
Abstract
We studied the effects of the cAMP-hydrolyzing enzyme phosphodiesterase type-4 (PDE4) on the L-type Ca2+ channels (LTCCs) and Ca2+-dependent secretion in mouse chromaffin cells (MCCs). The selective PDE4 inhibitor rolipram (3 μM) had a specific potentiating action on Ca2+ currents of MCCs (40% increase within 3 min). A similar effect was produced by the selective β1-AR agonist denopamine (1 μM) and by the unselective PDEs inhibitor IBMX (100 μM). Rolipram and denopamine actions were selective for LTCCs, and the Ca2+ current increase remained unchanged if the two compounds were applied simultaneously. This suggests that at rest, LTCCs in MCCs are down-regulated by the low levels of cAMP determined by PDE4 activity and that LTCCs can be up-regulated by either inhibiting PDE4 or activating β1-AR. No other PDEs are likely involved in this specific action. PDE4 inhibition had also a marked effect on the spontaneous firing of resting MCCs and catecholamine secretion. Rolipram up-regulated the LTCCs contributing to the “pace-maker” current underlying action potential (AP) discharges and accelerated the firing rate, with no significant effects on AP waveform. Acceleration of AP firing was also induced by the LTCC-agonist Bay K (1 μM), while nifedipine (3 μM) reduced the firing frequency, suggesting that LTCCs and intracellular cAMP play a key role in setting the pace-maker current regulating MCCs excitability. Rolipram increased also the size of the ready-releasable pool and the quantal content of secretory vesicles without affecting their probability of release. Thus, rolipram acts on MCCs by up-regulating both exocytosis and AP firings. These two processes are effectively down-regulated by PDE4 at rest and can dramatically increase the quantity of released catecholamines when PDE4 is inhibited and/or cAMP is raised.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
L-type Ca2+ channels (LTCCs) sustain catecholamine secretion [8, 16, 17] and contribute to shape the action potential firing [50], thus playing a major role in the control of chromaffin cell excitability. LTCC contribution is particularly effective in rodent and human chromaffin cells [5, 24, 27] and can be up- or down-modulated by a variety of locally released neurotransmitters. This occurs either through membrane-delimited pathways [7, 25] or through intracellular second messenger cascades, such as those mediated by cGMP/PKG [4] or cAMP/PKA [9]. The cAMP/PKA pathway is particularly effective in up-regulating LTCCs within minutes in rat chromaffin cells (RCCs) and to increase down-stream vesicle secretion during prolonged exposures with permeable cAMP or β1-AR agonists in bovine chromaffin cells (BCCs) [5]. Thus, cAMP/PKA acts on adrenal chromaffin cells by both potentiating LTCCs and up-regulating catecholamine release to a different degree.
In BCCs, intracellular cAMP and catecholamine secretion can be effectively raised by either applying neurotransmitters, such as nicotine [1], PACAP [51], histamine [37], and VIP [63] or by inhibiting cAMP degradation mediated by cyclic nucleotide phosphodiesterares (PDEs) [33, 36]. Inhibition of PDEs is very effective in elevating the basal levels of cAMP, suggesting high PDE enzymatic activity in chromaffin cells. This is interesting since there is clear evidence for the existence of intracellular compartmentalized pools of cAMP strictly controlled by PDEs and acting on multiple targets in a variety of cells [28]. Such a highly organized intracellular compartmentation provides a tool for regulating cAMP levels in regional areas of the cell and for driving cAMP-mediated effects to specific targets [57].
To date, little is known about the PDE isoforms effectively expressed in chromaffin cells, except that the PDE type-4 (PDE4) inhibitor rolipram raises cAMP levels [36, 38] and increases the net granule content of catecholamines [33]. Even more striking is the lack of information concerning the effects of PDEs on LTCCs and the parameters controlling secretion (ready-releasable pool, probability of vesicle release, membrane area of unitary vesicle, Ca2+ dependence). This is of great interest since LTCCs are the Ca2+ channels most sensitive to cAMP and PKA [3, 5, 9] and control a large fraction of secretion in chromaffin cells [16, 32, 41, 48], and preliminary findings [35] indicate them as possible candidates for pace-making chromaffin cell activity.
Here, we studied the effects of rolipram on Ca2+ channels in mouse chromaffin cells (MCCs) and found that the specific PDE4 inhibitor selectively up-regulates cAMP levels and the size of L-type currents. Elevation of cAMP and potentiation of LTCCs by rolipram were comparable in time and size to the effects of the selective β1-AR agonist denopamine [54], suggesting similar efficacy of cAMP molecules elevated by either β1-AR stimulation or PDE4 inhibition. Current-clamp and action potential-clamp measurements showed that inhibition of PDE4 by rolipram increased the size of inward L-type currents sustaining the interpulse pace-maker” potential and the frequency of spontaneous action potential (AP) firing, thus, furnishing a rationale to the hypothesized critical role of LTCC in controlling AP firing in MCCs. Rolipram had also a marked effect on catecholamine secretion, which was partially unrelated to the cAMP-dependent increase of LTCCs’ activity. The PDE4 inhibitor increased the size of the RRP and the unitary membrane area and quantal size of secretory vesicles without affecting their probability of release. Thus, by up-regulating the exocytosis and AP firing, rolipram produces a dual synergistic action on MCCs that can markedly increase the quantity of released catecholamines, highlighting the critical role that cAMP and PDE4 play in the control of L-type channels and secretion in MCCs.
Materials and methods
Isolation and culture of mouse adrenal medulla chromaffin cells
All experiments were performed in accordance with the guidelines established by the National Council on Animal Care and were approved by the local Animal Care Committee of Turin University. Chromaffin cells were obtained from young C57BL/6J mice, which were killed by cervical dislocation and cultured following the method of Sorensen et al. [56], with minimal modifications. After removal, the adrenal glands were placed in Ca2+- and Mg2+-free Locke’s buffer containing (in millimolar): 154 NaCl, 3.6 KCl, 5.6 NaHCO3, 5.6 glucose, and 10 HEPES, pH 7.2, at room temperature. The glands were decapsulated, and the medullas were precisely separated from the cortical tissue. Medulla digestion was achieved for 60 min at 37°C in the Locke’s buffer mentioned before containing 20 U/ml of papain (Worthington Biochemical, Lakewood, NJ, USA). The cell suspension was then centrifuged for 5 min at 900 rpm, washed two times, and resuspended in 2 ml DMEM supplemented with 15% fetal calf serum (FCS). Cells were plated in four-well plastic dishes treated with poly-L-ornithine (0.5 mg/ml) and laminin (10 μg/ml in L-15 carbonate) by placing a drop of concentrated cell suspension in the center of each well. After 1 h, 1.8 ml of DMEM supplemented with 15% FCS (Invitrogen, Grand Island, NY, USA), 50 IU/ml penicillin, and 50 μg/ml streptomycin (Invitrogen) was added to the wells. Cells were then incubated at 37°C in a water-saturated atmosphere with 5% CO2 and used within 2–6 days after plating.
Voltage-clamp and current-clamp recordings
Ca2+ currents were recorded in perforated-patch recording conditions using an Axopatch 200-A amplifier and pClamp 9.0 software programs (Axon Instruments Inc., USA) [6]. Patch pipettes were made of thin borosilicate glass (Kimax 51; Witz Scientific, Holland, OH, USA) and filled with an internal solution containing (in millimolar): 135 CsMeSO3, 8 NaCl, 2 MgCl2, 20 HEPES, pH 7.3, with CsOH plus amphotericin B (Sigma). The external bath contained (in millimolar): 145 TEACl, 5 CaCl2, 2 MgCl2, 10 glucose, 10 HEPES, pH 7.4, with CsOH. Amphotericin B was dissolved in dimethyl sulfoxide stored at −20°C in stock aliquots of 50 mg/ml and used at a final concentration of 500 μg/ml. To facilitate the sealing, the pipette was first dipped in a beaker containing the internal solution and then back-filled with the same solution containing amphotericin B. Pipettes with series resistance of 1–2 MΩ were used to form giga-seals. Recording of Ca2+ currents started when the access resistance decreased below 15 MΩ, which usually happened within 10 min after sealing [9]. Series resistance was compensated by 80% and monitored throughout the experiment.
Ca2+ currents were evoked by step depolarization of 10–100 ms to a fixed potential (+10 mV), ramp commands (1 V/s), or action potential trains (“action potential-clamp”). In the latter case, the cell was voltage-clamped using a pattern of three action potentials previously recorded in current-clamp conditions. This allowed studying the time course of Ca2+ currents underlying an action potential train [58]. Extra- and intracellular solutions in this case were the same as those used for current-clamp measurements. The holding potential was −70 mV throughout the experiments. Fast capacitative transients during step depolarization were minimized online by the patch-clamp analog compensation. Uncompensated capacitative currents were further reduced by subtracting the averaged currents in response to P/4 hyperpolarizing pulses. All of the experiments were performed at room temperature (22–24°C).
