumour suppressors, or the administration of synthetic antisense oligonucleotides that repress mature oncogenic miRNAs might effectively slow tumour growth. The sensitivity of glioma CSCs to the regulation by certain miRNAs is also very promising. Considering the proven resistance of these cells to traditional therapies, miRNA approaches could be of great help to improve current clinical trials against malignant gliomas.
We believe that functional studies will open the door to the use of microRNA-based strategies of potential clinical relevance, and that miRNA expression profiling of GBM-CSCs and of human gliomas will lead to the identification of useful signatures correlating with tumour diagnosis and response to treatment. It is important, however, to realise that we still ignore which are the defects underlying the imbalance of miRNAs in glioma cells. Chromosome and genetic alterations and/or failure of post-transcriptional control might cause the deregulation of miRNAs subsets, but epigenetic alterations may also be playing a role. Overall, although for long it has been believed that non-coding RNA may be transcriptional noise, new evidence suggest a role for miRNA in the cancer and stem cell fields that may be of major relevance in the nearby future.
References
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A et al (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21:2683–2710
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414:105–111
Read TA, Hegedus B, Wechsler-Reya R, Gutmann DH (2006) The neurobiology of neurooncology. Ann Neurol 60:3–11
Zaidi HA, Kosztowski T, DiMeco F, Quinones-Hinojosa A (2009) Origins and clinical implications of the brain tumor stem cell hypothesis. J Neurooncol 93:49–60
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S et al (2004) Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 64:7011–7021
Mazzoleni S, Politi LS, Pala M, Cominelli M, Franzin A, Sergi SL et al (2010) Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res 70:7500–7513
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760
Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G et al (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444:761–765
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Altaba A (2007) HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol 17:165–172
Stockhausen MT, Kristoffersen K, Poulsen HS (2010) The functional role of Notch signaling in human gliomas. Neuro Oncol 12(2):199–211
Li Z, Wang H, Eyler CE, Hjelmeland AB, Rich JN (2009) Turning cancer stem cells inside out: an exploration of glioma stem cell signaling pathways. J Biol Chem 284:16705–16709
Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170:1445–1453
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Soeda A, Inagaki A, Oka N, Ikegame Y, Aoki H, Yoshimura S et al (2008) Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem 283:10958–10966
Ciccolini F, Mandl C, Holzl-Wenig G, Kehlenbach A, Hellwig A (2005) Prospective isolation of late development multipotent precursors whose migration is promoted by EGFR. Dev Biol 284:112–125
Pastrana E, Cheng LC, Doetsch F (2009) Simultaneous prospective purification of adult subventricular zone neural stem cells and their progeny. Proc Natl Acad Sci U S A 106:6387–6392
Li L, Dutra A, Pak E, Labrie JE III, Gerstein RM, Pandolfi PP et al (2009) EGFRvIII expression and PTEN loss synergistically induce chromosomal instability and glial tumors. Neuro Oncol 11:9–21
Griffero F, Daga A, Marubbi D, Capra MC, Melotti A, Pattarozzi A et al (2009) Different response of human glioma tumor-initiating cells to epidermal growth factor receptor kinase inhibitors. J Biol Chem 284:7138–7148
Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S et al (2006) PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 51:187–199
Liu KW, Feng H, Bachoo R, Kazlauskas A, Smith EM, Symes K et al (2011) SHP-2/PTPN11 mediates gliomagenesis driven by PDGFRA and INK4A/ARF aberrations in mice and humans. J Clin Invest 43690
Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF et al (2006) Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci 26:1107–1116
Meletis K, Wirta V, Hede SM, Nister M, Lundeberg J, Frisen J (2006) p53 suppresses the self-renewal of adult neural stem cells. Development 133:363–369
