
Abstract
L-type Ca2+ channels (LTCCs, Cav1) open readily during membrane depolarization and allow Ca2+ to enter the cell. In this way, LTCCs regulate cell excitability and trigger a variety of Ca2+-dependent physiological processes such as: excitation–contraction coupling in muscle cells, gene expression, synaptic plasticity, neuronal differentiation, hormone secretion, and pacemaker activity in heart, neurons, and endocrine cells. Among the two major isoforms of LTCCs expressed in excitable tissues (Cav1.2 and Cav1.3), Cav1.3 appears suitable for supporting a pacemaker current in spontaneously firing cells. It has steep voltage dependence and low threshold of activation and inactivates slowly. Using Cav1.3−/− KO mice and membrane current recording techniques such as the dynamic and the action potential clamp, it has been possible to resolve the time course of Cav1.3 pacemaker currents that regulate the spontaneous firing of dopaminergic neurons and adrenal chromaffin cells. In several cell types, Cav1.3 is selectively coupled to BK channels within membrane nanodomains and controls both the firing frequency and the action potential repolarization phase. Here we review the most critical aspects of Cav1.3 channel gating and its coupling to large conductance BK channels recently discovered in spontaneously firing neurons and neuroendocrine cells with the aim of furnishing a converging view of the role that these two channel types play in the regulation of cell excitability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spontaneous electrical activity in excitable cells is fundamental to the control of basic physiological functions. Usually, spontaneous firings originate in narrow areas and spread to neighboring cells to drive rhythmic patterns of activity such as circadian rhythms in the central nervous system, activity-dependent hormone release in neuroendocrine cells, and heart beating [1, 2]. To undertake these duties, spontaneously firing cells are equipped with a set of ion channels that turn on at subthreshold membrane potentials and carry sufficient inward current to drive the action potential (AP) upstroke [3, 4]. In many cells, a spontaneous subthreshold depolarization is associated with Na+ entry through either hyperpolarization-activated cation channels (HCN) or voltage-gated Na+ channels [5]. There is, however, an increasing number of neurons and neuroendocrine cells in which subthreshold Ca2+ channels are shown to contribute to pacemaking. Besides the low voltage-activated T-types that are by definition sub-threshold Ca2+ channels [6, 7], Cav1.3 L-type calcium channels (LTCCs) are shown to possess all the prerequisites for pacemaking excitable cells: low threshold and steep voltage dependence of activation in addition to slow inactivation kinetics [8–11]. Cav1.3 channels drive the spontaneous firing of dopaminergic neurons in substantia nigra pars compacta (SNc) [12, 13] and mid-spiny striatal neurons [14]. They also contribute to the pacemaker current that generates the heartbeat at the cardiac sinoatrial node [15] and the spontaneous firing activity of adrenal chromaffin cells [16–18] and cochlear immature inner hair cells (IHCs) [19]. Finally, LTCCs contribute to the spontaneous AP firing of suprachiasmatic nucleus neurons [20, 21], locus coeruleus neurons [22], pituitary cells [23], and immortalized hypothalamic neurons [24, 25] and sustain the plateau potentials in spinal cord motoneurons [26]. In these cells, however, the direct involvement of Cav1.3 in pacemaking has not yet been proven.
Here, we will review the features that confer to LTCCs its pacemaker properties in a variety of cells and how, by using Cav1.3−/− KO mice [8], it has been possible to identify Cav1.3 as the L-type channel responsible for pacemaking spontaneously firing neurons, neuroendocrine, and cardiac cells. We also review how the selective coupling of Cav1.3 to BK channels affects the spontaneous activity of a number of cells with particular emphasis on the adrenal chromaffin cells and their implications in adrenal gland function.
L-Type Calcium Channels: Expression and Function
LTCCs are hetero-oligomers consisting of a pore-forming α1 subunit interacting with several accessory subunits (β and α2-δ) to form a functional complex. LTCCs belong to the large family of voltage-gated Ca2+ channels that are permeable to Ca2+ and include the N, P/Q, R, and T types [27]. The identification of L-types from the other Ca2+ channel currents (N, P/Q, R, and T) in different cells is facilitated by the availability of a class of organic molecules that possess high affinity and selectivity for LTCCs [27]. Among those, the dihydropyridines (DHPs) are available either as LTCC blockers (so-called Ca2+ antagonists, e.g., nifedipine) or LTCC activators (e.g., BayK 8644). They allow the unique isolation and characterization of L-type currents, although they are unable to distinguish between different LTCC isoforms.
Presently, LTCCs comprise of four genes that code for the α1S, α1C, α1D, and α1F subunits, now renamed Cav1.1, Cav1.2, Cav1.3, and Cav1.4, respectively [27]. Of these, Cav1.1 and Cav1.4 exhibit specific expression profiles that are restricted to the skeletal muscle and the retina, whereas Cav1.2 and Cav1.3 are widely expressed throughout the central nervous system, sensory and endocrine cells, atrial myocytes, and cardiac pacemaker cells [11]. In central neurons and neuroendocrine cells, Cav1.2 and Cav1.3 are often co-expressed, and the general rule is that Cav1.2 expression prevails on Cav1.3 expression in the mouse (80% vs. 20%) [28]. An exception to this rule is the distribution of Cav1 at the IHCs from the mouse cochlea, chromaffin cells of the adrenal medulla, and SNc neurons where Cav1.3 expression takes the overhand [8, 18, 29, 30].
Cav1.2 Versus Cav1.3 in Cell Activity Regulation
LTCCs are commonly defined as high voltage-activated Ca2+ channels since they require strong membrane depolarization to reach the threshold of activation (−30 mV in physiological Ca2+) and differ markedly from the low voltage-activated T-type channels that activate, at 20 to 30 mV, more negative potentials [6, 31, 32]. LTCCs are characterized by relatively fast activation kinetics, large single-channel conductances, and inactivation gating which is both sensitive to voltage and Ca2+ and varies among the different Cav1 subtypes [27]. Concerning the neuronal and neuroendocrine LTCCs (Cav1.2 and Cav1.3), a number of studies have shown that the two LTCCs possess quite different activation properties, voltage- and Ca2+-dependent inactivation (CDI), and sensitivity to DHPs which underlie the different role that the two channels are likely to play in cell function [9, 33]. Most of the currently existing information comes from studies using heterologously expressed channels, cell preparations with a predominant expression of one of the two channel isoforms and knock-out animal models [9, 18, 30, 33, 34].