Action potentials were recorded in perforated-patch-clamp conditions using an intracellular solution containing (in millimolar): 135 KAsp, 8 NaCl, 20 HEPES, 2 MgCl2, and 5 EGTA. The external bath contained (in millimolar): 137 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4, with NaOH. Spontaneous action potentials could be stably recorded in most chromaffin cells at resting conditions without passing any current.
Membrane capacitance changes
Ca2+ currents and the corresponding depolarization-evoked capacitance changes were measured in the perforated-patch configuration using an EPC-9 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany) and an extracellular solution containing (millimolar): 135 NaCl, 2 MgCl2, 10 HEPES, 10 glucose, and 5 CaCl2 (pH 7.3 with NaOH), as previously described [5, 18]. Ca2+ currents were sampled at 10 kHz and filtered at 2 kHz. Depolarization-evoked exocytosis was measured as membrane capacitance increases by applying a sinusoidal wave function on the holding potential (±25 mV amplitude, 1 kHz). Fast capacitive transients due to depolarizing pulses were minimized online by the patch-clamp analog compensation. Series resistance was compensated by 80% and monitored during the experiment.
Amperometric detection of exocytosis
Amperometric recordings were performed using a HEKA EPC-10 amplifier. Carbon fibers were purchased from ALA Scientific Instruments Inc. (Westbury, NY, USA). The carbon fiber microelectrode (5-μm diameter) was polarized at +800 mV and then gently positioned adjacent to the cell membrane (see [6]). Detection of amperometric spikes was performed by initially keeping the cells in a KCl-free solution containing (millimolar): 130 NaCl, 2 MgCl2, 10 HEPES, 10 glucose, and 10 CaCl2. The solution was then switched to one containing 30 mM KCl and proportionally less NaCl. Amperometric currents were sampled at 4 kHz, low-pass-filtered at 1 kHz, and monitored during 2-min recordings. Data were analyzed by IGOR macros (WaveMetrics, Lake Oswego, OR, USA) as described elsewhere [55].
Solutions, cAMP immunoassay, and data analysis
External solutions were exchanged by gravity using a multi-barreled pipette with a single outlet and five inlets. Nifedipine, Bay K 8644, denopamine, and rolipram were purchased from Sigma and dissolved daily in the extracellular solution. Tetrodotoxin citrate (TTX) was purchased from Tocris (Northpoint, Fourth Way Avonmouth, UK) and prepared to the final concentration.
The levels of cAMP in isolated chromaffin cells were measured using an enzymatic immunoassay (EIA) according to the manufacturer’s instructions (Biotrak, Amersham-Pharmacia Biotech, Amersham, UK). Briefly, chromaffin cells were plated in a 96-well microplate at a density of 60,000 cells/well and challenged for 10 min at 37°C with denopamine and rolipram. Cells were then lysed with the supplied buffer, and the supernatant was transferred to the EIA microplate. After processing, absorbance was read with a 450-nm filter microplate reader (BioRad, Hercules, CA, USA).
Data are given as mean ± SEM for n number of cell. Statistical significance was calculated by either using Student’s paired t test or one-way analysis of variance (ANOVA) followed by a Bonferroni post hoc test when multiple data comparisons were required (Figs. 2d, 3d, and 8). Values of p ≤ 0.05 were considered significant.
Results
The percentage of LTCCs expressed in mouse chromaffin cells was evaluated in a preliminary series of experiments. MCCs had mean cell capacitance of 9.3 ± 0.3 pF (n = 110) and Ca2+ currents, determined by ramp commands, had mean peak currents of 21.0 ± 2.9 pA/pF (n = 36; Fig. 1a) at around +10 mV in 5 mM external Ca2+. Nifedipine (3 μM) blocked 45.8 ± 4.6% of the total Ca2+ current (n = 12 cells), causing a slight shift of the I/V curve toward more positive potentials (Fig. 1a), in good agreement with what Hernandez-Guijo et al. [24] reported on the same cell preparation. L-type channels were expressed in all the MCCs tested.

Potentiating effects of rolipram on L-type currents. a I/V relationships recorded from a voltage-clamped MCC using a ramp command in control condition and in the presence of 3 μM nifedipine. In the inset are reported the percentage of L- and non-L-type current density normalized to the total Ca2+ current. The non-L-type component was determined by applying 3 μM nifedipine (n = 12), and the L-type component was calculated as the control minus the nifedipine-resistant fraction (**p < 0.01 vs. control using Student’s paired t test). b Time course of peak Ca2+ current at +10 mV before, during and after addition of 3 μM rolipram. In the inset are shown the current traces recorded at the time indicated by the numbers (1–3). Onset and offset of rolipram action were best fitted with single exponential functions with time constants: τ on = 2.8 min and τ off 1.6 min. c I/V relationships of total Ca2+ currents before and during rolipram application obtained by 10-ms step depolarizations to the indicated voltages for n = 10 MCCs (*p < 0.05 vs. control using Student’s paired t test). d Time course of peak Ca2+ currents before, during, and after nifedipine (3 μM) application. Addition of rolipram in the presence of nifedipine does not produce any current increase
Selective L-type Ca2+ current potentiation by rolipram
We then studied the acute effects of rolipram (3 μM) on Ca2+ currents (Fig. 1b). Cells were depolarized every 5 s at +10 mV, with pulses lasting 10 ms. We found that in a subpopulation of cells (60%, n = 17), the PDE4-inhibitor rolipram enhanced the currents up to 40.1 ± 6.1% over basal values (Fig. 1b), while in the remaining cells, there was no significant effect. We considered significant only those MCCs which responded to rolipram or to any other compound with a Ca2+ current increase >10% over control values. Maximal potentiation was observed after about 3 min of acute application, and return to control conditions occurred slightly faster. The onset of potentiation had mean time constant of 3.4 ± 1.0 min (τ on) while washout (offset) had mean τ off of 2.1 ± 0.9 min (white boxes in Fig. 2d). Time constants were evaluated by exponential fits to the data points of current amplitudes vs. time, as shown in Fig. 1b. Ca2+ current increase occurred in the whole range of tested potentials (from −50 to +30 mV; Fig. 1c) and was maximal at −30 mV (2.4-fold increment over control). No significant changes to the activation–inactivation kinetics could be detected (inset in Fig. 1b), suggesting that inhibition of PDE4 mainly causes an increased probability of channel opening with little or no changes to channel gating [3, 7].

Potentiating effects of denopamine on Ca2+ currents. a I/V relationships from a MCC in control conditions and in the presence of 1 μM denopamine. b Time course of peak Ca2+ current at +10 mV before, during, and after addition of 1 μM denopamine. Onset and offset of denopamine action was best fitted with single exponential functions with time constants: τ on = 2.5 min and τ off = 2.2 min. c Time course of peak Ca2+ currents before, during, and after nifedipine (3 μM) application. Addition of denopamine (1 μM) in the presence of nifedipine does not produce any Ca2+ current increase. d Mean values of τ on and τ off, determined for the potentiating action of rolipram, denopamine, rolipram + denopamine, and IBMX for the number of cells indicated inside the bars. The data sets were compared using one-way ANOVA followed by a Bonferroni post hoc test and were found not to be statistically different
A preferential potentiating effect of rolipram at very negative potentials (−30 mV) is indicative of a specific action on LTCCs, which activate at relatively low voltages. For this reason, we tested whether LTCCs were the specific target of rolipram and repeated the experiments in the presence of nifedipine (3 μM; Fig. 1d). In all the cells tested (n = 10), the DHP antagonist fully prevented the action of rolipram, confirming specific effects of PDE4 on L-type currents.