Ferron SR, Marques-Torrejon MA, Mira H, Flores I, Taylor K, Blasco MA et al (2009) Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J Neurosci 29:14394–14407
Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang P, McKeever PE et al (2009) Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell 15:514–526
Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ et al (2008) p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455:1129–1133
Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ (2003) Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425:962–967
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J et al (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443:448–452
Kippin TE, Martens DJ, van der Kooy D (2005) p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev 19:756–767
MacNicol MC, Cragle CE, MacNicol AM (2011) Context-dependent regulation of Musashi-mediated mRNA translation and cell cycle regulation. Cell Cycle 10(1):39–44
Ambros V (2004) The functions of animal microRNAs. Nature 431:350–355
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Kim VN (2005) MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol 6:376–385
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D et al (2005) MicroRNA expression profiles classify human cancers. Nature 435:834–838
Hammond SM (2006) MicroRNAs as oncogenes. Curr Opin Genet Dev 16:4–9
Barbarotto E, Schmittgen TD, Calin GA (2008) MicroRNAs and cancer: profile, profile, profile. Int J Cancer 122:969–977
Calin GA (2009) MicroRNAs and cancer: what we know and what we still have to learn. Genome Med 1:78
Kim TM, Huang W, Park R, Park PJ, Johnson MD (2011) A developmental taxonomy of glioblastoma defined and maintained by microRNAs. Cancer Res
Zhi F, Chen X, Wang S, Xia X, Shi Y, Guan W et al (2010) The use of hsa-miR-21, hsa-miR-181b and hsa-miR-106a as prognostic indicators of astrocytoma. Eur J Cancer 46:1640–1649
Chan JA, Krichevsky AM, Kosik KS (2005) MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 65:6029–6033
Zhou X, Zhang J, Jia Q, Ren Y, Wang Y, Shi L et al (2010) Reduction of miR-21 induces glioma cell apoptosis via activating caspase 9 and 3. Oncol Rep 24:195–201
Chen Y, Liu W, Chao T, Zhang Y, Yan X, Gong Y et al (2008) MicroRNA-21 down-regulates the expression of tumor suppressor PDCD4 in human glioblastoma cell T98G. Cancer Lett 272:197–205
Zhou X, Ren Y, Moore L, Mei M, You Y, Xu P et al (2010) Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab Invest 90:144–155
Ren Y, Zhou X, Mei M, Yuan XB, Han L, Wang GX et al (2010) MicroRNA-21 inhibitor sensitizes human glioblastoma cells U251 (PTEN-mutant) and LN229 (PTEN-wild type) to taxol. BMC Cancer 10:2727
Papagiannakopoulos T, Shapiro A, Kosik KS (2008) MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res 68:8164–8172
Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS et al (2008) MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol 28:5369–5380
Kwak HJ, Kim YJ, Chun KR, Woo YM, Park SJ, Jeong JA et al (2011) Downregulation of Spry2 by miR-21 triggers malignancy in human gliomas. Oncogene
Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K (2007) MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res 67:8994–9000
Ren Y, Kang CS, Yuan XB, Zhou X, Xu P, Han L et al (2010) Co-delivery of as-miR-21 and 5-FU by poly(amidoamine) dendrimer attenuates human glioma cell growth in vitro. J Biomater Sci Polym Ed 21:303–314
Ren Y, Zhou X, Mei M, Yuan XB, Han L, Wang GX et al (2010) MicroRNA-21 inhibitor sensitizes human glioblastoma cells U251 (PTEN-mutant) and LN229 (PTEN-wild type) to taxol. BMC Cancer 10:27
Shi L, Chen J, Yang J, Pan T, Zhang S, Wang Z (2010) MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res 1352:255–264
Li Y, Li W, Yang Y, Lu Y, He C, Hu G et al (2009) MicroRNA-21 targets LRRFIP1 and contributes to VM-26 resistance in glioblastoma multiforme. Brain Res 1286:13–18
Conti A, Aguennouz M, La Torre D, Tomasello C, Cardali S, Angileri FF et al (2009) miR-21 and 221 upregulation and miR-181b downregulation in human grade II–IV astrocytic tumors. J Neurooncol 93:325–332
Gillies JK, Lorimer IA (2007) Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 6:2005–2009
Zhang C, Kang C, You Y, Pu P, Yang W, Zhao P et al (2009) Co-suppression of miR-221/222 cluster suppresses human glioma cell growth by targeting p27kip1 in vitro and in vivo. Int J Oncol 34:1653–1660
