
Abstract
It has long been recognised that malignant tumours favour aerobic glycolysis to generate ATP and contain abnormalities of the intrinsic, mitochondria-dependent, apoptotic pathway, suggesting the involvement of dysfunctional mitochondria in tumour pathophysiology. However, the mechanisms underlying such processes in gliomas are poorly understood. Few recent studies have evaluated mitochondrial ultrastructure and proteomics in the pathophysiology of malignant gliomas. However, aberrant energy metabolism has been reported in gliomas and mitochondrial dysfunction links to glioma apoptotic signalling have been observed. Mitochondrial structural abnormalities and dysfunction in malignant gliomas is a neglected area of research. Definition of abnormalities in mitochondrial proteomics, membrane potential regulation, energy metabolism and intrinsic apoptotic pathway signalling in gliomas may open novel therapeutic opportunities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The worldwide incidence of primary brain tumours is estimated as 7.2–12.5 per 100,000 persons per year, encompassing ∼1–2% of all adult primary tumours and 23% of childhood cancers. Primary brain tumours are responsible for approximately 13,000 deaths per year and account for 2% of adult and 26% of childhood cancer deaths [1–6]. The most common type of tumour is glioma, found in 49% of cases, with an age-standardised incidence rate of ∼5 per 100,000 persons per year [2, 4, 7]. The resulting individual, social, healthcare and economic burden arising from the diagnosis and management of this group of cancers is significantly extensive.
The glial cell type of origin allows histological classification of gliomas into subtypes, with the most common type being astrocytoma [2]. Further histopathological analysis is then used to grade the level of malignancy from I to IV using the World Health Organization grading system (Table 1) [6, 8–11].
Glioblastoma multiforme (GBM) is the most malignant and most frequently found gliomal tumour type in adults, diagnosed in up to 80% of glioma cases [2, 4, 6, 8]. It can arise via one of two possible pathways: de novo or through evolvement of an existing lower-grade astrocytoma [3, 6, 11, 12]. Outlook from diagnosis is extremely poor, with a median survival of 9–14 months depending on age, performance status and treatment given. Over the last 40 years, outlook has improved only modestly despite technological and therapeutic improvements in surgical resection, radiotherapy and chemotherapy [3, 12–14].
It has been well reported that malignant tumours, including gliomas, favour abnormal energy production via aerobic glycolysis (non-oxidative metabolism of glucose in the presence of oxygen) and show an inherent resistance to apoptosis (programmed cell death) [6, 15, 16]. This suggests the underlying involvement of mitochondrial dysfunction in malignant tumour pathophysiology, which is not well understood.
Mitochondria are oval-shaped, membrane-enclosed intracellular organelles of 0.5- to 10-μm diameter which contain their own DNA. They have distinct structural compartments that are essential for performing specialised functions [17]. The most recognised of which is the generation of adenosine triphosphate (ATP), the chemical form of cellular energy, via aerobic respiration. This is the main energy-producing pathway of normal glial cells and the ensuing step following glycolysis in the presence of oxygen [17, 18]. Mitochondria are also involved in the regulation of cellular proliferation and apoptosis, two other important areas of dysfunction in glioma.
This paper aims to review and consolidate knowledge of the function and dysfunction of mitochondria in energy generation and apoptosis in gliomas. Insight into aberrant mitochondrial function in gliomas is essential to provide new directions in the development of novel research approaches that translate into improved therapies.
Energy Metabolism
Mitochondrial Decoupling
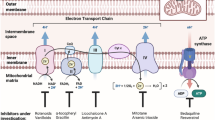
Mitochondrial aerobic respiration is the main source of cellular energy in glial cells [19]. It begins with enzymatic metabolism of acetyl-CoA in the mitochondrial matrix, via the tricarboxylic acid (TCA) cycle, which causes reduction of co-enzymes nicotinamide adenine dinucleotide (NAD+) and flavin adenine dinucleotide (FAD). The reduced forms (NADH and FADH2) then undergo oxidative phosphorylation by the mitochondrial respiratory chain. This is a series of five (I–V) transmembrane protein complexes spanning the inner mitochondrial membrane. Complexes I–IV act in a chain to transfer the electrons liberated by oxidation, which drives H+ (protons) into the intermembrane space, creating a proton motive gradient across the inner membrane. Complex V (ATP synthase) utilises the energy obtained from the movement of protons along this gradient to generate ATP (Fig. 1) [17, 18].