Denopamine selectively potentiates L-type currents
RCCs express sufficient densities of β1-ARs and β2-ARs. The former are remotely coupled to LTCCs via a potentiating cAMP/PKA-mediated pathway, while the latter are negatively coupled to LTCCs via PTX-sensitive Gi proteins [9]. We tested therefore if β1-ARs were also expressed in MCCs and how their action is coupled to the potentiating effects of rolipram. For this, we used the selective β1-AR agonist denopamine and found that in a large fraction of cells (58%, n = 12), addition of the agonist significantly enhanced the Ca2+ currents up to 41.0 ± 8.0% over basal values (Fig. 2a,b), whereas in the remaining cells, there was no action. As for the effects of isoprenaline on RCCs [9], the onset and offset of β1-AR stimulation was slow and comparable to that of rolipram (τ on 2.9 ± 1.8 min; τ off 2.5 ± 1.5 min n = 5; gray boxes in Fig. 2d). As for rolipram, the action of denopamine was fully prevented by nifedipine (Fig. 2c), suggesting a selective remote action of β1-AR on LTCCs. When denopamine and rolipram were applied together, Ca2+ currents increased to 39.0 ± 9.0% over basal values in 50% of the cells (n = 14; Fig. 3a), and this was not significantly different from what was observed after administration of denopamine or rolipram alone (Fig. 3d). The remaining cells did not respond, and sequential application of the two compounds could not be accurately tested since MCCs did not last for prolonged perfusion periods. We were, thus, unable to establish whether the subpopulation of cells not responding to rolipram were sensitive or not to denopamine and vice versa. We could nevertheless establish that the onset and the offset of the action caused by both compounds applied together (rolipram plus denopamine; black boxes in Fig. 2d) were not significantly different from those obtained by applying one compound at the time. Taken together, these data suggest that (1) β1-AR is expressed and functionally active in MCCs and their stimulation up-regulates LTCCs activity; (2) the cAMP-dependent transduction pathway controlled by PDE4 is equally effective to that activated by β1-AR, suggesting close coupling of the two pathways to membrane compartments where LTCCs are located; (3) both systems produce saturating effects on L-type currents; and (4) the lack of effects of denopamine and/or rolipram may be derived from the heterogeneous distribution of the two LTCCs expressed in MCCs (Cav1.2 and Cav1.3; Marcantoni, Carbone, Striessnig, unpublished observation), which may be differently sensitive to cAMP/PKA (see “Discussion”).

Potentiating effects of denopamine plus rolipram and IBMX on Ca2+ currents. a Time course of peak Ca2+ current at +10 mV before, during, and after addition of 1 μM denopamine plus 3 μM rolipram. Onset and offset of denopamine + rolipram action was best fitted with single exponential functions with time constants: τ on = 2.5 min and τ off = 2 min. b Time course of peak Ca2+ current at +10 mV before, during, and after addition of 100 μM IBMX. Onset and offset of IBMX action was best fitted with single exponential functions with time constants: τ on = 2.8 min and τ off = 2.7 min. c Mean I–V curves obtained from MCCs in control conditions (n = 15) or incubated for 20 min with 500 μM IBMX (n = 5; *p < 0.05; **p < 0.01 vs. control using Student’s paired t test). The dashed curve of rolipram effects was taken from Fig. 1c. d Percentage of Ca2+ current increase induced by rolipram (3 μM), denopamine (1 μM), a mixture of the two compounds, and IBMX (100 μM) for the number of cells indicated. The data sets were compared using one-way ANOVA followed by a Bonferroni post hoc test and were found not to be statistically different
The above data, however, do not exclude the possibility that other PDEs could be involved in the cAMP-dependent LTCCs potentiation. In mammals, of the 11 types of PDEs currently described [11], only three are cAMP specific (PDE4, PDE7, PDE8). All the remaining are either specific for cGMP (PDE5, PDE6, PD9) or partially sensitive to both cGMP and cAMP (PDE1, PDE2, PDE3, PDE10, PDE11). Since cGMP down-modulates the activity of single LTCCs in RCCs [4] and there are no available specific inhibitors for PDE7 and PDE8, we found it unreasonable to undertake a detailed investigation of the role of other PDEs on LTCCs. Just as a general test, we studied the effects of the unspecific PDE inhibitor, IBMX, on LTCCs. We found that in 40% of the cells (n = 16), acute application of 100 μM IBMX caused on average a 38.3 ± 6.0% increase of peak L-type current (Fig. 3b,d), with an “on” and “off” potentiation kinetics comparable to that of rolipram and denopamine (dashed boxes in Fig. 2d). Higher doses (500 μM IBMX) and prolonged incubation (20 min) of IBMX gave no better results when compared to rolipram (see I–V curves in Fig. 3c).
Effect of rolipram on action potential firing frequency
The effects of rolipram on spontaneous APs were studied in current-clamp mode using bath and intracellular solutions close to physiological conditions (see “Materials and methods”). The cells had mean resting potentials (V r) of −49.0 ± 1.5 mV (n = 24) that, after correction for the liquid junction potential (−15.8 mV), resulted to be more negative and in good agreement (after appropriate compensation) with other V r estimated using patch pipettes (from −50 to −70 mV [13, 27, 61]). The average input resistance measured using small hyperpolarizing current pulses (5–10 pA) was very high (1.2 ± 0.2 GΩ; n = 7) and in good agreement with previously reported values in other chromaffin cell types (0.6 to 5 GΩ) [13, 27, 35].
Cultured MCCs normally exhibited spontaneous AP firings (85% of the cells), which lasted for several minutes (Fig. 4a) and had mean firing frequency of 0.7 ± 0.1 Hz. The frequency of the spontaneous APs, however, was not constant over time for any given cell and varied between cells. AP waveforms were analyzed by measuring several parameters both in control conditions and after rolipram administration. To characterize the AP firings, we measured the frequency, the potential at half the interpulse duration, the maximum rate of raise, the width at 0 mV, the overshoot, and undershoot. We observed that all these parameters remained unchanged after rolipram administration (Table 1), except for the frequency that increased by about a factor 2 (1.5 ± 0.2 Hz, n = 14; *p < 0.05; Figs. 4b and 5b and d). Thus, tonic activation of PDE4 mainly contributes to maintain a low frequency firing at rest, while the block of the enzyme increases the firing rate. This also suggests that LTCCs control the duration of interpulse intervals in MCCs and may effectively contribute to the pace-maker current sustaining AP spontaneous activity.

The accelerating effects of rolipram on action potentials firings in MCCs. a Spontaneous AP recordings in current-clamp mode from a MCC before, during, and after exposure to rolipram (3 μM). The thick horizontal bar indicates the interval of rolipram application. On top, 18-s intervals of recordings are shown at a more expanded time scale. Notice the mean increased frequency of firing when rolipram is added to the bath. b Overlapping of two consecutive APs in control condition (dark trace) and with rolipram (gray trace) to highlight the change in the slope of the pace-maker potential during the interpulse interval. c Two overlapped APs in control conditions and with rolipram at a more expanded time scale. The PDE4 inhibitor causes only a slight decrease of the AP amplitude with no changes to the time course. The overshoot depression is not significative on average (see Table 1)

Opposing effects of nifedipine and Bay K 8644 on action potentials firing in MCCs. a 1 Spontaneous AP recordings from a MCC before, during, and after exposure to nifedipine (3 μM). a 2 , a 3 Overlapped APs at two expanded time scales to highlight the prolonged pace-maker potential (a 2 ) and the changes of AP waveform (a 3 ) in the presence of nifedipine. b Spontaneous AP recordings in control conditions, with rolipram alone and with rolipram plus nifedipine. Notice the increased AP discharges with rolipram alone and their full block after addition of the DHP antagonist. c Spontaneous AP recordings under basal conditions and sequential exposure to Bay K 8644 (1 μM) and nifedipine (3 μM). On the top are illustrated 4-s intervals of recordings at a more expanded time scale. The thick horizontal bars indicate the interval of drug application. d Percentage of frequency increase induced by rolipram (n = 9) and Bay K 8644 (n = 6) and percentage of frequency decrease induced by nifedipine (n = 14). Notice the potent action of Bay K 8644 on AP frequency, which increases about four-fold (*p < 0.05 vs. control using Student’s paired t test)
We tested this hypothesis by checking whether blocking LTCCs by nifedipine could have the opposite effects of rolipram. In 14 MCCs, we found that 3 μM nifedipine could either decrease the rate of firing (Fig. 5a1, n = 5) or fully block the spikes’ activity (Fig. 5b, n = 9). This gives a mean frequency of about 1/10 of the control value (91% decrease; Fig. 5d). Figure 5b shows an example of how nifedipine can prevent the increased firings induced by rolipram (n = 3) by fully blocking AP generation. In either case, the washout of nifedipine required some time before restoring the normal firing (20–40 s). The partial or full-blocking action of nifedipine could depend on the density and isoform of LTTCs expressed in MCCs (see “Discussion”). Most interesting, when nifedipine had a mild action, the AP amplitude and duration increased (Fig. 5a3) as expected from the block of BK channels, which are shown to be strongly coupled to LTCCs in RCCs [50] and MCCs (Marcantoni et al., unpublished observations). The less pronounced hyperpolarization due to the LTCCs block induces a slower depolarization, which prolongs the interpulse duration (Fig. 5a2). Further support for a role of LTCCs in the control of firing frequency comes from the observation that Bay K 8644 (1 μM) increases AP discharges in MCCs by about a factor of 4 over basal value (392% increase; p < 0.05; Fig. 5c,d).