Medina R, Zaidi SK, Liu CG, Stein JL, van Wijnen AJ, Croce CM et al (2008) MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res 68:2773–2780
le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A et al (2007) Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J 26:3699–3708
Zhang J, Han L, Ge Y, Zhou X, Zhang A, Zhang C et al (2010) miR-221/222 promote malignant progression of glioma through activation of the Akt pathway. Int J Oncol 36:913–920
Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH et al (2009) The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev 23:1327–1337
Kim H, Huang W, Jiang X, Pennicooke B, Park PJ, Johnson MD (2010) Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc Natl Acad Sci U S A 107:2183–2188
Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M et al (2008) microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res 68:3566–3572
Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V (2004) Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 5:R13
Zhang Y, Chao T, Li R, Liu W, Chen Y, Yan X et al (2009) MicroRNA-128 inhibits glioma cells proliferation by targeting transcription factor E2F3a. J Mol Med 87:43–51
Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M et al (2008) miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med 6:14
Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G et al (2008) Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res 68:9125–9130
Ciafre SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G et al (2005) Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun 334:1351–1358
Cui JG, Zhao Y, Sethi P, Li YY, Mahta A, Culicchia F et al (2010) Micro-RNA-128 (miRNA-128) down-regulation in glioblastoma targets ARP5 (ANGPTL6), Bmi-1 and E2F-3a, key regulators of brain cell proliferation. J Neurooncol 98:297–304
Adlakha YK, Saini N (2010) MicroRNA-128 downregulates Bax and induces apoptosis in human embryonic kidney cells. Cell Mol Life Sci
Itahana K, Zou Y, Itahana Y, Martinez JL, Beausejour C, Jacobs JJ et al (2003) Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol 23:389–401
Venkataraman S, Alimova I, Fan R, Harris P, Foreman N, Vibhakar R (2010) MicroRNA 128a increases intracellular ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS One 5:e10748
Shi L, Cheng Z, Zhang J, Li R, Zhao P, Fu Z et al (2008) hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res 1236:185–193
Slaby O, Lakomy R, Fadrus P, Hrstka R, Kren L, Lzicarova E et al (2010) MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma 57:264–269
Chen G, Zhu W, Shi D, Lv L, Zhang C, Liu P et al (2010) MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol Rep 23:997–1003
Wurdinger T, Tannous BA, Saydam O, Skog J, Grau S, Soutschek J et al (2008) miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 14:382–393
Chen C, Ridzon D, Lee CT, Blake J, Sun Y, Strauss WM (2007) Defining embryonic stem cell identity using differentiation-related microRNAs and their potential targets. Mamm Genome 18:316–27
Houbaviy HB, Murray MF, Sharp PA (2003) Embryonic stem cell-specific MicroRNAs. Dev Cell 5:351–358
Suh MR, Lee Y, Kim JY, Kim SK, Moon SH, Lee JY et al (2004) Human embryonic stem cells express a unique set of microRNAs. Dev Biol 270:488–498
Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS (2003) A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 9:1274–1281
Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Sestan N, Rakic P et al (2004) Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol 5:R68
Lavon I, Zrihan D, Granit A, Einstein O, Fainstein N, Cohen MA et al (2010) Gliomas display a microRNA expression profile reminiscent of neural precursor cells. Neuro Oncol 12:422–433
Cao X, Pfaff SL, Gage FH (2007) A functional study of miR-124 in the developing neural tube. Genes Dev 21:531–536
Makeyev EV, Zhang J, Carrasco MA, Maniatis T (2007) The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 27:435–448
Cheng LC, Pastrana E, Tavazoie M, Doetsch F (2009) miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci 12:399–408
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T (2002) Identification of tissue-specific microRNAs from mouse. Curr Biol 12:735–739
Li KK, Pang JC, Ching AK, Wong CK, Kong X, Wang Y et al (2009) miR-124 is frequently down-regulated in medulloblastoma and is a negative regulator of SLC16A1. Hum Pathol 40:1234–1243