The biophysical and pharmacological differences between Cav1.2 and Cav1.3 can be summarized as follows: (1) Cav1.3 activates with steep voltage dependence at voltages significantly more negative than Cav1.2 (up to −20 mV) [9, 18, 33]. The lower threshold of activation does not depend on co-expression with the diverse β or α2δ accessory subunits and is thus an intrinsic property of the pore-forming α1 subunit [33]; (2) Cav1.3 has faster activation but slower and less complete voltage-dependent inactivation as compared to Cav1.2 [9, 18]. Inactivation is nearly absent when recording the Ca2+ currents of IHCs of the cochlea where 90% of the total Ca2+ current is carried by Cav1.3 [8, 29]. This is partly due to an unusually slow voltage-dependent inactivation besides an almost complete lack of CDI. The absence of CDI in IHCs is partly due to the high expression of Ca2+-binding proteins CaBP4 in this tissue [35]. Long-lasting Ca2+ currents persisting for 500 ms at positive potentials are also evident in patch-perforated mouse chromaffin cells (MCCs), which express high densities of Cav1.3 channels [18]. (3) Cav1.3 and Cav1.2 have equal Ca2+/Ba2+ permeability ratio and similar high single-channel conductances [27, 36]. (4) Cav1.3 isoforms are less sensitive to DHPs as compared with Cav1.2, requiring about ten times more DHPs for full block at normal holding potentials (V h −70 mV). The IC50 of DHP for Cav1.3 current block decreases markedly at less negative holding potentials (V h −50 mV), reaching comparable IC50 values of Cav1.2 [9, 18]. Saturating concentrations of DHPs that block Cav1.2 at −50 mV also ensure full block of Cav1.3. This is important when assaying the role of Cav1.3 during spontaneous AP recordings in firing cells that spend most of their time at the interspike potential around −50 mV.
Following the above-mentioned properties, it is evident that Cav1.3 and Cav1.2 play quite distinct roles in neuronal, endocrine, and muscle cell physiology, depending critically on the degree of expression of both isoforms. Cav1.3 is a suitable candidate for supporting pacemaker currents, while Cav1.2 recruited at more positive potentials plays a major role in sustaining AP upstrokes. The critical issues characterizing Cav1.3 and its functions will be clarified in the following chapters.
Cav1.3 Expression and Pacemaking
Convincing evidence that Cav1.3 is a suitable channel for pacemaking started to accumulate recently in neuronal, cardiac, and neuroendocrine cells in which Cav1.3 is highly expressed. In many cells, LTCCs fulfill the role of pacemaker channels although a direct involvement of Cav1.3 is still lacking (see Table 1). We will review here the three most convincing cases in which Cav1.3 has been proven to control pacemaking.
Cav1.3 in Pacemaking Neostriatal SNc Dopaminergic Neurons
In vivo, midbrain dopaminergic (DA) neurons display intrinsic pacemaker firing, which is thought to set background dopamine levels and receptor signaling at synaptic targets. Pharmacological and molecular studies have shown that the regular pacemaker activity of DA neuron in the SNc depends on LTCCs: (1) SNc DA neurons express LTCCs, predominantly Cav1.3 [37] and (2) micromolar concentrations of DHPs effectively silence neuronal activity in SNc slice preparations [38, 39]. The identification of a sub-threshold L-type current dominating the pacemaker current came only after using the action potential clamp technique [13]. The L-type pacemaker current was found to rise continuously during the interspike interval to reach maximal values just before the AP upstroke. P/Q-type Ca2+ and TTX-sensitive Nav1 channels contribute as well to pacemaking SNc neurons, but to a minor degree [13].
Convincing evidence for the existence of voltage-gated Ca2+ channels with gating properties similar to Cav1.3 has been found in SNc slices [40, 41], while direct evidence of Cav1.3 involvement in pacemaking SNc DA neurons and burst firing in striatal medium spiny neurons (MSNs) came from works using Cav1.3−/− KO mice [12, 14]. In MSNs, the absence of Cav1.3 interfered with burst firing. Spontaneous bursts could be recovered only after adding the DHP Ca2+ agonist BayK 8644 [14]. In SNc DA neurons [12], somatodendritic Cav1.3 channels gain importance after the second postnatal week. Spontaneous firing in these mature neurons is fully controlled by LTCCs, while in the same aged neurons of mice lacking Cav1.3 the pacemaking is controlled by Nav1 and HCN-gated channels, which are the pacemaker channels during the “juvenile” state. Chan et al. [12] proposed that the switch from a Cav1.3-independent to a Cav1.3-dependent pacemaking mode during aging may be at the basis of the Ca2+-mediated SNc degeneration in Parkinson’s disease (PD) [42].
Notice that cells exhibiting a Ca2+-dependent pacemaker are subjected to an increased ATP consumption in order to keep up with the physiological cytosolic Ca2+ levels. This elevated metabolic rate facilitates the accumulation of mitochondrial DNA damage and reactive oxygen species production that leads to cellular aging [42]. Interestingly, locus coeruleus and tuberomammillary neurons that fire in a clock like pattern and allow considerable amounts of Ca2+ entry during the pacemaker potential show an increased vulnerability to aging and cell death [43]. On the contrary, dopaminergic neurons of the ventral tegmental area and neurons of the olfactory bulb that use Na+ for their pacemaker do not seem to suffer from aging to the same degree [12, 44]. It is therefore reasonable that Cav1.3 channel blockers are expected to reduce the aging processes associated with Ca2+-dependent pacemaker mechanisms, sparing dopaminergic neurons from Ca2+-dependent cell death and delaying the clinical appearance and progression of PD. These findings prompted ongoing clinical trials based on the therapeutical use of DHP Cav1.3 blockers for the treatment of PD in humans [45]. The potential usage of Cav1 blockers in PD has been confirmed by epidemiological studies showing a decreased risk in humans treated for hypertension with DHPs [46].
Although the above-mentioned findings support the idea of Cav1.3 as the pacemaker channel in SNc dopaminergic neurons, new evidence suggest the involvement of other ion channels on the pacemaker mechanism [47]. Low concentrations of isradipine (5 μM) clearly slow down or block the dendritic spontaneous oscillatory potentials (SOP) while preserving the somatic spontaneous firing [47]. Furthermore, the SOP frequency does not coincide with the somatic spiking frequency. Experimental as well as simulation-based studies show that, in order to obtain a full block of spontaneous activity, the simultaneous block of HCN and Cav1.3 channels is required. Thus, pacemaking of SNc dopaminergic neurons seems more robust than expected and not entirely depending on Cav1.3 but rather to a multichannel system, which includes HCN [47], Cav1.3 [12], and Nav1 channels [13].
A confirmation that Cav1.3 plays a role in setting the spontaneous firing of SNc neurons derives from a recent study on SNc slices using the dynamic clamp [48]. With this approach, native LTCCs are first pharmacologically blocked by high doses of DHPs and then virtually replaced by imposing an ad-hoc simulating Cav1.3 conductance model to the voltage-clamped neuron (for details on the technique, see [49]). The great advantage of this approach is that it allows one to identify the key parameters of channel gating that regulate neuronal firing. Curiously enough, pacemaker recovery was obtained in the absence of Ca2+ ions (replaced by other conductive ions), indicating that some other feature of Cav1.3 is responsible for its pacemaker properties besides the Ca2+-mediated SOP generated at the SNc dendrites [47]. Indeed the only parameter that seems critical for Cav1.3 to exert its pacemaking role turns out to be its unique subthreshold voltage dependence of activation [48]. In fact, simulation to replace the normal pacemaking with a 20-mV more negative Cav1.3 voltage dependence to reach maximal currents at rest (−60 mV) failed to recover the firing. This suggests that it is more important how much the pacemaker channel conductance increases with increasing voltages rather than how much its maximal value is at the interspike potential (−50 mV). In conclusion, the somatodendritic Cav1.3 is a subthreshold channel that effectively contributes to SNc pacemaking activity.