L-type channels contribute to the pace-maker current generating spontaneous APs
As shown in Fig. 1, LTCCs in MCCs start activating at relatively low voltages (around −50 mV in 5 mM Ca2+) and are good candidates for carrying the inward current that generates the pace-maker potential of spontaneous firings. In order to test this hypothesis, we recorded MCCs Ca2+ currents in voltage-clamp conditions using either single or trains of APs as voltage command (action potential-clamp) [58]. Ca2+ currents were isolated by adding TEA (20 mM) and TTX (300 nM) to the extracellular solution in order to block K+ and Na+ channels.
Figure 6a shows the shape of the single AP used as voltage command at two different time scales recorded during spontaneous activity. The cell was held at −70 mV before and after the AP clamp. Under these conditions, Ca2+ currents activated soon after the beginning of the AP stimulus, had slow rise during the early depolarizing phase (−50 to −40 mV), and showed no sign of inactivation (slow phase). The current accelerated to reach a first negative peak at the beginning of the AP upstroke and then decreased during the rapid rise of the AP (due to the lowered driving force for Ca2+) to become slightly outward at the peak of the AP. The inward Ca2+ current increased again during the repolarizing phase and reached a second negative peak, which results from the increased driving force and the large Ca2+ channel conductance at these potentials (fast phase). Addition of 200 μM Cd2+ (n = 6) blocked both the early and late phase of the currents, leaving a residual transient component associated with a fraction of uncompensated leakage current (Fig. 6b).

Ca2+ currents elicited by an AP waveform. a Two different views of the AP waveform used to voltage-clamp MCCs. The arrow indicates the time when the membrane potential reaches −45 mV, corresponding to the time at which the amplitude of the slow Ca2+ current component was estimated. b Ca2+ currents recorded in control conditions and during exposure to 200 μM Cd2+. c Nifedipine blocks most potently the slow Ca2+ current component preceding the AP (pace-maker current) and to a minor extent, the second current component during the spike. d, e Effects of rolipram (3 μM) on the inward Ca2+ current sustaining the AP from two MCCs displaying different Ca2+ current components during the slow depolarizing phase in control conditions and responding differently to rolipram (see text). In both cases, rolipram potentiates the slow early phase of the Ca2+ current but causes a partial decrease of the second fast peak current (see text). Inset Percentage variation of Ca2+ currents induced by rolipram (positive changes) and nifedipine (negative changes) at the time indicated by the arrows (see panels above; *p < 0.05 vs. control using Student’s paired t test)
The slow early phase of the action potential was characterized by an inward Ca2+ current dominated by LTCCs (Fig. 6c). Nifedipine reduced by 75.6 ± 6.6% this slow phase at the time indicated by the arrow when the potential is −45 mV (from −54.7 ± 16.5 to −14.6 ± 6.3 pA; n = 8; inset in Fig. 6e) and had minor effects on the second negative peak (Fig. 6c—right). On average, nifedipine blocked by 31.1 ± 4.3% the second negative peak. Rolipram had also a marked action on the inward Ca2+ current, although opposite to nifedipine. It increased 3.8-fold the slow early phase of the current (from −11.8 ± 5.4 to −38.7 ± 7.9 pA at −45 mV; n = 5; inset in Fig. 6e) and partially depressed the second negative peak (average 22%; Fig. 6d). The recordings in Fig. 6d and e were obtained from two MCCs with different control Ca2+ currents. In Fig. 6d, the slow early phase was 29.6 pA at −45 mV and increased to 53.3 pA (80% increase over control value) with rolipram. In Fig. 6e, the control current was smaller (13.6 pA at −45 mV) and increased to 68.5 pA (403% increase) with rolipram. The unexpected decrease of the second peak with rolipram occurred most likely for three reasons: (1) the little or no sensitivity of rolipram for the non-LTCCs that dominate this component, (2) a decreased driving force for Ca2+ due to a lowered reversal potential for Ca2+ (E Ca) caused by the increased Ca2+ entry and accumulation induced by the PDE4 inhibitor. The lower E Ca is suggested by the increased outward current with rolipram at the peak of AP (Fig. 6d) and (3) a partial rundown of Ca2+ channels with time (10–15%).
The up-regulating action of rolipram was fully prevented by nifedipine (3 μM). We never observed a rolipram-induced Ca2+ current increase in the presence of nifedipine (n = 16). This suggests that the slow early phase of inward Ca2+ currents is mostly carried by LTCCs while the late fast phase by non-LTCCs. The L-type component is thus relevant for the generation of spontaneous APs, whereas the nifedipine-resistant Ca2+ currents contribute mainly to the fast rise and duration of the AP.
We also tested whether the potentiation of the pace-maker current by rolipram was preserved during a train of three APs and not limited to a single AP generated after holding the cell to −70 mV (Fig. 7a). In this case, the percentage of Ca2+ channels available for opening is higher than after holding the cell at resting potential. As shown in Fig. 7b, the size of the rolipram-potentiated current remained high and persistent also before the second and third AP generated after the long depolarization preceding the two pulses. Nifedipine blocked the potentiating action of rolipram also when using the AP train command (Fig. 7b), confirming that LTCCs are the main targets of rolipram effects.

Ca2+ currents elicited by a train of three APs. a The AP train waveform used to measure Ca2+ currents during the interspike intervals and responsible for the pace-maker potential, as indicated. b Ca2+ currents recorded in control conditions and during exposure to rolipram (3 μM) and nifedipine (3 μM). Notice how the potentiating effects of rolipram are preserved after the first and second AP
Rolipram, denopamine, and IBMX increase the basal levels of cAMP in MCCs
We also evaluated the effects of rolipram, denopamine, and IBMX on basal intracellular cAMP in cultured MCCs, using an EIA system on microtiter plates (96 wells). The amount of cAMP under basal conditions was very low (about 33.6 ± 5.3 fmol/well, n = 8, Fig. 8). Considering that each well contained on average 60,000 cells, a single MCC had a mean cAMP quantity of ∼5.6 × 10−4 fmol, which corresponds to a mean [cAMP] of 2.2 mM, assuming a mean cell diameter of 17 μm that gives a mean cell capacitance of 9.3 pF. In the presence of denopamine, the cAMP level nearly doubled (62.7 ± 6.7 fmol/well, n = 7), but with rolipram and rolipram + denopamine, cAMP increased nearly three-fold (91.2 ± 4.3 and 83.9 ± 6.8 fmol/well, respectively; n = 6). IBMX increased even further the resting cAMP levels (121.2 ± 6.8 fmol/well; n = 4), suggesting that at variance with the cAMP-dependent LTCC potentiation (mainly regulated by PDE4), the overall cytosolic cAMP is only partially affected by PDE4 and that other PDEs down-regulate cAMP levels at rest.

Rolipram, denopamine, and IBMX up-regulate the intracellular cAMP concentration. Mean intracellular cAMP concentration measured in populations of cultured MCCs in the presence of denopamine (1 μM), rolipram (3 μM), denopamine + rolipram, and IBMX (100 μM) using an enzyme immunoassay. Values are expressed as fentomoles per well, and the cells were plated at concentration of 60,000 cells/well. The data sets were compared using one-way ANOVA followed by a Bonferroni post hoc test (*p < 0.05, ***p < 0.001 vs. control). Moreover, the intracellular cAMP increase by rolipram was higher than that induced by denopamine (#p < 0.05) and the effect by IBMX was higher than that induced by rolipram, denopamine, and rolipram + denopamine (#p < 0.05; ##p < 0.01; ###p < 0.001; indicated by horizontal bars)
Rolipram increases the size of the RRP and the magnitude of single secretory events without altering the probability of vesicle release
To complete our analysis on the effects of rolipram on MCCs activity, we studied how the PDE-4 inhibitor affects the secretion by measuring the depolarization-induced capacitance changes [5, 18]. Figure 9a shows fast capacitance changes (ΔC) recorded following pulses of 100 ms to +10 mV from different MCCs maintained in control solution (n = 52) and after incubation for 10 min with rolipram (3 μM). The voltage protocol was designed to attain maximal Ca2+ current amplitudes and robust secretion during single pulses. In 48 MCCs tested, rolipram caused well-resolved fast capacitance changes that increased on average by 25% with respect to control (from 32.5 ± 2.0 to 40.6 ± 3.3 fF; p < 0.05; Fig. 9b), while the total Ca2+ charge density entering during the pulse increased by only 16% (from 3.6 ± 0.3 to 4.2 ± 0.3 pC/pF; p < 0.05). The small increase of Ca2+ entry was mainly due to the fact that (1) rolipram effects were tested on a set of cells different from control and thus comprehensive of those cells responding and not responding to rolipram; (2) the longer pulses used in these experiments (100 ms) attenuated significantly the large current increases induced by rolipram on pulses of 10 ms. The Ca2+ charge increase is calculated over the total Ca2+ current area and not up the current peak; (3) in the experiments of Fig. 9, rolipram was incubated for 10 min, while in those of Figs. 1, 2, 3, and 4, rolipram was applied acutely. Apart from this, the results of Fig. 9 suggest that rolipram produces a percentage increase of exocytosis, which is higher than that expected from the increased amount of Ca2+ entry during voltage-clamp pulses.