Pathways of energy metabolism in glial cells. Cytoplasmic glycolysis has a net yield of two ATP per molecule of glucose, whilst acetyl-CoA metabolism, via the TCA cycle and mitochondrial respiratory chain (complexes I–V), requires oxygen and has a net yield of 36 ATP per molecule of acetyl-CoA. Lactate is formed in the absence of oxygen and is recycled into glucose via the Cori cycle in the liver. GLUT1 glucose transporter 1, LDH lactate dehydrogenase, PDH pyruvate dehydrogenase, TCA cycle tricarboxylic acid cycle, NAD + nicotinamide adenine dinucleotide, FAD flavin adenine dinucleotide, ATP adenosine triphosphate, ADP adenosine diphosphate, Pi inorganic phosphate, β-OHB β-hydroxybutyrate, MCT1 monocarboxylate transporter 1, β-HBDH β-hydroxybutyrate dehydrogenase, SCOT succinyl-CoA-acetoacetate-CoA transferase (modified from figures in [18, 25])
Acetyl-CoA is mainly derived from glucose by cytoplasmic glycolysis to pyruvate and subsequent metabolism by pyruvate dehydrogenase. However, when blood glucose becomes low following prolonged fasting, the blood levels of the liver-produced ketone β-hydroxybutyrate (β-OHB) are increased, which becomes an alternative substrate for acetyl-CoA generation in glial cells [20]. It is transported across the blood–brain barrier by monocarboxylic transporters (MCTs) and is metabolised within the mitochondrial matrix directly, allowing rapid energy production capable of sustaining the brain’s metabolic demands (Fig. 1) [21–24].
In contrast, gliomas show reliance on persistent aerobic glycolysis as their main source of ATP production, as exhibited by animal models, human glioma-derived cell cultures and in vivo human studies [25–27]. In fact, toxic inhibition of the mitochondrial respiratory chain in glioma cell lines has no effect on cell viability or ATP production in the presence of adequate glucose [28]. This failure of malignant tumour cells to progress to more efficient aerobic respiration for energy production was first described by the Nobel laureate Warburg and subsequently termed the Warburg hypothesis [29].
Glioma-derived cell lines demonstrate the presence of ketone energy metabolism pathways, but despite this, animal glioma models cannot sustain tumour growth in the presence of reduced glucose and elevated β-OHB, presumably due to dysfunction of ketone-derived ATP production [25, 30–32]. This difference in normal glial cell versus glioma cell metabolism has been exploited as a potential therapeutic strategy in two female paediatric cases of malignant glioma (grades III and IV) which were non-responsive to aggressive radiotherapy and chemotherapy [33]. These patients were maintained on a ketogenic diet for 8 weeks, which lowered blood glucose and produced ketosis (elevated circulating ketones) within 7 days. This resulted in marked symptomatic improvement, long-term tumour management (one patient remaining free of disease progression at 12 months with continued consumption of the ketogenic diet) and a 21.8% average reduction in tumour glucose uptake on positron emission tomography [33]. No adverse effects were reported.
Mouse, in vivo, glioma model studies (using implanted mouse and human glioma cell lines) indicate additional anti-inflammatory, anti-angiogenic and pro-apoptotic tumour effects of ketosis induced by dietary glucose restriction [25, 32, 34, 35]. The exact mechanisms underlying these beneficial results remain to be uncovered. Some insight has been provided by glial cell cultures which release pro-inflammatory cytokines in response to lactate, the end product of obligatory glycolytic energy production [36]. This could feasibly be a contributory process to tumour growth, oedema, angiogenesis and apoptosis resistance in the lactate-rich environment of gliomas, which glucose restriction would counter. Furthermore, immunohistochemistry of human CNS tissue shows increased expression of MCT 1 transporters in high-grade gliomas, potentially facilitating increased ketone uptake and making any direct, ketone-induced anti-tumour effects an appealing target for novel therapeutics [22].
Overall, the Warburg hypothesis is supported by the lack of mitochondria-dependent ketone utilisation for energy metabolism. Both point to the presence of dysfunctional mitochondria, which essentially decouple from the normal, cellular energy-generating pathways, thus making glioma dependent on cytosolic glycolysis. Further evidence for this is seen in glioblastoma cell cultures where constitutive activation of the Akt oncogene, a frequent mutation in malignant tumours responsible for overactivity of the cell proliferation pathway, results in a clear shift from normal aerobic respiration to abnormal, persistent aerobic glycolysis for cell survival [37].
Cardiolipin Dysfunction
The mitochondrial membrane has a unique structure due to the phospholipid cardiolipin which is specific to mitochondria and bacteria. Cardiolipin is highly expressed on the inner mitochondrial membrane and at contact points between the inner and outer mitochondrial membranes [38, 39]. It is a dimeric phospholipid with four variable acyl groups, mostly of C18 chain length in mammals, whose different combinations give rise to the multitude of cardiolipin molecular species [38, 40, 41]. It is synthesised through the common phospholipid biosynthetic pathway, with the final distinct step, catalysed by the inner mitochondrial membrane-bound enzyme cardiolipin synthase. The end result is the production of cardiolipin within the mitochondrial matrix, which then undergoes further acyl group modification [38, 42].