Cav1.3 Expression in Cardiac Pacemaker Cells
Heart beating in vertebrates is controlled by a number of channels that contribute to the diastolic pacemaker current [3, 34]. A major role is played by the HCN-gated channel but there is also evidence that a sustained inward current (I st), a T-type channel, an L-type channel and a Na+–Ca2+ exchanger activated by sarcolemmal Ca2+ release during diastolic depolarisations contribute to pacemaking sino-atrial node (SAN) cells [34]. Concerning the role of LTCCs as pacemaker channels, there is evidence that nicardipine induces bradycardia in anesthetized mice in vivo [50] and that nifedipine slows down the pacemaker activity of isolated rabbit SAN cells in vitro [51].
Cav1.3 is expressed in sinoatrial and atrioventricular nodes but not in ventricular cardiac myocytes [15], and its deletion in Cav1.3−/− mice causes bradycardia and arrhythmia [8]. Cav1.3−/− mice display a significant prolongation of the atrioventricular conduction time and suffer from episodes of second-degree AV block [8]. This peculiar phenotype of the Cav1.3−/− mouse persists after the block of sympathetic and parasympathetic tones and thus is considered to be a direct consequence of an altered SAN pacemaker activity [8]. By comparing the amplitude and the voltage dependence of the Ca2+ currents in WT and Cav1.3−/− SAN cells, it became evident that WT cells expressed Ca2+ currents that activated steeply at 22 mV more negative potentials and were 70% larger than those of Cav1.3−/− SAN cells [15, 52]. In addition, non-saturating concentrations of isradipine blocked more effectively the Ca2+ currents of Cav1.3−/− SAN cells, indicating that the only LTCCs available in Cav1.3−/− SAN cells (Cav1.2) are more sensitive to DHPs. In conclusion, Cav1.3 possesses the right gating properties for controlling the diastolic pacemaking current in SAN cells [15, 52].
Cav1.3 as Pacemaker Channel of Adrenal Chromaffin Cells
Bovine, rat, mouse, and human adrenal chromaffin cells fire spontaneously when cultured in vitro [16–18, 53–61] or prepared in slices of the adrenal gland [62, 63]. Spontaneous firing is not observed in all chromaffin cells, a feature that may point to a different function of the cells. The fraction of firing cells is highly variable (20% to 80%) depending on the animal species, the intracellular physiological solution used, and the cell preparation methods. In spite of this, chromaffin cells fire spontaneously regardless of the patch–clamp technique used for AP recording: whole cell, cell attached, and perforated–patch recording modes. The spontaneous activity of chromaffin cells can also be monitored by extracellular recording methods. When dispersed on a multi-electrode array (MEA) recording system [16], spontaneous activity is maintained, proving that spontaneous AP firing is a genuine phenomenon of chromaffin cells and does not derive from cell damage by the recording electrode. With the MEA system, the cell remains intact. It simply adheres to the metallic microelectrodes and AP firing is monitored extracellularly (Fig. 1a). In addition, firing frequencies and firing modes (tonic versus bursts) monitored with the MEA are not significantly different from those intracellularly recorded in patch-perforated conditions (Fig. 1b)

Distinct firing modes recorded from spontaneously firing rat chromaffin cells. a Two types of extracellularly recorded AP firings using MEAs. Note the microvolt vertical scale and the different firing modes: tonic (top trace) and bursting (bottom trace). To the right, single AP events are shown. b Two modes of AP firing intracellularly recorded from cultured RCCs in perforated-patch conditions. In both recording conditions (MEA and perforated-patch), RCCs were bathed in Tyrode solution (2 mM Ca2+)
Given that chromaffin cells fire spontaneously, an open issue is why adrenal chromaffin cells possess spontaneous AP activity although they are contacted by multiple cholinergic innervations that effectively control their activity by splanchnic nerve discharges. Indeed the chromaffin cells are packed together and electrically coupled by gap junctions [63] to form groups of cells that release synchronously the content of secretory granules in nearby blood capillaries [64]. Under these conditions, the spontaneous firing of one or a group of chromaffin cells could warrant the basal release of catecholamines of several electrically coupled cells. Tonic or burst firing activities as shown in Fig. 1 could be at the basis of the adrenal gland response to increased blood levels of histamine, acetylcholine, angiotensin II (ATII), and K+ ions. Histamine and acetylcholine (muscarine) are shown to increase the firing rate of spontaneous APs [57, 59, 60] and the same is likely to occur during postprandial hyperkaliemia and enhanced blood levels of ATII that is known to increase the amount of circulating catecholamines. These physiological responses coming from the periphery (adrenal medulla capillaries) could occur independently of the central sympathetic control of chromaffin cell activity and could be regulated by the electrical synchronism of firing cells coupled by gap junctions [63].
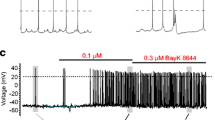
All these arguments justify a detailed analysis of the pacemaker current sustaining chromaffin cell firing, but surprisingly enough there are only a few reports on the ion channels controlling chromaffin cell pacemaking. The few studies available focus on the fundamental role of TTX-sensitive Na+ channels in sustaining the AP upstroke [53, 55] and highlight the additional role of voltage-gated Ca2+ channels in maintaining AP activity [53, 58, 62] or shaping APs through the coupling to BK channels [65]. The critical role of voltage-gated LTCCs in pacemaking MCCs is illustrated in Fig. 2a where addition of 300 nM TTX is shown to reduce the AP overshoot and undershoot with little effect on the firing frequency. In the presence of TTX, there are oscillatory potentials of smaller amplitude that do not overshoot and are effectively blocked by cadmium (200 μM) (Fig. 2b). This is similar to what is reported for dopaminergic neurons and is indicative of a role of voltage-gated Ca2+ channels in sustaining both the AP upstroke and the firing frequency. In the case of dopaminergic neurons, the block of firing after cobalt application causes hyperpolarization due to the block of a dominant subthreshold Ca2+ current [13]. In MCCs, this is not always the case; in half of the cells the block of firing by cadmium or nifedipine causes slight depolarizations, as illustrated in Fig. 2b, c, which indicates a parallel block of Ca2+-activated K+ currents. Figure 2c shows how the firing persists in the presence of TTX and can be either accelerated by BayK 8644 (1 μM) or blocked by nifedipine (3 μM).

a Spontaneous firing in MCCs persists in the presence of TTX (0.3 μM). b, c AP firing is blocked by Cd2+ (200 μM) and nifedipine (3 μM). The (±)-BayK 8644 (1 μM) increases the firing rate and the afterhyperpolarization. The recording conditions are as described in [18]
Evidence for an L-type pacemaker current in MCCs has been obtained by action potential–clamp experiments [17] and, strikingly, a similar subthreshold L-type current is shown to exist in rat chromaffin cells (RCCs) when Na+ TTX-sensitive and K+ channels are blocked. Figure 3 shows the time course of these currents which is very similar to that recorded in SNc DA neurons [13]. As for dopaminergic and cardiac SAN cells, a direct role of Cav1.3 in pacemaking MCCs was uncovered by using Cav1.3−/− KO mice [18]. Deletion of Cav1.3 reduces drastically the amplitude of the pacemaker L-type current and the fraction of firing MCCs (from 80% to 30%). In conclusion, Cav1.3 contributes to the pacemaker current in MCCs (and possibly in RCCs) and pacemaking is affected by BK channels that are highly expressed in these cells [18, 65]. The peculiarities of Cav1.3-BK channel coupling in pacemaking neurons and chromaffin cells will be discussed below.