Rolipram potentiates the quantity of Ca2+ charge and corresponding secretion by increasing the RRP without affecting the probability of release (p r). a Capacitance changes (top) and Ca2+ current recordings (bottom) during step depolarizations to +10 mV in two representative MCCs: one in control condition and the other in the presence of rolipram (3 μM; 10 min incubation). b The mean quantity of charge normalized to cell capacitance was 3.6 ± 0.3 pC/pF (n = 52) in control MCCs and 4.2 ± 0.2 pC/pF (n = 48) with rolipram. Mean capacitance changes (ΔC) were 32.5 ± 2.0 fF in control MCCs and 40.6 ± 3.3 with rolipram. Both sets of data were significantly different (*p < 0.05 vs. control using Student’s paired t test). c Double pulse protocol used to estimate the size of the RRP and p r. The two consecutive pulses were applied at 0 and +10 mV in order to have the same amount of Ca2+ ions entering the cell during the pulse (see [5]). ΔC 1 and ΔC 2 indicate the capacitance changes measured during the first and second pulse, respectively. d Left Mean values of B max calculated from (ΔC 1 + ΔC 2)/(1 − (ΔC 2/ΔC 1)2) (corresponding to the upper limit of the RRP). They were 43.6 ± 3.7 for control MCCs (n = 18) and 64.2 ± 13.5 with rolipram (n = 6; *p < 0.05 vs. control using Student’s paired t test). Middle Mean values of ΔC 1 (corresponding to the lower limit of the RRP): 28.0 ± 2.8% for control MCCs and 36.1 ± 4.6% with rolipram. Right Mean values of p r calculated from 1 − ΔC 2/ΔC 1. Mean values were 65 ± 5% for control MMCs and 57 ± 6% with rolipram. These values were not statistically different
To obtain more detailed information on the molecular parameters controlling secretion, we ran a series of experiments to estimate the maximal size of the pool of readily releasable vesicles (RRP) and probability of release (p r). Using the double-pulse protocol illustrated in Fig. 9c (see figure legend), we found that rolipram caused a 47% increase of B max (from 43.6 ± 3.7 to 64.2 ± 13.5 fF; p < 0.05; Fig. 9d), while the capacitance change induced by the first pulse (ΔC 1) increased by 25%, in good agreement with the result of Fig. 9b. It is interesting to notice that B max and ΔC 1 set the upper and lower limits for the true value of the RRP (see [19]). In addition, we found that p r was not affected by rolipram (0.65 ± 0.05 vs. 0.57 ± 0.06; p > 0.05; Fig. 9d), suggesting that as for the effects of cAMP on RCCs [5], rolipram increases the depolarization-evoked exocytosis by mainly affecting the size of the RRP but not p r.
To complete this analysis, we also studied the action of rolipram on the Ca2+ dependence of secretion and on the size of unitary secretory events (Δc). In the first case, we measured the amplitude of fast capacitance changes following voltage pulses of fixed amplitude (+10 mV) and increasing length (10 to 200 ms) and plot the values of ΔC vs. the density of Ca2+ charges entering the cell. The results are shown in Fig. 10a, in which it is evident that rolipram (filled squares) increased by nearly 50% the slope of the linear Ca2+ dependence of secretion of control MCCs (empty squares). This implies that for the same quantity of Ca2+ entering the cell, pretreatment with rolipram increases vesicle secretion by about 50%, in good agreement with the increased estimate of B max in the presence of rolipram (Fig. 9d).

Rolipram enhances the Ca2+ dependence of exocytosis and the quantal size of elementary exocytic events. a Ca2+ dependence of exocytosis determined by plotting ΔC vs. the corresponding Ca2+ charge density. Pulses of variable length (10, 20, 50, 100, 150, 200 ms) were applied at +10 mV either in control or in the presence of 3 μM rolipram, incubated for 10 min (n = 22 cells in both cases). The slope of the linear regression line is 8.4 ± 0.6 fF/(pC/pF) for control MCCs (empty squares) and 12.9 ± 1.3 fF/(pC/pF) with rolipram (filled squares). The two slopes were statistically different (*p < 0.05 vs. control using Student’s paired t test). b Estimate of the quantal capacitance change (Δc) obtained by plotting sample variances (σ 2) vs. averaged sample means (<ΔC>; see “Results” for details). The secretory responses were triggered by brief depolarization pulses of 20 ms to +10 mV and repeated at a frequency of 0.3 Hz. The linear fit gave 1.6 ± 0.1 fF for control MCCs and 2.2 ± 0.2 with rolipram. Inset Mean values of Δc were 1.6 ± 0.1 and 2.3 ± 0.2 as indicated (*p < 0.05 vs. control using Student’s paired t test)
In the second case, we estimated the capacitance change associated to individual exocytotic events (membrane area of a single vesicle) by using a statistical analysis of trial-to-trial variations during short depolarization-induced capacitance increases repeated quickly [40]. Using this approach, we measured the ΔC increases after repetitive depolarizations of 20 ms to +10 mV, applied at 0.3 Hz. We collected 150 to 180 depolarizations over a period of 9–10 min and measured the corresponding ΔC values plotted vs. time (see [5] for further details). After having calculated the means and variances using a four-bin analysis moving forward point-to-point, the sample variances (σ 2) were plotted vs. the averaged sample means (<ΔC>) and fitted by a regression line. The slope of the linear regression gave the mean exocytic size (Δc) according to the equation: σ 2 = Δc <ΔC> [40]. As shown in Fig. 10b, rolipram significantly increased the size of Δc (from 1.56 ± 0.11 to 2.28 ± 0.23 fF; p < 0.05; inset), suggesting that the PDE-4 inhibitor increases the size of elementary exocytic events by 46%.
Rolipram increases the quantal content of secretory granules without affecting the frequency of amperometric spikes
The results of Figs. 9 and 10 lead to two main conclusions: (1) rolipram acts on the secretory apparatus of MCCs by increasing the size of the RRP without affecting the probability of vesicle release, and (2) most of the RRP increase (47%) is likely associated to an augmented size of single exocytic events (46%). To confirm this possibility, we studied the exocytosis of single chromaffin granules using the carbon fiber detection method, which allows measuring of the quantal release of catecholamine as fast amperometric spikes [10, 62]. Catecholamine release was evoked by cell depolarizations using solutions containing 30 mM KCl and 10 mM CaCl2. As shown in Fig. 11a, control MCCs responded on average with a sequence of exocytotic bursts, with mean frequency of 34 spikes/min, which lasted for the entire duration of the KCl-induced depolarization (2 min). Histograms of the cubic root of the charge associated to each spike (Q 1/3) was best fit with a double Gaussian function, suggesting that the MCCs used in these experiments possess two populations of vesicles, one with small and one with large diameter, which is in good agreement with the findings of Grabner et al. [21] (Fig. 11a, top-middle). Notice that due to the spherical shape of chromaffin granules, Q 1/3 is commonly associated to their diameter (Q 1/3 ∝ d). The mean Q 1/3 was 0.67 pC1/3, which increased to 0.76 pC1/3 with rolipram. The corresponding mean quantal charge (Q) was 0.47 ± 0.04 pC (Fig. 11a, top right), which markedly increased to 0.67 ± 0.09; p < 0.05) without affecting the burst frequency (32 ± 3 spikes/min) and the proportion of the two Gaussian distribution fitting the Q 1/3 histogram (see legend of Fig. 11b). The estimated value of Q at control is in-line with those reported by other groups in MCCs: 0.67 pC [39], 0.45 pC [14], and 0.3 pC [20].

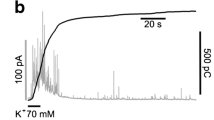
Rolipram enhances the charge content of individual secreting granules without affecting their frequency of occurrence in amperometric recordings. Amperometric spikes detection of catecholamine release in a representative control (a) and a rolipram-treated MCC (b). The spikes were elicited by applying an external solution containing 30 mM KCl and 10 mM CaCl2 (see “Materials and methods”). Data were acquired for 2 min. Top panels Enlarged view of a representative spike and histogram of Q 1/3 values. Data are fitted by the sum of two Gaussian distributions (two white traces), peaking at 0.55 (55%) and 0.77 (45%) pC1/3 for control MCCs (n = 59) and at 0.57 (54%) and 0.85 (46%) pC1/3 with rolipram (n = 24). The sum of the two Gaussians is indicated by dark traces. The spike frequency was not statically different: 33 ± 3 spike/min (control) and 32 ± 3 spike/min (rolipram). To the top right of a are reported the mean values of Q: 0.47 ± 0.04 pC (control) and 0.67 ± 0.09 pC (rolipram; *p < 0.05). To the top right of b are illustrated the time course of the mean cumulative secretion in control and with rolipram, obtained by averaging over 2 min the cumulative secretion of each amperometric recordings. Notice the marked increased final amplitude in the presence of rolipram (42.4 vs. 27.1 pC)
The nearly 43% increase of Q with rolipram was confirmed by calculating the cumulative secretion curve, which represents the sum of the quantity of charge of all the spikes recorded during a 2-min depolarization. As shown in the top-right panel of Fig. 11b, rolipram increased the cumulative secretion by 56% (from 27.1 to 42.4 pC). Since the frequency of spikes was nearly constant, this suggests that the enhanced catecholamine secretion is mainly due to the increased quantal size of secretory granules.