Cardiolipin has an essential role in mitochondrial function through its interactions with inner mitochondrial membrane proteins. Its different molecular species are closely associated with the quaternary structure of several metabolite carriers and complexes I, III, IV and V of oxidative phosphorylation (Table 2).
As well as incorporation into individual proteins, cardiolipin is required for structural support and maintenance in respirasome supercomplexes, which are the functional units of oxidative phosphorylation formed by an amalgamation of complexes I–IV [49]. Furthermore, mitochondrial structure, membrane stability, production of the proton motive gradient and role in apoptosis (discussed later, page 15) depend on the presence of normal cardiolipin [38, 50].
Loss of mitochondrial function in glioma could be attributed to deficiencies of cardiolipin. This has recently been explored by shotgun lipidomics comparing glial mitochondria of mice highly susceptible to spontaneous gliomas with those of mice refractory to spontaneous tumours. Both strains showed almost equal levels of total cardiolipin; however, the number of molecular species was reduced by ∼50% in the susceptible strain even prior to the development of glioma. In addition, the distribution of these molecular species was altered, showing an asymmetrical pattern across the inner mitochondrial membrane, overall leading to an observed reduction in the activity of complexes I, II and III [51]. These data suggest that altered cardiolipin differentiation into molecular species and organisation may be an early component of glioma development and consequently responsible for mitochondrial decoupling from energy production. This is supported by shotgun lipidomics of mitochondria from mouse gliomas which again show fewer cardiolipin molecular species and an associated, significant reduction in the activity of the mitochondrial respiratory chain complexes. Furthermore, the presence of abnormal and poorly developed cardiolipin molecular species was seen [52]. This implies that defective cardiolipin biosynthesis and subsequent failure of acyl group remodelling may play a central part in mitochondrial dysfunction in glioma. Unfortunately, the regulatory mechanisms and pathways for molecular species formation in cardiolipin synthesis are not well understood and require further research.
Oxidative Stress
Normal energy production through aerobic respiration generates reactive oxygen species (ROS) in the mitochondria. At physiological levels, ROS are non-lethal and act as intracellular messengers; however, prolonged excess of ROS results in cellular damage and death by apoptosis [53, 54]. Glial cells contain a pool of glutathione (GSH) and its oxidised form glutathione disulfide (GSSG) which act as a redox buffer system that prevents the buildup of ROS to dangerous levels [55].
In human glioma samples, the levels of GSH/GSSG buffer are significantly lower in the highly proliferative tumour periphery compared to the necrotic centre, even though mitochondrial content is the same [56]. This is important as glucose withdrawal from glioma cell cultures is shown to increase ROS production and induce apoptosis due to insufficient redox buffering [28, 57]. In these glioma cells, glucose withdrawal is also accompanied by an increased breakdown of fatty acids within the mitochondria, presumably as part of an alternative pathway for generating ATP [57]. However, loss of normal mitochondrial structure, stability and function in glioma is likely to result in abnormalities of the attempted fatty acid metabolism, causing the high output of ROS and lethal oxidative stress. In addition, studies of cardiolipin in rat mitochondria highlight increased levels of ROS as a direct cause of cardiolipin dysfunction through oxidation and, as a result, loss of mitochondrial function [58–60]. This detrimental cycle may be contributing to further mitochondrial damage and decoupling from normal cellular function in glioma (Fig. 2).

Effects of metabolic shift from glycolysis to dysfunctional fatty acid metabolism for attempted ATP production due to glucose withdrawal in glioma. Plus sign denotes stimulation. Minus sign denotes inhibition/buffering. ROS reactive oxygen species, GSH glutathione, GSSG glutathione disulfide
Therefore, the proliferating tumour periphery, with its high energy demands and low GSH/GSSG buffer levels, is highly susceptible to apoptosis induced by the metabolic effects of insufficient glucose, once again highlighting the potential of targeting energy metabolism to stop glioma progression.
Apoptosis
Apoptosis is a cascade of biochemical reactions culminating in cellular and nuclear fragmentation, followed by packaging of cellular contents into small vesicles known as apoptotic bodies and subsequent phagocytosis of these vesicles by surrounding cells. This process can be triggered via extrinsic and intrinsic apoptotic pathways (Fig. 3).