Pacemaker Ca2+ currents recorded in spontaneously firing chromaffin cells of mouse (left) and rat (right), before and during the application of nifedipine (3 μM). On top, the AP waveforms used for AP clamp are illustrated. K+ and Na+ currents were blocked by adding 135 mM TEA and 0.3 μM TTX to the bath solution containing 2 mM Ca2+. The left panel is adapted from [18]
Cav–BK Crosstalk Affects Cell Firing and Regulates AP Shape in Neurons and Neuroendocrine Cells
Paradoxically, Cav1.3−/− MCCs have more depolarized interspike (resting) potentials and exhibit prolonged plateau depolarizations in response to BayK 8644: two properties that cannot be explained by simply silencing Cav1.3. These findings have a rationale if assuming that Cav1.3 is effectively coupled to Ca2+-activated BK channels, responsible for fast AP repolarization.
The large conductance BK channels need a concerted binding of Ca2+ to the so-called Ca2+ bowl and strong membrane depolarizations for opening the pore [66, 67]. Increasing the cytoplasmic free Ca2+ concentration from nanomolar to micromolar levels shifts the voltage-dependent activation curve of BK currents from very positive to negative resting potentials (from +100 to −50 mV) [68]. To activate during physiological depolarization, BK channels need an intracellular Ca2+ concentration of at least 10 μM. Considering the diverse and tightly regulated intracellular Ca2+ buffering systems, such high concentrations occur only within “Ca2+ nanodomains” in the close vicinity of Ca2+ sources, mainly near Cav channels [67, 69]. Experiments with the Ca2+ chelators BAPTA and EGTA prove that BK channels must be within nanometer distances from their Ca2+ source [69]. In fact, BK channels are often functionally coupled to a voltage-gated Ca channel such as L-type [18, 29, 65], P/Q-type [70], or N-type [71] in different cells. Proteomic approaches that combined affinity purification with mass spectrometry showed that BK channels in the mammalian brain may exist in high molecular weight complexes (~1.6 MDa) [72]. Among the proteins co-immunoprecipitated with BK, there are Cav1.2; Cav2.1 (P/Q), or Cav2.2 (N) [72]. Heterologous expression studies support the idea that the repolarizing responses of Cav–BK complexes are distinctly shaped by their Cav subunits [72].
Below we will focus on two examples in which LTCCs are functionally coupled to BK channels to regulate AP shape and firing frequency. They are only part of the many existing examples of Cav–BK crosstalk (for reviews, see [67]) but are representative for the heterogeneous way of how coupling occurring in a membrane nanodomain may contribute to the fine-tuning of different cells’ excitability.
BK and LTCCs Control Circadian Rhythms in Suprachiasmatic Nucleus Neurons
Neurons from the suprachiasmatic nucleus (SCN) transmit circadian output to other brain regions by modulating their diurnal spontaneous oscillatory firing patterns. It is well known that these neurons fire faster during daytime and slower at night [73]. When TTX is applied to fast-firing neurons during daytime, the resting membrane potential is depolarized but spontaneous oscillations are preserved. These subthreshold oscillations are completely blocked by cadmium or nifedipine and are insensitive to T-, P/Q-, and N-type channel blockers. Current–voltage relationships indicate that the L-type current is up-regulated during daytime, giving rise to elevated total Ca2+ currents as compared to neurons recorded during nighttime [20]. The sustained DHP-sensitive Ca2+ currents during daytime activates at very negative membrane potentials and thus is likely to be carried by Cav1.3 LTCCs. Interestingly, SCN neurons that express the Per1 clock gene depolarize in the afternoon and go into a hyperpolarized state during nighttime. The depolarized state in the afternoon is accompanied by a rise of the input resistance due to the closure of BK and SK channels. Indeed the firing cells recorded in the morning could be brought into a depolarized state as observed in the afternoon by applying either TEA (30 mM) or nimodipine (2 μM), and the depolarization could be mimicked by the co-application of apamine and iberiotoxin (IbTX) [74]. Thus, it seems that LTCCs can effectively regulate SCN pacemaking by partly activating BK currents. Fast-firing neurons, in fact, are characterized by afterhyperpolarizations (AHPs) of shorter duration typically driven by BK channels [75]. A tight coupling between pacemaking L-type currents and BK channels is also evident in AP clamp recordings of isolated SCN neurons [21]. Interestingly, Cav1.3 KO mice are shown to have a normal day–night cycle as measured by spontaneous homecage activities [76]. This might indicate a major contribution by Cav1.2 to activate BK/SK or that some compensatory mechanism to the day–night pacemaker has taken place.
BK and Cav1.3 Control Chromaffin Cell Firing
In RCCs, charybdotoxin (ChTX) significantly broadens the AP half-width, suggesting that BK channels control the fast AP repolarization phase [77]. RCCs and MCCs express two different BK channel subtypes that can be distinguished according to their inactivation kinetics: a fast-inactivating and a slowly inactivating subtype [77]. The fast-inactivating BK channel is typically expressed in chromaffin cells and is specifically involved in tonic cell firing. The slowly inactivating BK channel has gating properties similar to the central neurons and smooth muscle BK channels and gives rise to phasic firings [77]. The chromaffin cells expressing fast-inactivating BK channels are further characterized by deeper AHPs and higher ChTX sensitivity [77]. Fast-inactivating BK channels possess slower deactivation kinetics that might contribute to long-lasting AHP necessary for recovering voltage-gated Na+ and Ca2+ channels to initiate the following AP during sustained firing [77]. In MCCs, we have observed that blocking BK channels by paxilline significantly augmented the firing frequency by delaying AP repolarization and slightly reducing the early phases of the AHP [18]. Nifedipine affected the AP repolarization phase just like paxilline, proving the existence of a strong L-type–BK channel coupling in WT MCCs as well as in RCCs [18, 65]. Figure 4 shows the comparable effects of nifedipine and paxilline on the AP falling phase in RCCs and MCCs and highlights the stronger effects in RCCs which express higher densities of fast-inactivating BK channels as compared to MCCs [18, 65]. In MCCs, however, deletion of Cav1.3 drastically alters the L-type–BK channel coupling in a way that the loss of Cav1.3 prevents the expression of BK channels [18]. This issue will be discussed in the following chapters.