Discussion
We provided evidence that PDE4 inhibition exerts a dual action on MCCs functioning: It up-regulates the activity of LTCCs and increases the amount of released catecholamines. A second important finding is that LTCCs act as pace-maker channels and control AP generation in resting MCCs. This has a direct consequence on the action of PDE4 or of any other compound controlling intracellular cAMP, such as nicotine [1], histamine [37], PACAP [51], VIP [63], and β1-AR agonists (Fig. 2). The increased cAMP markedly increases the quantity of secreted adrenaline and noradrenaline by enhancing the quantal size of vesicles and the rate of AP firing. The third important observation is that PDE4 inhibition and the consequent elevated local cAMP increase exocytosis by mainly altering the size and content of single secretory vesicles without affecting the probability of vesicle release. The increased exocytosis is only partially due to the enhanced Ca2+ passing through open LTCCs, while most of the action occurs down-stream the Ca2+-entry and acts directly on the quantal size of vesicle secretion. A corollary of our findings is that there is an effective cAMP production at rest in MCCs and PDE4 keeps cAMP levels low. Under these conditions, the functioning LTCCs and exocytosis are down-regulated and both increase when cAMP hydrolysis by PDE4 is blocked by rolipram.
L-type channels as pace-makers of MCCs firing activity
An important issue of our findings is the specific role that LTCCs play in the regulation of AP activity in isolated MCCs (Figs. 4 and 5). LTCCs do not only contribute to the broadening of the AP waveform, as in most excitable cells [26], but participate to the inward pace-maker current, which underline the “slow depolarization potential” preceding the fast upstroke. This derives from three main properties of LTCCs: (1) their high densities of expression in MCCs (45% of the total Ca2+ currents are carried by LTCCs; Fig. 1c), (2) their low-threshold of activation (LTCCs open at about −50 mV in 2 mM Ca2+ and carry tens of picoamperes at −40 mV, Fig. 7), and (3) their slow kinetics of inactivation [29], which allows to maintain long depolarization during interpulse intervals of 300–600 ms (Figs. 6 and 7). All three conditions are necessary for generating slowly inactivating inward Ca2+ currents of 5 to 10 pA that, passing through an input resistance of 1–2 GΩ, can depolarize the cell by 5 to 20 mV. This small amount of current is sufficient to drive the resting voltage from the mean interpulse potential (−49 mV) to the threshold of AP firing (−40 mV) and generate spikes. Rolipram and Bay K 8644 increase this inward current and accelerate the slow depolarizing phase (pace-maker potential), which sets the frequency of firing discharges. Nifedipine blocks LTCCs and thus prolongs interspikes intervals or even completely blocks spontaneous firing.
Our data suggest a new uncovered role of LTCCs in the control of chromaffin cell activity. These channels do not only contribute to the Ca2+ entry during an AP but also set the action potential firing frequency and modulate catecholamines release [17]. LTCCs carry significant current during the slow rising phase that precedes the spike and, thus, any change of their gating and availability will strongly condition chromaffin cells functioning. This might partially explain why LTCCs are so critically linked to adrenaline and noradrenaline release in chromaffin cell preparations where LTCCs contribute very little to the total Ca2+ currents [15]. In BCCs, which express small densities of LTCCs, the DHP antagonists block about 80% of the total catecholamines release during prolonged KCl-induced cell depolarizations [32]. It is also interesting to notice that LTCCs dominate catecholamine secretion not only on BCCs but also on MCCs [48] and RCCs [41] when using repeated electrical stimulations. The unifying rationale for this is most likely associated with the slower time-dependent inactivation and the favored availability of LTCCs at low holding potentials with respect to the other high-threshold channels (N, P/Q, R).
Cav1.3 as possible candidate for pace-making MCCs: contribution of other ion channels
Given that LTCCs affect the frequency of AP discharge, the next question is: What types of Cav1 channels are involved in this process? RCCs express two LTCC isoforms, Cav1.2 and Cav1.3 [2], and the same is true for MCCs (Marcantoni, Sinnegger-Brauns, Striessnig, Carbone; unpublished observations). Cav1.2 channels activate at membrane voltages more negative than N, P/Q, and R-type channels [31], but the Cav1.3 isoform activates at even more negative potentials (about −50 mV), making these channels good candidates for supporting pace-maker currents in spontaneously firing neurons and sino-atrial cells [12, 34, 47, 64]. Preliminary experiments performed comparing the density of LTCCs of MCCs in wild and Cav1.3 KO mice suggest that a large fraction of cells possess functional Cav1.3 channels with low-threshold of activation and incomplete inactivation during step depolarizations of 600 ms to +10 mV in 2 mM Ca2+ (Marcantoni, Striessnig, Carbone, unpublished observations). Thus, the present information supports the view that MCCs are equipped with a set of “low-threshold” LTCCs that may sustain spontaneous AP discharges.
Cav1 channels are unlikely to be the only ion channels controlling the rhythmic activity of MCCs, but our present data do not support a role for other channels carrying ionic currents near resting conditions. The hyperpolarization-activated cationic current (I h) seems not directly involved in MCCs AP firings (Marcantoni, Vandael, Carbone, unpublished observations). The low-threshold T-type channel Cav3.2 could be another good candidate for pace-making cells, but in physiological conditions, this channel is weakly expressed in RCCs [6, 46] and MCCs (Figs. 1 and 2). Other candidates are also the BK, SK, and ERG K+ channels, which are expressed in RCCs [22, 44] and whose outward currents counterbalance the inward L-type currents flowing during interspike intervals. However, we do not have enough data either to prove or disprove their involvement in controlling AP frequency. We also cannot exclude the contribution of Na+ TTX-sensitive inward currents as in central neurons [58], although it seems unlikely that these fast inactivating Na+ channels can carry sufficient inward currents during interspike intervals of 300–600 ms.
Rolipram and denopamine up-regulate L-type channels in MCCs
Our findings on the potentiating action of rolipram on LTCCs are in good agreement with the reported data on adult rat ventricular myocytes, in which basal L-type currents are effectively up-regulated by selective inhibition of PDE4 [30]. Among the various PDE isoforms expressed in cardiac myocytes, PDE4 appears the most directly involved in the control of intracellular cAMP and cardiac contraction. Inhibition of PDE4 by rolipram causes marked potentiation of cardiac LTCCs requiring 3–4 min to reach maximal effects [60], similar to the up-regulation produced in MCCs (Fig. 1). PDE4 is also expressed in chromaffin cells, but its role has not yet been properly investigated. The few available data show that rolipram increases the basal levels of intracellular cAMP in BCCs [36] and produces a 25% increase of catecholamines content inside the granules [33]. To our knowledge, there are no data on the action of PDE4 and on the gating and availability of LTCCs in chromaffin cells. This is surprising, given the high density of expression of Cav1 channels in rodent chromaffin cells [17] and the existing coupling with cAMP [7].
The present data are in good agreement with previous findings on the up-regulatory effects of β1-AR on the LTCCs of RCCs [9]. β1-AR stimulation in chromaffin cells up-regulates LTCCs by increasing the open channel probability through a cAMP/PKA-dependent pathway [3]. MCCs appear to possess the same modulatory cascade (Figs. 2 and 3). The selective β1-AR agonist denopamine potentiate LTCCs in the majority of cells and rolipram and IBMX mimic this action, indicating the specific involvement of PDE4, which selectively hydrolyzes cAMP with high affinity [11, 23]. Our results also reveal that rolipram and IBMX generate high intracellular cAMP levels than that produced by denopamine (Fig. 8), suggesting good coupling between β1-ARs and LTCCs. Simultaneous application of denopamine and rolipram increases further the intracellular cAMP concentration but not the size of Cav1 currents and the onset of potentiation, suggesting that the inhibition of PDE4 by rolipram produces alone saturating levels of cAMP. Most likely both modulators (rolipram and denopamine) act independently on the same pool of cAMP surrounding LTCCs. It is also interesting to notice that PDE4 is not the only PDE affecting the basal levels of cAMP since IBMX largely elevates the resting cAMP (Fig. 8). The lack of selective inhibitors for the two other PDEs specific for cAMP (PDE7 and PDE8) [11] did not allow any better conclusion about the involvement of other PDEs on LTCCs modulation (see “Results”).