Extrinsic/intrinsic apoptotic pathways and their interactions. TRAIL TNF-related apoptosis-inducing ligand, TNF-α tumour necrosis factor-α, DR death receptor, DISC death-inducing signalling complex, EGF epidermal growth factor, PDGF platelet-derived growth factor, CXCL12 chemokine stromal cell-derived factor 12, RTK receptor tyrosine kinase, CXCR4 chemokine receptor 4, PI3K phosphoinositide 3-kinase, PIKE-A PI3K enhancer activating Akt, mTOR mammalian target of rapamycin, MDM2 murine double minute 2, PTEN phosphatase and tensin homolog, ROS reactive oxygen species, DNA deoxyribonucleic acid, p53 protein 53, PUMA p53 upregulated modulator of apoptosis, NOXA phorbol-12-myristate-13-acetate-induced protein 1, BH3 BCL-2 homology domain 3 protein, BCL-2 B cell lymphoma-2, BCL-xL B cell lymphoma-extra large, MCL-1 myeloid cell leukaemia sequence 1 (BCL2-related), BAD BCL-2-associated death promoter, BAK BCL-2 antagonist/killer, BAX BCL-2-associated X protein, MPTP mitochondrial permeability transition pore, SMAC second mitochondria-derived activator of caspases, IAP inhibitors of apoptosis, Cyt C cytochrome C, APAF-1 apoptotic peptidase activating factor-1
The extrinsic apoptotic pathway is activated by the immune system following detection of abnormal surface markers on the cell. There is subsequent release of specific ligands that bind and activate cell surface death receptors (DR) which include the Fas and TNF receptor families. This results in intracellular signal transduction through the formation of a death-inducing signalling complex, causing activating cleavage of caspases 8 and 10. These in turn cleave caspases 3 and 7, which go on to begin the irreversible apoptotic cascade [61, 62]. In glioma, the extrinsic apoptotic pathway is suppressed, even though its triggers and constituents are present [63, 64]. Administration of the DR-specific TNF-related apoptosis-inducing ligand shows that this may be due to an inherent tumour resistance, with only a minority of glioma cell lines showing susceptibility to apoptosis through this pathway [65]. Further studies of glioma cell lines and human, glioma, tumour samples indicate that this resistance is related to overactivation of the Akt and mammalian target of rapamycin (mTOR) proliferative pathway [66, 67].
The intrinsic apoptotic pathway is triggered by direct damage to the cell from various insults, including hypoxia, oxidative stress, radiotherapy and chemotherapy, all of which can cause influx of Ca2+ into the cell, generation of ROS and direct DNA damage. As a result, protein 53 (p53) is activated and subsequently enhances DNA transcription of anti-proliferative and pro-apoptotic genes [6, 15, 61, 62]. The up-regulated pro-apoptotic gene products include p53 upregulated modulator of apoptosis (PUMA), phorbol-12-myristate-13-acetate-induced protein 1 (NOXA) and BCL-2 homology domain 3 proteins (BH3), which interact with the B cell lymphoma-2 (BCL-2) homology family of proteins. The members of this family have a close structural and functional relationship with the mitochondrial membranes and exert either membrane-stabilising or permeability-inducing effects, classifying them into anti-apoptotic and pro-apoptotic, respectively. PUMA, NOXA and BH3 stimulate the pro-apoptotic BCL-2 family members and, together with these, inhibit the anti-apoptotic family members [61, 62, 68]. It is believed that the resulting shift in balance towards apoptosis causes opening of a mitochondrial permeability transition pore (MPTP) through the interaction of several proteins (discussed later, page 14). This pore allows the rapid release of cytochrome C (Cyt C), second mitochondria-derived activator of caspases (SMAC) and Omi from the mitochondrion into the cytosol. Cyt C then binds with apoptotic peptidase activating factor-1, a p53 enhanced gene product, and caspase 9 to form an apoptosome which cleaves caspases 3 and 7 instigating the apoptotic cascade, whilst SMAC and Omi inhibit the function of the inhibitors of apoptosis (IAP) family of proteins which prevent apoptosome action (Fig. 3) [61, 62, 68–70].
Abnormalities of this pathway are frequent in human cancers, including glioma. Inactivating p53 gene mutations are expressed in 30–50% of human glioma tumour samples, which is seen as an early change in low-grade glioma. Perseverance of this mutation to malignant glioma is common and associated with significantly increased angiogenesis and a higher chance of recurrence following resection [71, 72].
The strong association of BCL-2 family members with the mitochondrial membranes makes them possible contributors to mitochondrial dysfunction in glioma. Immunohistochemistry for the anti-apoptotic BCL-2 protein shows that it is highly over-expressed in human glioma cell lines and that this protects the cell from undergoing apoptosis following chemically induced DNA damage [73]. In addition, immunohistochemical studies of human glioma tumour samples show elevated BCL-2 family expression in all glioma cells compared to normal astrocytes, with very highly expressed members found in all grades of malignancy (Table 3) [74, 75].
Paradoxical elevation of the pro-apoptotic BCL-2-associated X protein (BAX) could suggest that over-expression of functional BCL-2 proteins in itself is not the only factor conferring apoptosis resistance. However, mitochondrial membrane dysfunction, which results in ineffective BCL-2 family interactions and subsequent attempted compensation by up-regulation of its members, could also underlie the inherent apoptosis-resistant nature of malignant cancers. If this is the case, then expression of the BCL-2 family could be used as a marker for the degree of mitochondrial dysfunction present.