Paxilline (1 μM) and nifedipine (3 μM) produce a marked AP broadening on spontaneously firing RCCs and MCCs. The four recordings are representative of the effects of the two blockers. Notice the marked effects on RCCs which express higher densities of BK channels. In RCCs, paxilline caused a significant 2.2-fold increase of the mean AP width at 0 mV (from 2.8 to 6.1 ms, n = 5; p <0.05), and nifedipine caused a 1.5-fold increase (from 4.6 to 7.1 ms, n = 7; p < 0.05). The MCCs traces are adapted from Marcantoni et al. [17, 18]
A Molecular View to the Functional Coupling of Cav1.3 to BK Channels Using Transgenic Mice: the Case of Pacemaking Chromaffin Cells
In line with the above arguments, it is evident that the BK channels contributing to the repolarization phase of the AP are mostly activated by the Ca2+ entering the cell during the interspike. Since this current is mainly carried by Cav1.3, this explains why nifedipine can effectively control both the frequency and the shape of the AP in spontaneously firing chromaffin cells. A better view to this functional coupling is obtained by directly measuring the K+ currents passing during an AP and testing their block by nifedipine (Fig. 5). The K+ current rises and falls very quickly during the AP. The majority of this current is carried by voltage-gated K+ channels and BK channels and the latter are activated by both the AP and the Ca2+ entering the cytoplasm during the interspike. This Ca2+ is mainly carried by Cav1.3 in WT cells and by Cav1.2 in Cav1.3−/− cells. Thus, the percentage of K+ current blocked by nifedipine furnishes a direct estimate of the effective coupling between Cav1.3 and BK channels. Figure 5 shows that, in the case of WT MCCs, nifedipine blocks more than 60% of the outward K+ currents while in Cav1.3−/− MCCs the DHP blocks only a small fraction of BK current in spite of the similar inward Ca2+ current passing during the spike (blue trace).

Different block by nifedipine of the BK currents activated during an action potential in WT and Cav1.3 KO MCCs. The two MCCs were voltage-clamped to the same AP waveform (top trace) in the presence of 300 nM TTX. In WT MCCs, nifedipine (3 μM) blocks most of the K+ outward current, while in Cav1.3 KO the current amplitude and the block is strongly attenuated (red traces). The blue traces are Ca2+ currents measured in solutions containing 135 mM TEA and 300 nM TTX (adapted from [18])
Given the strong coupling between Cav1.3 and BK channels, a second interesting issue to solve is how close the two channel subunits are. This can be done using well-calibrated Ca2+ buffers that limit the diffusion of Ca2+ ions beyond membrane nano- or microdomains in which voltage-gated Ca2+ channels and BK channels operate. Marty and Neher [78] used this approach in bovine chromaffin cells and found that the internal solutions containing BAPTA were more effective in blocking the Ca2+-dependent K+ currents than EGTA-containing solutions. Following the same approach and using a double-pulse protocol to quantify the amount and type of BK currents, Chris Lingle and coworkers could formulate a quite realistic picture of how L-type and BK channels are coupled in RCCs [79, 80]. The experimental data are consistent with a model in which BK channels are located between 160 and 50 nm from the Ca2+ channels that fuel them [81]. More precisely, 30% to 40% of fast-inactivating BK channels in RCCs are insensitive to EGTA buffers and are therefore positioned sufficiently close to LTCCs (between 50 and 160 nm) to become influenced by the Ca2+ influx through these channels during brief depolarization steps [81]. The remaining channels are far apart (>160 nm) and their activation is fully prevented by saturating EGTA.
We followed a similar approach to evaluate the coupling between Cav1.3 and BK channels in MCCs, measuring the BK currents by means of a voltage clamp protocol that consists of two consecutive pulses. During the first pulse of 400 ms, the cell is stepped from a negative holding potential (−70 mV) to a positive test potential (+80 mV) that overrates the Ca2+ reversal potential and produces large driving forces for K+ ions. In this way, the voltage-gated K+ current (Kv) is fully activated for the entire duration of the pulse. During the second pulse, the cell is first stimulated by a Ca2+ preloading step to 0 mV for 10 to 90 ms to achieve maximal Ca2+ entry and then stepped to the positive test potential. The difference between the peak K+ currents with and without Ca2+ preloading represents the BK current [77, 79].
In the case of MCCs, prolongation of the Ca2+ preloading step has different effects on WT and Cav1.3 KO MCCs (Fig. 6). WT MCCs possess BK currents with relatively fast-inactivating kinetics that increase in amplitude with increasing preloading steps [18]. In contrast, KO MCCs mainly show slowly inactivating BK currents which turn on with long preloading steps [18]. When the same experiments are repeated in the presence of 20 μM EGTA-AM, WT MCCs display their fast-inactivating BK currents while the slowly inactivating channels are apparently lost [18]. This suggests that, in agreement with Prakriya and Lingle’s model, fast-inactivating BK channels are preferentially coupled to Cav1.3 (between 50 and 160 nm) while the slow-inactivating BK channels are more distant (>160 nm; [18]). Incubating WT and KO MCCs with 20 μM of BAPTA-AM, the BK currents are functionally abolished, indicating that the fast-inactivating BK channels are not physically coupled to Cav1.3 [18] (see Fig. 6, bottom).

Effects of EGTA/BAPTA buffers on Cav1.3-BK channels coupling. Membrane-permeable EGTA-AM is unable to prevent the coupling in WT MCCs but is effective on Cav1.3 KO MCCs. BAPTA-AM is effective on both WT and Cav1.3−/− MCCs (adapted from [18]). At the bottom, the main conclusions of the experiments are illustrated, i.e., Cav1.3 is closely coupled to fast inactivating BK channels while Cav1.2 and the other Cav channels are weakly coupled to the slowly inactivating BK channels
BK−/− and Cav1.3−/− KO Mouse Chromaffin Cells Reveal Similar Firing Responses to BayK 8644
The loss of Cav1.3 coupling to BK channels is particularly evident when the DHP Ca2+ agonist BayK 8644 is applied to spontaneously firing MCCs. BayK 8644 prolongs the open state of the LTCCs [82] and, in normal conditions, typically accelerates the pacemaking frequency, producing slight hyperpolarizations and increased AHPs [17, 18]. When BayK 8644 is applied to spontaneously firing Cav1.3−/− MCCs, in the majority of cases the cells respond with a long-lasting membrane depolarization (Fig. 7, middle). In our view, this paradoxical response can be explained by assuming that Cav1.3−/− MCCs are lacking both Cav1.3 and a considerable fraction of BK channels. The Ca2+ fluxes passing through the BayK-modified Cav1.2 channels are sufficient to preserve the firing, but the lack of fast-inactivating BK channels does not allow fast and complete AP repolarizations. Without an effective repolarization, the cell accumulates Ca2+, depolarizes, and stays depolarized until Ca2+ is removed by Ca2+ pumps or Na+/Ca2+ exchangers (see [83] for a review).

Paradoxical effects of the (±)-BayK 8644 (1 μM) on spontaneous firing MCCs lacking either Cav1.3 or BK channels. Each panel shows typical spontaneous firings (top), single AP waveforms (middle), and responses to BayK 8644 (bottom). In WT MCCs (bottom left), the (±) racemic mixture BayK 8644 causes increased firing frequency and enhanced AHP. In Cav1.3−/− and BK−/− MCCs (bottom middle and right), BayK 8644 causes prolonged plateau depolarizations due to the lack of the K+ channels controlling most of the AP repolarization: BK and the Cav channel that opens BK (Cav1.3). For details on BK−/− KO mice, see [84]
A prediction to this hypothesis is that MCCs lacking BK channels should respond to BayK 8644 similarly to Cav1.3−/− MCCs. Indeed MCCs lacking BK channels show remarkable changes of the AP shape and exhibit some similarities to the spontaneous firing of Cav1.3−/− MCCs. Paradoxically, BK−/− MCCs have a reduced frequency of firing that is apparently in contrast to the potentiating effects induced by paxilline in WT MCCs. However, this is similar to what occurs in Purkinje cells from BK−/− mice, which have lower AP activity than WT cells while IbTX increases the firing frequency [84]. Regarding BK−/− MCCs, most striking is the broadening of the AP half-width and the significant prolongation of the interspike interval, a feature that confirms not only the important role of BK channels in MCCs AP repolarization but also its role on cell firing. There is a significant reduction of the AHP amplitude and we found a slightly depolarized resting membrane potential in BK−/− MCCs. These changes are similar to those of Cav1.3−/− MCCs with the main difference that the number of silent cells does not increase in BK−/− MCCs as it does in Cav1.3−/−, suggesting that BK channels are potent modulators but are not conditional as Cav1.3 in pacemaking chromaffin cells.