An unresolved issue of the present work is related to the observation that only a fraction of MCCs respond to rolipram. This could be due to various causes: (1) a different degree of expression of PDE4, (2) a different degree of coupling of PDE4 to the two Cav1 channels expressed in MCCs (Cav1.2 and Cav1.3), and (3) a different sensitivity of Cav1 isoforms to cAMP/PKA. The latter, however, is the most unlikely since, in cardiac tissues, both isoforms can be up-regulated by cAMP–PKA [43, 52]. Experiments using Cav1.3 and Cav1.2 KO mice will certainly clarify this issue.
Rolipram increases the membrane area and catecholamine content of unitary vesicle without affecting the probability of release
Our conclusions on the potentiating action of rolipram on catecholamine secretion could be derived by combining membrane capacitance [19, 45] and amperometric measurements [10, 62]. As shown in Fig. 9d, rolipram increases more the size of the RRP (47%) than the quantity of Ca2+ charges entering the cell (16%). This suggests that about 30% of the increased secretion is Ca2+ independent and occurs down-stream of Ca2+ elevation through LTCCs, most likely by affecting directly the secretory apparatus. Since the probability of release remains unaltered (Fig. 9d) and the size of single capacitance changes Δc increases (46%; Fig. 10b), most of the Ca2+-independent capacitance enhancement is likely associated to an augmented vesicle membrane area resulting in an increased vesicle diameter (d). Although large (46%), the Δc increase is in fairly good agreement with the expected 30% increase of the RRP, particularly if considering that the estimate of Δc is complex and strongly biased by the critical experimental conditions and specific assumptions of the method (see [40]).
Assuming that the Ca2+-independent increase of RRP associated to an increased diameter of secretory vesicle is likely in the order of 30%, this allows clear predictions on how much should increase the quantal content of each vesicle if the concentration of catecholamine remains constant [20]. Indeed, a 30% increase of membrane capacitance would predict a 15% increase of d (ΔC/C ∝ 2Δd/d) and a 45% increase of vesicle volume (V) and quantal content Q (ΔV/V ∝ ΔQ/Q ∝ 3Δd/d). The amperometric data of Fig. 11 are in good agreement with this prediction. They show an average 43% increase of Q with rolipram, confirmed by the increased amount of cumulative secretion (56%). As the frequency of spikes is unaffected by rolipram, the increased cumulative secretion (Fig. 11b) is entirely attributable to the increased quantal size.
Our data are in good agreement and give a rationale to previously reported findings on the action of cAMP and rolipram. cAMP increases the size of the RRP by 75% and nearly doubles the membrane area of unitary vesicles in RCCs [5], while it increases the quantal size by 38% in RCCs [59] and by 50% in BCCs ([33]; in this work, cAMP was raised by applying forskolin). Rolipram has been tested only in BCCs and causes a quantal content increase of 25% [33]. Our data are also in excellent agreement with recent observations showing that an increase in quantal size in BCCs is usually associated with a proportional increase in vesicle membrane area [20]. In other words, vesicles undergo swelling or shrinking in response to changes in transmitter content in order to preserve the intravesicular concentration of transmitter. This is an interesting mechanism of “membrane dynamics” that applies also to vesicles of other neuroendocrine cells and presynaptic terminals [49]. Concerning the possible mechanisms by which cAMP or rolipram can increase the quantal size and vesicle membrane area in RCCs, there are various possibilities (see [20, 33, 49]). Those most likely are linked to actions that increase the transport rate of neurotransmitter inside the vesicle, but our present analysis does not allow any reasonable discrimination among them.
The functional role of PDE4 in MCCs
In chromaffin cells, different pools of compartmentalized cAMP control a variety of processes, including CREB-mediated protein synthesis [53], vesicle recruitment and fusion [42], catecholamine secretion [5], ion channel gating [3], and ion channel recruitment [18, 46]. Thus, cAMP represents a clear example of how a single messenger regulates multiple transduction pathways. All these processes require specific stimuli, which elevate cAMP from low basal levels. Under these conditions, the basal hydrolysis of cAMP mediated by PDE4 is crucial for triggering cAMP signaling, which regulate specific cellular functions. This holds true also for the cAMP-sensitive LTCC modulation [3, 7, 9] and catecholamine secretion [5]. As LTCCs are shown to control the spontaneous firing discharge, the hydrolyzing action of PDE4 appears critical for the control of basal MCCs electrical activity. If cAMP rises in an uncontrolled manner inside the cell, the increased firing frequency and catecholamine release [65] may trigger an autocrine feedback loop that lead to an undesired cell overstimulation. The increased release of catecholamines may in fact boost cAMP levels through the activation of β1-AR and drive the cells to a progressively increasing firing frequency. PDE4 activity thus works as a “brake” for cell excitability, allowing chromaffin cells to fire and release catecholamines regularly during resting activity even when cAMP is moderately produced by various stimuli.
References
Anderson K, Robinson PJ, Marley PD (1992) Cholinoceptor regulation of cyclic AMP levels in bovine adrenal medullary cells. Br J Pharmacol 106:360–366
Baldelli P, Hernández-Guijo JM, Carabelli V, Novara M, Cesetti T, Andrés-Mateos E, Montiel C, Carbone E (2004) Direct and remote modulation of L-channels in chromaffin cells: distinct actions on alpha1C and alpha1D subunits? Mol Neurobiol 29:73–96
Carabelli V, Hernández-Guijo JM, Baldelli P, Carbone E (2001) Direct autocrine inhibition and cAMP-dependent potentiation of single L-type Ca2+ channels in bovine chromaffin cells. J Physiol 532:73–90
Carabelli V, D’Ascenzo M, Carbone E, Grassi C (2002) Nitric oxide inhibits neuroendocrine Ca(V)1 L-channel gating via cGMP-dependent protein kinase in cell-attached patches of bovine chromaffin cells. J Physiol 541:351–366
Carabelli V, Giancippoli A, Baldelli P, Carbone E, Artalejo AR (2003) Distinct potentiation of L-type currents and secretion by cAMP in rat chromaffin cells. Biophys J 85:1326–1337
Carabelli V, Marcantoni A, Comunanza V, de Luca A, Díaz J, Borges R, Carbone E (2007) Chronic hypoxia up-regulates alpha1H T-type channels and low-threshold catecholamine secretion in rat chromaffin cells. J Physiol 584:149–165
Carbone E, Carabelli V, Cesetti T, Baldelli P, Hernandez-Guijo JM, Giusta L (2001) G-protein- and cAMP-dependent L-channel gating modulation: a manyfold system to control calcium entry in neurosecretory cells. Pflugers Arch 442:801–813
Ceña V, Nicolas GP, Sanchez-García P, Kirpekar SM, García AG (1983) Pharmacological dissection of receptor-associated and voltage-sensitive ionic channels involved in catecholamine release. Neuroscience 10:1455–1462
Cesetti T, Hernandez-Guijo JM, Baldelli P, Carabelli V, Carbone E (2003) Opposite action of beta1- and beta2-adrenergic receptors on Ca(V)1 L-channel current in rat adrenal chromaffin cells. J Neurosci 23:73–83
Chow RH, Rüden LV, Neher E (1992) Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature 356:60–63
Conti M, Beavo J (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76:481–511
Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ (2006) Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci 9:251–259
Fenwick EM, Marty A, Neher E (1982) A patch-clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. J Physiol 331:577–597
Fulop T, Smith C (2006) Physiological stimulation regulates the exocytic mode through calcium activation of protein kinase C in mouse chromaffin cells. Biochem J 399:111–119
Gandía L, Albillos A, García AG (1993) Bovine chromaffin cells possess FTX-sensitive calcium channels. Biochem Biophys Res Commun 194:671–676
García AG, Sala F, Reig JA, Viniegra S, Frías J, Fontériz R, Gandía L (1984) Dihydropyridine BAY-K-8644 activates chromaffin cell calcium channels. Nature 309:69–71
García AG, García-De-Diego AM, Gandía L, Borges R, García-Sancho J (2006) Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol Rev 86:1093–1131
Giancippoli A, Novara M, de Luca A, Baldelli P, Marcantoni A, Carbone E, Carabelli V (2006) Low-threshold induced by cAMP-recruited Cav 3.2 (alpha1H) channels in rat chromaffin cells. Biophys J 90:1830–1841
Gillis KD, Mossner R, Neher E (1996) Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron 16:1209–1220