In view of this, GBM recurrences following resection, radiotherapy and chemotherapy have been shown to contain significantly increased levels of BCL-2, BCL-xL and MCL-1, with decreased BAX compared to the initially resected tumour [75, 76]. Further studies in human malignant glioma cell lines show that high expression of BCL-xL and low expression of BAX is also associated with radiotherapy resistance and that down-regulation of BCL-xL and BCL-2 with antisense complimentary DNA (cDNA) transfection produces chemosensitivity [77–80]. Overall, suggesting that current treatment modalities select out resistant tumours, which appear to have a high expression of the mitochondria-associated anti-apoptotic BCL-2 family members, and that targeting this over-expression may provide a novel approach to glioma treatment.
Abnormalities in the intrinsic apoptotic pathway also occur downstream to mitochondria. Over 95% of human GBM samples show increased expression of the anti-apoptotic BCL-2-like 12 (BCL2L12) protein which inhibits caspase 3 through αB-crystallin and caspase 7 directly. Modified rat primary cortical astrocyte cell lines also indicate that BCL2L12 may be responsible for the terminal shift from apoptosis to necrosis, potentially accounting for the highly necrotic nature of gliomas [81]. Finally, IAP family proteins are also constitutively over-expressed in human malignant glioma cell lines. This can, however, be overcome by antisense cDNA transfection and SMAC mimetics which have shown therapeutic potential by increasing sensitivity to radiotherapy and chemotherapy and by potentiation of the extrinsic apoptotic pathway [82–86].
The cell-proliferative pathway is an important modulator of both extrinsic and intrinsic apoptotic pathways. It is triggered by the binding of growth factors to their respective receptor tyrosine kinases (RTKs) found on the cell surface. Activated receptors recruit phosphoinositide 3-kinase (PI3K) which phosphorylates a multitude of signalling proteins, including the protein kinase Akt. This causes activation of Akt, which then phosphorylates further proteins that potentiate the action of anti-apoptotic BCL-2 family members, suppress the intrinsic apoptotic pathway by negative regulation of p53 by murine double minute 2 (MDM2) and inhibit the extrinsic apoptotic pathway, whilst promoting protein synthesis for cell survival, growth and proliferation, by mTOR complexes (Fig. 3) [6, 15, 62, 87].
The constituents of the proliferative pathway are also adversely affected in glioma, producing an overall pro-proliferative and anti-apoptotic stimulus. There is over-expression of RTKs for epidermal growth factor in ∼20–40% of high-grade gliomas and for platelet-derived growth factor (PDGF) in ∼25% of cases [88–90]. Furthermore, immunohistochemistry has shown the presence of PDGF autocrine and paracrine loops in glioma tumour samples, which suggests that glioma exhibits a malignant ability to promote its own growth [91]. In addition, the proliferative pathway can be triggered by chemokine stromal cell-derived factor 12 (CXCL12) acting on its chemokine receptor (CXCR4), which are both found to be highly expressed in human malignant glioma cell lines and mouse glioma models [92]. Further down the pathway, as mentioned previously, there is often constitutive activation of Akt and enhancement of its anti-apoptotic action by the increased expression of PI3K enhancer activating Akt (PIKE-A) in GBM [37, 93, 94]. Finally, normal inhibition of the proliferative pathway by p53-enhanced transcription of the phosphatase and tensin homolog (PTEN) tumour suppressor gene is disrupted by inactivating PTEN gene mutations in 30–40% of glioblastomas [95].
Mitochondrial Permeability Transition Pore
The release of apoptosis effectors (Cyt C, SMAC and Omi) from mitochondria relies on the permeabilisation of inner and outer mitochondrial membranes. This process occurs through apoptotic-pathway-triggered cleavage of the cytosol expressed, pro-apoptotic BCL-2 family member known as BH3 interacting domain death agonist (BID). The resulting truncated form of the protein (tBID) translocates from the cytosol to the mitochondria where it attracts and activates cytosolic BAX and the outer mitochondrial membrane-bound BCL-2 antagonist/killer (BAK). The currently favoured theory is that recruitment of these pro-apoptotic BCL-2 family members facilitates the opening of a multi-protein pore complex, known as MPTP, found at contact sites between the mitochondrial membranes. This provides a transmembrane channel for the release of Cyt C, SMAC and Omi whilst also causing dissipation of the mitochondrial transmembrane potential and proton gradient (Fig. 4) [96–99].