As expected, BK−/− and Cav1.3−/− MCCs exhibit similar responses to BayK 8644 (Fig. 7, bottom). A fraction of the Cav1.3−/− and BK−/− MCCs responds to BayK 8644 with a sustained membrane depolarization (Fig. 7, bottom), suggesting that BK channels are critical to counterbalance the Cav1.3-mediated AP depolarization. In a number of MCCs, the absence of Cav1.3 induces a loss of BK channels, proving the tight coupling between these two channels and an existing adaptive phenomenon regulating their simultaneous expression or membrane incorporation.
A Molecular Approach to the Cav1.3-BK Channel Coupling: Deletion of Cav1.3 Prevents BK Channel Expression
An existing tight coupling of Cav1.3 and BK channels in MCCs can be proved also by immunostaining, using monoclonal antibodies against Cav1.3 and BK channels (Fig. 8). Well-identified dot-like patterns corresponding to Cav1.3α1 (Fig. 8a, red) and BKSlo1 subunits (Fig. 8a, green) are clearly visible after direct double-staining in slices of the adrenal medulla. The merge signal (Fig. 8a) reveals close proximity of the two channels, where often BK (green) appears surrounded by various Cav1.3 (red) (Fig. 8b; [85]). A broad green puncta (BK) appears surrounded by a number of red puncta (Cav1.3), favoring the view that, as predicted by electrophysiological reasoning in RCCs [81] and MCCs [18], Ca2+ and BK channels are most likely organized within membrane Ca2+ nanodomains rather than in a one-to-one Ca2+–BK channel complex. Under this hypothesis, one large conductance BK channel is surrounded by several L-type channels whose Ca2+ fluxes produce the rapid-activating K+ currents, sustaining the fast repolarization phase of the AP (Fig. 5).

Immunostaining of Cav1.3α1 (red) and BKSlo1 (green) on mouse adrenal gland cryosections using antibody tested for their specificity in Cav1.3−/− and BK−/− tissues (those on BK−/− are not shown). Chromaffin cells in adrenal medulla are imaged over a distance of 8 μm in an image stack along the z-axis (z-stack). Images are illustrated as a composite image, which represents the maximum intensity projection over all the layers of the z-stack (for methods, see [85]). a Arrows indicate the typical dot-like pattern of both Cav1.3α1 and BKSlo1. The merge image shows that most Cav1.3α1 dots are in close localization with BKSlo1 (square), which becomes evident after enlargement (b). Dotted circles indicate the presumptive cell boundary. c, d Immunohistochemistry of Cav1.3α1 and BKSlo1 channel on Cav1.3 WT (c) and Cav1.3−/− (d) mouse adrenal gland cryosections. Arrows indicate the typical dot-like staining of Cav1.3α1 and BKSlo1. In d, the absence of Cav1.3α1 and the strong reduction of BKSlo1 are evident. Nuclei are counterstained with DAPI (blue). Scale bar, 10 μm; n = 3
Immunostaining also supports the view that the deletion of Cav1.3 reduces the expression and functioning of BK channels. This property appears to be an adaptive phenomenon first discovered in cochlear IHCs [29, 86, 87]. IHCs express large densities of Cav1.3 and BK channels [19] and the deletion of Cav1.3 clearly decreases the quantity of functional BK channels activated during Ca2+ loading. Loss of BK also prevents the generation of AP trains induced by current injections [29] and affects glutamate release. A lack of plasma membrane BK channel immunoreactivity in Cav1.3-deficient IHCs has been also reported [87]. The same observation is valid for spontaneously firing MCCs. Deletion of Cav1.3 strongly attenuates the immunoreactivity of BK channels in the plasma membrane of chromaffin cells as shown by comparing the density of green dots in WT and Cav1.3−/− adrenal glands (Fig. 8c, d). Thus, deletion of Cav1.3 reduces the expression of BK channels, confirming the electrophysiological results discussed above [18].
Conclusions
Pacemaking is a cell signaling mechanism widely used in cortical, midbrain, and sensory neurons for regulating vital physiological functions just like controlling the heartbeat and hormone release in neuroendocrine cells. Among the ion channels supporting subthreshold currents, Cav1.3 appears particularly suitable for pacemaking SNc neurons, cardiac sino-atrial node cells, chromaffin cells, and immature cochlear inner hair cells. Although structurally and functionally distinct from the most ubiquitous Cav1.2 isoform, Cav1.3 is always co-expressed at variable ratios with Cav1.2 and is often tightly coupled to BK channels. Its role in the control of vital functions demands for new DHP compounds that selectively block either one of the two major L-type channel isoforms. A selective blocker for Cav1.3 would be beneficial for the therapeutic treatment of Parkinson’s disease, cardiac heart rate, chronic stress disorders, and other neuro- and cardiovascular pathologies in which Cav1.3 is likely to be involved.
New Cav1.3-specific DHP blockers may become available after a better understanding of the specific structural features defining Cav1.3 channel gating. A lot remains to be discovered concerning the role of the different existing splice variants of Cav1.3. In line with this, it will be interesting to clarify the function of the newly identified short splice variant of Cav1.3 (Cav1.342A) which lacks the C-terminal modulator domain that typically prevents the Ca2+-dependent inactivation and shifts the activation voltage to more positive potentials [88]. It could possibly be that the short form (Cav1.342A) is in charge of starting the cellular pacemaking and the long form (Cav1.342) to sustain it (Zuccotti et al., submitted for publication). Splice variant-specific drugs might create new opportunities to target precise problems without possible harmful side effects.