Le MT, Xie H, Zhou B, Chia PH, Rizk P, Um M et al (2009) MicroRNA-125b promotes neuronal differentiation in human cells by repressing multiple targets. Mol Cell Biol 29:5290–5305
Shi L, Zhang J, Pan T, Zhou J, Gong W, Liu N et al (2010) MiR-125b is critical for the suppression of human U251 glioma stem cell proliferation. Brain Res 1312:120–126
Xia HF, He TZ, Liu CM, Cui Y, Song PP, Jin XH et al (2009) MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cell Physiol Biochem 23:347–358
Gal H, Pandi G, Kanner AA, Ram Z, Lithwick-Yanai G, Amariglio N et al (2008) MIR-451 and Imatinib mesylate inhibit tumor growth of Glioblastoma stem cells. Biochem Biophys Res Commun 376:86–90
Godlewski J, Bronisz A, Nowicki MO, Chiocca EA, Lawler S (2010) microRNA-451: a conditional switch controlling glioma cell proliferation and migration. Cell Cycle 9:2742–2748
Godlewski J, Nowicki MO, Bronisz A, Nuovo G, Palatini J, De LM et al (2010) MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol Cell 37:620–632
Nan Y, Han L, Zhang A, Wang G, Jia Z, Yang Y et al (2010) MiRNA-451 plays a role as tumor suppressor in human glioma cells. Brain Res 1359:14–21
Nass D, Rosenwald S, Meiri E, Gilad S, Tabibian-Keissar H, Schlosberg A et al (2009) MiR-92b and miR-9/9* are specifically expressed in brain primary tumors and can be used to differentiate primary from metastatic brain tumors. Brain Pathol 19:375–383
Fineberg SK, Kosik KS, Davidson BL (2009) MicroRNAs potentiate neural development. Neuron 64:303–309
Fasano CA, Phoenix TN, Kokovay E, Lowry N, Elkabetz Y, Dimos JT et al (2009) Bmi-1 cooperates with Foxg1 to maintain neural stem cell self-renewal in the forebrain. Genes Dev 23:561–574
Luan S, Sun L, Huang F (2010) MicroRNA-34a: a novel tumor suppressor in p53-mutant glioma cell line U251. Arch Med Res 41:67–74
Guessous F, Zhang Y, Kofman A, Catania A, Li Y, Schiff D et al (2010) microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 9
Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E et al (2009) MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res 69:7569–7576
Kefas B, Comeau L, Floyd DH, Seleverstov O, Godlewski J, Schmittgen T et al (2009) The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J Neurosci 29:15161–15168
Malzkorn B, Wolter M, Liesenberg F, Grzendowski M, Stuhler K, Meyer HE et al (2009) Identification and functional characterization of microRNAs involved in the malignant progression of gliomas. Brain Pathol
Laneve P, Di Marcotullio L, Gioia U, Fiori ME, Ferretti E, Gulino A et al (2007) The interplay between microRNAs and the neurotrophin receptor tropomyosin-related kinase C controls proliferation of human neuroblastoma cells. Proc Natl Acad Sci U S A 104:7957–7962
Ferretti E, De Smaele E, Po A, Di Marcotullio L, Tosi E, Espinola MS et al (2009) MicroRNA profiling in human medulloblastoma. Int J Cancer 124:568–577
Sasayama T, Nishihara M, Kondoh T, Hosoda K, Kohmura E (2009) MicroRNA-10b is overexpressed in malignant glioma and associated with tumor invasive factors, uPAR and RhoC. Int J Cancer 125:1407–1413
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E et al (2002) Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99:15524–15529
Xia H, Qi Y, Ng SS, Chen X, Chen S, Fang M et al (2009) MicroRNA-15b regulates cell cycle progression by targeting cyclins in glioma cells. Biochem Biophys Res Commun 380:205–210
Rao SA, Santosh V, Somasundaram K (2010) Genome-wide expression profiling identifies deregulated miRNAs in malignant astrocytoma. Mod Pathol 23:1404–1417
Ferretti E, De Smaele E, Miele E, Laneve P, Po A, Pelloni M et al (2008) Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J 27:2616–2627
Pierson J, Hostager B, Fan R, Vibhakar R (2008) Regulation of cyclin dependent kinase 6 by microRNA 124 in medulloblastoma. J Neurooncol 90:1–7
Song L, Huang Q, Chen K, Liu L, Lin C, Dai T et al (2010) miR-218 inhibits the invasive ability of glioma cells by direct downregulation of IKK-beta. Biochem Biophys Res Commun 402:135–140
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
González-Gómez, P., Sánchez, P. & Mira, H. MicroRNAs as Regulators of Neural Stem Cell-Related Pathways in Glioblastoma Multiforme. Mol Neurobiol 44, 235–249 (2011). https://doi.org/10.1007/s12035-011-8196-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-011-8196-y