Structure and interaction of the mitochondrial permeability transition pore (MPTP) in apoptosis. Plus sign denotes stimulation. VDAC voltage-dependent anion channel, ANT adenine nucleotide translocator, PBR peripheral benzodiazepine receptor, CK creatine kinase, HK hexokinase, Cyp D cyclophillin D, BID BH3 interacting domain death agonist, tBID truncated BID, BAX BCL-2-associated X protein, BAK BCL-2 antagonist/killer, Cyt C cytochrome C, SMAC second mitochondria-derived activator of caspases (modified from [19])
Cardiolipin is essential for maintaining the normal function of the mitochondrion during induction of apoptosis. Studies using mouse liver isolated mitochondria and liposome mitochondrial membrane models show that cardiolipin is necessary for targeting of tBID to mitochondria and for its subsequent binding, which occurs at the cariolipin-rich contact sites between the inner and outer mitochondrial membranes [100–102]. Interaction of tBID at these sites results in the redistribution of cardiolipin on the inner and outer mitochondrial membranes, as observed in haematological cancer cell lines prior to apoptosis [103]. This is likely to alter the structure and function of the mitochondrion to favour membrane permeabilisation. Furthermore, isolated mitochondrial systems suggest that cardiolipin is also required to facilitate the interaction of BAX with the MPTP and result in its opening following pro-apoptotic signalling [98]. Finally, embryonic mouse mitochondria indicate that specific molecular species of cardiolipin are bound to cyt C within the intermembrane space, forming an oxygenase complex. This complex catalyses cardiolipin peroxidation in response to apoptogenic signalling, which induces cyt C and SMAC release [104]. Therefore, it is reasonable to assume that the abnormalities of cardiolipin found in mouse glioma studies of abnormal mitochondrial energy metabolism (described previously, page 8) [51, 52] could also represent an underlying mechanism for mitochondrial dysfunction resulting in intrinsic apoptosis resistance in glioma.
Targeting the MPTP separately from its cardiolipin-dependent activation may provide a useful way of bypassing the mitochondrial dysfunction. Specific ligands to the peripheral benzodiazepine receptor, an over-expressed component of the MPTP in human glioma tumour samples, have shown promise by inducing apoptosis in rat glioma cell lines [105, 106]. In addition, direct MPTP-inducing agents promote apoptosis in human malignant glioma cell lines, suggesting that the pore itself is still functional whilst its activation by pro-apoptotic BCL-2 family members may be interrupted by cardiolipin abnormalities within the mitochondrial membranes [107].
Concluding Remarks/Future Perspectives
This review has highlighted the existence of mitochondrial dysfunction in glioma. Glioma tumour cells generate ATP through glycolosis exclusively, even though the presence of oxygen should allow progression of energy metabolism to mitochondrial aerobic respiration. This decoupling of mitochondria from the energy-generating pathway is described as a fundamental feature in many malignant cancers of different origin [29, 108–110].
In contrast to normal glial cells, gliomas also exhibit an inability to metabolise ketones and fatty acids for energy production following glucose deprivation. Both of these alternative ATP-generating pathways are reliant on mitochondria and their enzyme systems. Additionally, attempted fatty acid metabolism by mitochondria of glioma cells results in the abnormal production of high levels of ROS and subsequent cell death by apoptosis. Therefore, glucose deprivation and high ketogenic diets may provide a tangible therapeutic approach to specifically target glioma tumour cells whilst sparing normal glia from damage.
Several abnormalities of the intrinsic, mitochondria-dependent apoptotic pathway are evident in glioma as well as other malignant tumours [111]. There is a clear over-expression of the BCL-2 homology protein family which is associated with radiotherapy and chemotherapy resistance in different malignant cancers [112–114]. These proteins are closely related to mitochondrial membranes and their interaction mediates mitochondrial release of apoptogenic factors, an interaction which requires cardiolipin, a mitochondrial membrane-specific phospholipid, to function. Abnormalities of cardiolipin are described in glioma cells and are perceived to cause dysfunction of the mitochondrial respiratory chain and energy metabolism. However, bearing in mind the necessity of cardiolipin for pro-apoptotic BCL-2 family members to exert their effects, it is reasonable to speculate that these abnormalities may also potentially account for the interruption of the apoptotic pathway.
In other malignant tumours, oncogene expression and mitochondrial DNA (mtDNA) mutations have been proposed as mechanisms of mitochondrial dysfunction [108–111]. Nevertheless, these mechanisms cannot account for the defective role of mitochondria in apoptosis. In addition, a recent study of mtDNA in chemically induced mouse gliomas showed the absence of any causative mutations [115].
Mitochondrial proteomics is a further approach which can provide a fresh perspective of mitochondrial dysfunction in glioma. Proteomics allows large-scale simultaneous analysis of more than a thousand proteins and is particularly potent in assessing alterations in levels but also in defining protein–protein interactions. Proteomics along with genetic analysis has been employed in glioma with considerable success [116]. Proteomic analysis of mitochondrial fractions from glioma (Fig. 5) may provide additional insight into the dynamics of mitochondrial protein alterations in glioblastomas.