References
Welsh DK, Logothetis DE, Meister M, Reppert SM (1995) Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron 14:697–706
Schultz W (2002) Getting formal with dopamine and reward. Neuron 36:241–263
Noble D (1984) The surprising heart: a review of recent progress in cardiac electrophysiology. J Physiol 353:1–50
Bevan MD, Wilson CJ (1999) Mechanisms underlying spontaneous oscillation and rhythmic firing in rat subthalamic neurons. J Neurosci 19:7617–7628
Bean BP (2007) The action potential in mammalian central neurons. Nature Rev 8:451–465
Pérez-Reyes E (2003) Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev 83:117–161
Carbone E, Marcantoni A, Giancippoli A, Guido D, Carabelli V (2006) T-type channels-secretion coupling: evidence for a fast low-threshold exocytosis. Pflügers Archiv 453:373–383
Platzer J, Engel J, Schrott-fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J (2000) Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102:89–97
Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Englel J, Striessnig J (2001) Alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem 276:22100–22106
Lipscombe D, Helton TD, Xu W (2004) L-type calcium channels: the low down. J Neurophysiol 92:2633–2641
Striessnig J, Koschak A (2008) Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2:233–251
Chan CS, Guzman JN, LLijic E, Mercer JN, RickC TT, Meredith GE, Surmeier DJ (2007) Rejuvenation protects neurons in mouse models of Parkinson’s disease. Nature 447:1081–1090
Puopolo M, Raviola E, Bean BP (2007) Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27:645–656
Olson PA, Tkatch T, Hernandez-Lopez US, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ (2005) G-protein coupled receptor modulation of striatal Cav1.3 L-type Ca2+ channels is dependent on a shank-binding domain. J Neurosci 25:1050–1062
Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J (2003) Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA 100:5543–5548
Marcantoni A, Baldelli P, Hernandez-Guijo J-M, Comunanza V, Carabelli V, Carbone E (2007) L-type calcium channels in adrenal chromaffin cells: role in pace-making and secretion. Cell Calcium 42:397–408
Marcantoni A, Carabelli V, Vandael DH, Comunanza V, Carbone E (2009) PDE type-4 inhibition increases L-type Ca2+ currents, action potential firing and quantal size of exocytosis in mouse chromaffin cells. Pflügers Archiv 457:1093–1110
Marcantoni A, Vandael DHF, Mahapatra S, Carabelli V, Sinneger-Brauns MJ, Striessnig J, Carbone E (2010) Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci 30:491–504
Marcotti W, Johnson SL, Rüsch A, Kros CJ (2003) Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol 552:743–761
Pennartz CMA, de Jeu MTG, Bos NPA, Schaap J, Geurtsen AMS (2002) Diurnal modulation of pacemaker potentials and calcium current in the mammalian circadian clock. Nature 416:286–290
Jackson AC, Yao GL, Bean BP (2004) Mechanism of spontaneous firing in dorsomedial suprachiasmatic nucleus neurons. J Neurosci 24:7985–7998
Filosa JA, Putnam RW (2003) Multiple targets of chemosensitive signaling in locus coeruleus neurons: role of K+ and Ca2+ channels. Am J Physiol Cell Physiol 284:145–155
Tsaneva-Atanasova K, Sherman A, van Goor F, Stojilkovic SS (2007) Mechanism of spontaneous and receptor-controlled electrical activity in pituitary somatotrophs: experiments and theory. J Neurophysiol 98:131–144
Van Goor F, Krsmanovic LK, Catt KJ, Stojilkovi SS (1999) Control of action potential-driven calcium influx in GT1 neurons by the activation status of sodium and calcium channels. Molec Endocrinol 13:587–603
Costantin JL, Charles AC (1999) Spontaneous action potentials initiate rhythmic intercellular calcium waves in immortalized hypothalamic (GT1–1) neurons. J Neurophysiol 82:429–435
Hounsgaard J, Kiehn O (1989) Serotonin-induced bistability of turtle motoneurones caused by a nifedipine-sensitive calcium plateau potential. J Physiol 414:265–282
Catterall WA, Perez-Reyez E, Snutch TP, Striessnig J (2005) Nomenclature and structure–function relationships of voltage-gated calcium channels. Pharmacol Rev 57:411–425
Calin-Jageman I, Lee A (2008) Ca(v)1 L-type Ca2+ channel signaling complexes in neurons. J Neurochem 105:573–583
Brandt A, Striessnig J, Moser T (2003) Cav1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci 23:10832–10840
Baldelli P, Hernández-Guijo JM, Carabelli V, Novara M, Cesetti T, Andrés-Mateos E, Montiel C, Carbone E (2004) Direct and remote modulation of L-channel in chromaffin cells: distinct actions on α1C and α1D subunits? Mol Neurobiol 29:73–96
Carbone E, Lux HD (1984) A low-voltage-activated, fully inactivating calcium channel in vertebrate sensory neurons. Nature 310:501–502
De Waard M, Gurnett CA, Campbell KP (1996) Structural and functional diversity of voltage activated calcium channels. Ion Channels 4:41–87
Xu W, Lipscombe D (2001) Neuronal Cav1.3 alpha1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21:5944–5951
Mangoni ME, Nargeot J (2008) Genesis and regulation of the heart automaticity. Physiol Rev 88:919–982
Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, Fuchs PA, Yue DT (2006) Switching of Ca2+-dependent inactivation of Cav1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci 26:10677–10689
Chahine M, Qu Y, Mancarella S, Boutjdir M (2008) Protein kinase C activation inhibits α1D L-type calcium channel: a single-channel analysis. Pflügers-Archiv 455:913–919
Takada M, Kang Y, Imanishi M (2001) Immunohistochemical localization of voltage-gated calcium channels in substantia nigra dopamine neurons. Europ J Neurosci 13:757–762
Nedergaard S, Flatman JA, Engberg I (1993) Nifedipine- and omegaconotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurons. J Physiol 466:727–747
Mercuri NB, Bonci A, Calabresi P, Stratta F, Stefani A, Bernardi G (1994) Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br J Pharmacol 113:831–838
Kang Y, Kitai ST (1993) Calcium spike underlying rhythmic firing in dopaminergic neurons of the rat substantia nigra. Neurosci Res 18:195–207
Wilson CJ, Callaway JC (2000) Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol 83:3084–3100
Surmeier DJ, Guzman JN, Sanchez-Padilla J (2010) Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium 47:175–182
German DC, Manaye KF, White CL 3rd, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM (1992) Disease-specific patterns of locus coeruleus cell loss. Ann Neurol 32:667–676
Puopolo M, Bean BP, Raviola E (2005) Spontaneous activity of isolated dopaminergic periglomerular cells of the main olfactory bulb. J Neurophysiol 94:3618–3627
Chan CS, Gertler TS, Surmeier DJ (2010) A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson’s disease. Mov Disord 25:S63–S70
Becker C, Jick SS, Meier CR (2008) Use of antihypertensives and the risk of Parkinson disease. Neurology 70:1438–1444
Guzman JM, Sanchez-Padilla J, Chan CS, Surmeier DJ (2009) Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29:11011–11019
Putzier I, Kullmann PHM, Horn JP, Levitan ES (2009) Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J Neurosci 29:15414–15419
Prinz AA, Abbott LF, Marder E (2004) The dynamic clamp comes of age. Trends Neurosci 27:218–224