Gong LW, Hafez I, Alvarez de Toledo G, Lindau M (2003) Secretory vesicles membrane area is regulated in tandem with quantal size in chromaffin cells. J Neurosci 23:7917–7921
Grabner CP, Price SD, Lysakowski A, Fox AP (2005) Mouse chromaffin cells have two populations of dense core vesicles. J Neurophysiol 94:2093–2104
Gullo F, Ales E, Rosati B, Lecchi M, Masi A, Guasti L, Cano-Abad MF, Arcangeli A, Lopez MG, Wanke E (2003) ERG K+ channel blockade enhances firing and epinephrine secretion in rat chromaffin cells: the missing link to LQT2-related sudden death? FASEB J F17:330–332
Gupta R, Kumar G, Kumar RS (2005) An update on cyclic nucleotide phosphodiesterase (PDE) inhibitors: phosphodiesterases and drug selectivity. Methods Find Exp Clin Pharmacol 27:101–118
Hernández-Guijo JM, de Pascual R, García AG, Gandía L (1998) Separation of calcium channel current components in mouse chromaffin cells superfused with low- and high-barium solutions. Pflugers Arch 436:75–82
Hernández-Guijo JM, Carabelli V, Gandía L, García AG, Carbone E (1999) Voltage-independent autocrine modulation of L-type channels mediated by ATP, opioids and catecholamines in rat chromaffin cells. Eur J Neurosci 11:3574–3584
Hille B (2001) Ion channels of excitable membranes, 3rd edn. Sinauer, Sunderland
Hollins B, Ikeda SR (1996) Inward currents underlying action potentials in rat adrenal chromaffin cells. J Neurophysiol 76:1195–1211
Houslay MD, Adams DR (2003) PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signaling cross-talk, desensitization and compartmentalization. Biochem J 370:1–18
Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J (2001) Alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem 276:22100–22106
Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechêne P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G (2008) Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res 102:1091–1100
Lipscombe D, Helton TD, Xu W (2004) L-type calcium channels: the low down. J Neurophysiol 92:2633–2641
López MG, Villarroya M, Lara B, Martínez Sierra R, Albillos A, García AG, Gandía (1994) L, Q- and L-type Ca2+ channels dominate the control of secretion in bovine chromaffin cells. FEBS Lett 349:331–337
Machado JD, Morales A, Gomez JF, Borges R (2001) cAMP modulates exocytotic kinetics and increases quantal size in chromaffin cells. Mol Pharmacol 60:514–520
Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J (2003) Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA 100:5543–5548
Marcantoni A, Baldelli P, Hernandez-Guijo JM, Comunanza V, Carabelli V, Carbone E (2007) L-type calcium channels in adrenal chromaffin cells: role in pace-making and secretion. Cell Calcium 42:397–408
Marley PD, Thomson KA (1992) Regulation of cyclic AMP metabolism in bovine adrenal medullary cells. Biochem Pharmacol 44:2105–2110
Marley PD (2003) Mechanisms in histamine-mediated secretion from adrenal chromaffin cells. Pharmacol Ther 98:1–34
Marley PD, Thomson KA, Jachno K, Johnston MJ (1991) Histamine-induced increases in cyclic AMP levels in bovine adrenal medullary cells. Br J Pharmacol 104:839–846
Montesinos MS, Machado JD, Camacho M, Diaz J, Morales YG, Alvarez de la Rosa D, Carmona E, Castañeyra A, Viveros HO, O’Connor DT, Mahata SK, Borges R (2008) The crucial role of chromogranins in storage and exocytosis revealed using chromaffin cells from chromogranin A null mouse. J Neurosci 28(13):3350–3358
Moser T, Neher E (1997) Estimation of mean exocytotic vesicle capacitance in mouse adrenal chromaffin cells. Proc Natl Acad Sci USA 94:6735–6740
Nagayama T, Matsumoto T, Kuwakubo F, Fukushima Y, Yoshida M, Suzuki-Kusaba M, Hisa H, Kimura T, Satoh S (1999) Role of calcium channels in catecholamine secretion in the rat adrenal gland. J Physiol 520:503–512
Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sørensen JB (2004) Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 41:417–429
Nargeot J, Lory P, Richard S (1997) Molecular basis of the diversity of calcium channels in cardiovascular tissues. Eur Heart J 18:A15–A26
Neely A, Lingle CJ (1992) Two components of calcium-activated potassium current in rat adrenal chromaffin cells. J Physiol 453:97–131
Neher E, Marty A (1982) Discrete changes of cell membrane capacitance cells observed under conditions of enhanced secretion in bovine adrenal chromaffin. Proc Natl Acad Sci USA 79:6712–6716
Novara M, Baldelli P, Cavallari D, Carabelli V, Giancippoli A, Carbone E (2004) Exposure to cAMP and beta-adrenergic stimulation recruits Ca(V)3 T-type channels in rat chromaffin cells through Epac cAMP-receptor proteins. J Physiol 558:433–449
Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J (2000) Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102:89–97
Polo-Parada L, Chan SA, Smith C (2006) An activity-dependent increased role for L-type calcium channels in exocytosisis regulated by adrenergic signaling in chromaffin cells. Neuroscience 143:445–459
Pothos EN, Mosharov E, Liu KP, Setlik W, Haburcak M, Baldini G, Gershon MD, Tamir H, Sulzer D (2002) Stimulation-dependent regulation of the pH, volume and quantal size of bovine and rodent secretory vesicles. J Physiol (Lond) 542:453–476
Prakriya M, Lingle CJ (1999) BK channel activation by brief depolarizations requires Ca2+ influx through L- and Q-type Ca2+ channels in rat chromaffin cells. J Neurophysiol 81:2267–2278
Przywara DA, Guo X, Angelilli ML, Wakade TD, Wakade AR (1996) A non-cholinergic transmitter, pituitary adenylate cyclase-activating polypeptide, utilizes a novel mechanism to evoke catecholamine secretion in rat adrenal chromaffin cells. J Biol Chem 271:10545–10550
Qu Y, Baroudi G, Yue Y, El-Sherif N, Boutjdir M (2005) Localization and modulation of α1D (Cav1.3) L-type Ca channel by protein kinase A. Am J Physiol Heart Circ Physiol 288:H2123–H2130
Sabban EL, Liu X, Serova L, Gueorguiev V, Kvetnansky R (2006) Stress triggered changes in gene expression in adrenal medulla: transcriptional responses to acute and chronic stress. Cell Mol Neurobiol 26:845–856
Sakuma T, Tuchihara C, Ishigaki M, Osanai K, Nambu Y, Toga H, Takahashi K, Ohya N, Kurihara T, Matthay MA (2001) Denopamine, a beta1-adrenergic agonist, increases alveolar fluid clearance in ex vivo rat and guinea pig lungs. J Appl Physiol 90:10–16
Segura F, Brioso MA, Gómez JF, Machado JD, Borges R (2000) Automatic analysis for amperometrical recordings of exocytosis. J Neurosci Methods 103:151–156
Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003) Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114:75–86
Steinberg SF, Brunton LL (2001) Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol 41:751–773
Swensen AM, Bean BP (2003) Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci 23:9650–9663
Tang KS, Tse A, Tse FW (2005) Differential regulation of multiple populations of granules in rat adrenal chromaffin cells by culture duration and cyclic AMP. J Neurochem 92:1126–1139
Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R (1999) Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol 127:65–74
Wallace DJ, Chen C, Philip D, Marley PD (2002) Histamine promotes excitability in bovine adrenal chromaffin cells by inhibiting an M-current. J Physiol 540:921–939
Wightman RM, Jankowski JA, Kennedy RT, Kawagoe DT, Schroeder TJ, Leszczyszyn DJ, Near JA, Diliberto Jr EJ, Viveros OH (1991) Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc Natl cad Sci USA 88:10754–10758
Wilson SP (1988) Vasoactive intestinal peptide elevates cyclic AMP levels and potentiates secretion in bovine adrenal chromaffin cells. Neuropeptides 11:17–21
Zhang ZY, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, Chiamvimonvat N (2002) Functional roles of Ca(v)1.3 [alpha(1D)] calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res 90:981–987
Zhou Z, Misler S (1995) Action potential-induced quantal secretion of catecholamines from rat adrenal chromaffin cells. J Biol Chem 270:3498–3505
Acknowledgments
This work was supported by the Marie Curie Research Training Network “CavNET” (Contract No MRTN-CT-2006-035367), the MIUR (grant COFIN no. 2005054435 to EC), the Regione Piemonte (grants No. A28-2005 to VC), and the Compagnia SanPaolo (grant to the NIS Center). We thank also Dr L. Anfossi for technical assistance in the cAMP immunoassays and Dr. C. Franchino for cell preparation
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Marcantoni, A., Carabelli, V., Vandael, D.H. et al. PDE type-4 inhibition increases L-type Ca2+ currents, action potential firing, and quantal size of exocytosis in mouse chromaffin cells. Pflugers Arch - Eur J Physiol 457, 1093–1110 (2009). https://doi.org/10.1007/s00424-008-0584-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0584-4