Representative 2D SDS-PAGE gels from control and glioblastoma mitochondrial fractions. Each spot represents an individual protein. Proteins are separated by charge (x dimension) and molecular weight (y dimension). Eighteen-centimetre nonlinear IPG strips with a pI range from 3 to 10 were used. Representative gels of control (left) and glioblastoma (right) mitochondrial fractions are shown. Alterations in the abundance of various mitochondrial proteins in glioblastomas can be discerned in various quadrants of the gels
Overall, this review shows that some attempts have been made to explain the precise nature of the mitochondrial abnormalities present in glioma; however, more work is needed to fully explore their mechanisms and provide effective novel therapeutics.
References
Legler JM, Ries LA, Smith MA, Warren JL, Heineman EF, Kaplan RS, Linet MS (1999) Cancer surveillance series: brain and other central nervous system cancers: recent trends in incidence and mortality. J Natl Cancer Inst 91:1382–1390
Wrensch M, Minn Y, Chew T, Bondy M, Berger MS (2002) Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro Oncol 4:278–299
DeAngelis LM (2001) Brain tumors. N Engl J Med 344:114–123
Radhakrishnan K, Mokri B, Parisi JE, O’Fallon WM, Sunku J, Kurland LT (1995) The trends in incidence of primary brain tumors in the population of Rochester, Minnesota. Ann Neurol 37:67–73
Landis SH, Murray T, Bolden S, Wingo PA (1999) Cancer statistics. CA Cancer J Clin 49:8–31
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21:2683–2710
Larjavaara S, Mäntylä R, Salminen T, Haapasalo H, Raitanen J, Jääskeläinen J, Auvinen A (2007) Incidence of gliomas by anatomic location. Neuro Oncol 9:319–325
Inskip PD, Linet MS, Heineman EF (1995) Etiology of brain tumors in adults. Epidemiol Rev 17:382–414
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO classification of tumours of the central nervous system, 4th edn. IARC, Lyon
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Rich JN, Bigner DD (2004) Development of novel targeted therapies in the treatment of malignant glioma. Nat Rev Drug Discov 3:430–446
Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lütolf UM, Kleihues P (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64:6892–6899
Anderson E, Grant R, Lewis SC, Whittle IR (2004) Randomized phase III controlled trials of therapy in malignant glioma: where are we after 40 years? Br J Neurosurg (2008) 22:339–349
Glioma Meta-Analysis Trialists (GMT) Group (2002) Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet 359:1011–1018
Ziegler DS, Kung AL, Kieran MW (2008) Anti-apoptosis mechanisms in malignant gliomas. J Clin Oncol 26:493–500
Seyfried TN, Mukherjee P (2005) Targeting energy metabolism in brain cancer: review and hypothesis. Nutr Metab 2:30
Benard G, Rossignol R (2008) Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid Redox Signal 10:1313–1342
Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80:315–360
Hertz L, Peng L, Dienel GA (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 27:219–249
Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr (1967) Brain metabolism during fasting. J Clin Invest 46:1589–1595
Pellerin L, Bergersen LH, Halestrap AP, Pierre K (2005) Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res 79:55–64
Froberg MK, Gerhart DZ, Enerson BE, Manivel C, Guzman-Paz M, Seacotte N, Drewes LR (2001) Expression of monocarboxylate transporter MCT1 in normal and neoplastic human CNS tissues. Neuroreport 12:761–765
Cahill GF Jr (2006) Fuel metabolism in starvation. Annu Rev Nutr 26:1–22
Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF Jr (2001) Ketone bodies, potential therapeutic uses. IUBMB Life 51:241–247
Seyfried TN, Sanderson TM, El-Abbadi MM, McGowan R, Mukherjee P (2003) Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br J Cancer 89:1375–1382
Galarraga J, Loreck DJ, Graham JF, DeLaPaz RL, Smith BH, Hallgren D, Cummins CJ (1986) Glucose metabolism in human gliomas: correspondence of in situ and in vitro metabolic rates and altered energy metabolism. Metab Brain Dis 1:279–291
Roslin M, Henriksson R, Bergström P, Ungerstedt U, Bergenheim AT (2003) Baseline levels of glucose metabolites, glutamate and glycerol in malignant glioma assessed by stereotactic microdialysis. J Neurooncol 61:151–160
Williams ZR, Goodman CB, Soliman KF (2007) Anaerobic glycolysis protection against 1-methy-4-phenylpyridinium (MPP+) toxicity in C6 glioma cells. Neurochem Res 32:1071–1080
Warburg O (ed) (1931) The metabolism of tumours. Richard R. Smith, New York
Roeder LM, Poduslo SE, Tildon JT (1982) Utilization of ketone bodies and glucose by established neural cell lines. J Neurosci Res 8:671–682
Patel MS, Russell JJ, Gershman H (1981) Ketone-body metabolism in glioma and neuroblastoma cells. Proc Natl Acad Sci U S A 78:7214–7218