Lande G, Demolombe S, Bammert A, Moorman AF, Charpentier F, Escande D (2001) Transgenic mice overexpressing human KvLQT1 dominant negative isoform. Part II: pharmacological profile. Cardiovasc Res 50:328–334
Kodama I, Nikmaram MR, Boyett MR, Suzuki R, Honjo H, Owen JM (1997) Regional differences in the role of the Ca2+ and Na+ currents in pacemaker activity in the sinoatrial node. Am J Physiol 272:H2793–H2806
Mangoni ME, Laurine CB, Margera L, Bourinet E, Striessnig J, Nargeot J (2006) Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Progr Biophys Molec Biol 90:38–63
Brandt B, Hagiwara S, Kidokoro Y, Miyazaki S (1976) Action potentials in the rat chromaffin cell and effects of acetylcholine. J Physiol 263:417–439
Biales B, Dichter M, Tischler A (1976) Electrical excitability of cultured adrenal chromaffin cells. J Physiol 262:743–753
Fenwick EM, Marty A, Neher E (1982) A patch–clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. J Physiol 331:577–597
Kubo Y, Kidokoro Y (1989) Potassium currents induced by muscarinic receptor activation in the rat adrenal chromaffin cell. Biomed Res 10:71–81
Akaike A, Mine Y, Sasa M, Takaori S (1990) Voltage and current clamp studies of muscarinic and nicotinic excitation of the rat adrenal chromaffin cells. J Pharm Exp Therap 255:333–339
Hollins B, Ikeda SR (1996) Inward currents underlying action potentials in rat adrenal chromaffin cells. J Neurophysiol 76:1195–1211
Wallace DJ, Chen C, Marley PD (2002) Histamine promotes excitability in bovine adrenal chromaffin cells by inhibiting an M-current. J Physiol 540:921–939
Gullo F, Ales E, Rosati B, Lecchi M, Masi A, Guasti L, Cano-Abad MF, Arcangeli A, Lopez MG, Wanke E (2003) ERG K+ channel blockade enhances firing and epinephrine secretion in rat chromaffin cells: the missing link to LQT2-related sudden death? FASEB J 17:330–332
De Diego AMG (2010) Electrophysiological and morphological features underlying neurotransmission efficacy at the splanchnic nerve–chromaffin cell synapse of bovine adrenal medulla. Am J Physiol Cell Physiol 298:397–405
Nassar-Gentina V, Pollard HB, Rojas E (1988) Electrical activity in chromaffin cells of intact mouse adrenal gland. Am J Physiol 254:C675–C683
Colomer C, Lafont C, Guerineau NC (2008) Stress-induced intercellular communication remodeling in the rat adrenal medulla. Ann NY Acad Sci 1148:106–111
Coupland RE (1965) Electron microscopic observations on the structure of the rat adrenal medulla. I. The ultrastructure and organization of chromaffin cells in the normal adrenal medulla. J Anat 99:231–254
Prakriya M, Lingle CJ (1999) BK channel activation by brief depolarizations requires Ca2+ influx through L- and Q-type Ca2+ channels in rat chromaffin cells. J Neurophysiol 81:2267–2278
Dai S, Hall DD, Hell JW (2007) Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev 89:411–452
Fakler B, Adelman JP (2008) Control of KCa channels by calcium nano/microdomains. Neuron 59:873–881
Orio P, Rojas P, Ferreira G, Latorre R (2002) New disguises for an old channel: maxiK channel β-subunits. News Physiol Sci 17:156–161
Augustine GJ, Santamaria F, Tanaka K (2003) Local calcium signaling in neurons. Neuron 40:331–246
Edgerton JR, Reinhart PH (2002) Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol 548:53–69
Marrion NV, Tavalin SJ (1998) Selective activation of Ca2+-activated K+channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395:900–905
Berkefeld H, Sailer CC, Bidl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U, Fakler B (2006) BKCa–Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314:615–620
Reppert SM, Weaver DR (2001) Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol 63:647–667
Belle MD, Diekman CO, Forger DB, Piggins HD (2009) Daily electrical silencing in the mammalian circadian clock. Science 326:281–284
Cloues RK, Sather WA (2003) Afterhyperpolarization regulates firing rate in neurons of the suprachiasmatic nucleus. J Neurosci 23:1593–1604
Busquet P, Nguyena NK, Schmid E, Tanimoto N, Seeliger MW, Ben-Yosef T, Mizuno F, Akopian A, Striessnig J, Singewald N (2010) Cav1.3 L-type Ca2+ channels modulate depression-like behaviour in mice independent of deaf phenotype. Int J Neuropsychopharm 13:499–513
Solaro CR, Prakriya M, Ding JP, Lingle CP (1995) Inactivating and noninactivating Ca2+- and voltage-dependent K+ current in rat adrenal chromaffin cells. J Neurosci 15:6110–6123
Marty A, Neher E (1985) Potassium channels in cultured bovine adrenal chromaffin cells. J Physiol 367:117–141
Herrington J, Solaro CR, Neely A, Lingle CJ (1995) The suppression of Ca2+- and voltage-dependent K+ current during mAChR activation in rat adrenal chromaffin cells. J Physiol 452:297–318
Prakriya M, Solaro CR, Lingle CJ (1996) [Ca2+]i elevations detected by BK channels during Ca2+ influx and muscarine-mediated release of Ca2+ from intracellular stores in rat chromaffin cells. J Neurosci 16:4344–4359
Prakriya M, Lingle CJ (2000) Activation of BK channels in rat chromaffin cells requires summation of Ca2+ influx from multiple Ca2+ channels. J Neurophysiol 84:1123–1135
Hess P, Lansman JB, Tsien RW (1984) Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 311:538–544
Garcia AG, Garcia de Diego AM, Gandia L, Borges R, Garcia-Sancho J (2006) Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol Rev 86:1093–1131
Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P (2004) Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci USA 101:9474–9478
Zampini V, Johnson SL, Franz C, Lawrence ND, Münkner S, Engel J, Knipper M, Magistretti J, Masetto S, Marcotti W (2010) Elementary properties of Cav1.3 Ca2+ channels expressed in mouse cochlear inner hair cells. J Physiol 588:187–199
Engel J, Braig C, Ruttiger L, Kuhn S, Zimmerman U, Blin N, Sausbier M, Kalbacher M, Munkner S, Rohbock K, Ruth P, Winter H, Knipper M (2006) Two classes of outer hair cells along the tonotopic axis of the cochlea. Neuroscience 143:837–849
Nemzou RMN, Bulankina AV, Khimich D, Giese A, Moser T (2006) Synaptic organization in cochlear inner hair cells deficient for the Cav1.3 (α1D) subunit of L-type Ca2+ channels. Neuroscience 141:1849–1860
Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, Engel J, Romanin C, Striessnig J, Koschak A (2008) Modulation of voltage- and Ca2+-dependent gating of Cav1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J Biol Chem 283:20733–20744
Acknowledgements
We wish to thank Prof. Valentina Carabelli and Dr. Daniela Gavello for the helpful discussions, Dr. Claudio Franchino for the technical assistance, and Prof. Joerg Striessnig for supplying the Cav1.3−/− KO mouse. This work was supported by the Marie Curie Research Training Network “CavNET” Contract MRTN-CT-2006-035367, the Italian M.I.U.R. Grant PRIN 2007SYRBBH_001 to EC, and the San Paolo Company Grant 2008.2191 to AM.
Author information
Authors and Affiliations
Corresponding author
Additional information
David Henry Vandael and Andrea Marcantoni contributed equally to this work.
Rights and permissions
About this article
Cite this article
Vandael, D.H., Marcantoni, A., Mahapatra, S. et al. Cav1.3 and BK Channels for Timing and Regulating Cell Firing. Mol Neurobiol 42, 185–198 (2010). https://doi.org/10.1007/s12035-010-8151-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-010-8151-3