Zhou W, Mukherjee P, Kiebish MA, Markis WT, Mantis JG, Seyfried TN (2007) The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr Metab 4:5
Nebeling LC, Miraldi F, Shurin SB, Lerner E (1995) Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr 14:202–208
Mukherjee P, Abate LE, Seyfried TN (2004) Antiangiogenic and proapoptotic effects of dietary restriction on experimental mouse and human brain tumors. Clin Cancer Res 10:5622–5629
Mukherjee P, El-Abbadi MM, Kasperzyk JL, Ranes MK, Seyfried TN (2002) Dietary restriction reduces angiogenesis and growth in an orthotopic mouse brain tumour model. Br J Cancer 86:1615–1621
Andersson AK, Rönnbäck L, Hansson E (2005) Lactate induces tumour necrosis factor-alpha, interleukin-6 and interleukin-1beta release in microglial- and astroglial-enriched primary cultures. J Neurochem 93:1327–1333
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64:3892–3899
Schlame M, Rua D, Greenberg ML (2000) The biosynthesis and functional role of cardiolipin. Prog Lipid Res 39:257–288
Ardail D, Privat JP, Egret-Charlier M, Levrat C, Lerme F, Louisot P (1990) Mitochondrial contact sites. Lipid composition and dynamics. J Biol Chem 265:18797–18802
LeCcocq J, Ballou CE (1964) On the structure of cardiolipin. Biochemistry 3:976–980
Schlame M, Brody S, Hostetler KY (1993) Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur J Biochem 212:727–735
Schlame M, Hostetler KY (1997) Cardiolipin synthase from mammalian mitochondria. Biochim Biophys Acta 1348:207–213
Fry M, Green DE (1981) Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J Biol Chem 256:1874–1880
Robinson NC, Zborowski J, Talbert LH (1990) Cardiolipin-depleted bovine heart cytochrome c oxidase: binding stoichiometry and affinity for cardiolipin derivatives. Biochemistry 29:8962–8969
Robinson NC (1993) Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr 25:153–163
Eble KS, Coleman WB, Hantgan RR, Cunningham CC (1990) Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J Biol Chem 265:19434–19440
Beyer K, Klingenberg M (1985) ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 24:3821–3826
Hoffmann B, Stöckl A, Schlame M, Beyer K, Klingenberg M (1994) The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J Biol Chem 269:1940–1944
Bogdanov M, Mileykovskaya E, Dowhan W (2008) Lipids in the assembly of membrane proteins and organization of protein supercomplexes: implications for lipid-linked disorders. Subcell Biochem 49:197–239
Jiang F, Ryan MT, Schlame M, Zhao M, Gu Z, Klingenberg M, Pfanner N, Greenberg ML (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem 275:22387–22394
Kiebish MA, Han X, Cheng H, Chuang JH, Seyfried TN (2008) Brain mitochondrial lipid abnormalities in mice susceptible to spontaneous gliomas. Lipids 43:951–959
Kiebish MA, Han X, Cheng H, Chuang JH, Seyfried TN (2008) Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the Warburg theory of cancer. J Lipid Res 49:2545–2556
Poli G, Leonarduzzi G, Biasi F, Chiarpotto E (2004) Oxidative stress and cell signalling. Curr Med Chem 11:1163–1182
Lee HC, Wei YH (2005) Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol 37:822–834
Almeida A, Delgado-Esteban M, Bolaños JP, Medina JM (2002) Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem 81:207–217
Santandreu FM, Brell M, Gene AH, Guevara R, Oliver J, Couce ME, Roca P (2008) Differences in mitochondrial function and antioxidant systems between regions of human glioma. Cell Physiol Biochem 22:757–768
Jelluma N, Yang X, Stokoe D, Evan GI, Dansen TB, Haas-Kogan DA (2006) Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol Cancer Res 4:319–330
Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G (2003) Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J 17:714–716
Petrosillo G, Di Venosa N, Ruggiero FM, Pistolese M, D’Agostino D, Tiravanti E, Fiore T, Paradies G (2005) Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochim Biophys Acta 1710:78–86
Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, Paradies G (2007) Mitochondrial dysfunction in rat with nonalcoholic fatty liver involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta 1767:1260–1267
Degterev A, Boyce M, Yuan J (2003) A decade of caspases. Oncogene 22:8543–8567
Klein S, McCormick F, Levitzki A (2005) Killing time for cancer cells. Nat Rev Cancer 5:573–580
Frank S, Köhler U, Schackert G, Schackert HK (1999) Expression of TRAIL and its receptors in human brain tumors. Biochem Biophys Res Commun 257:454–459
Kuijlen JM, Mooij JJ, Platteel I, Hoving EW, van der Graaf WT, Span MM, Hollema H, den Dunnen WF (2006) TRAIL-receptor expression is an independent prognostic factor for survival in patients with a primary glioblastoma multiforme. J Neurooncol 78:161–171
Hao C, Beguinot F, Condorelli G, Trencia A, Van Meir EG, Yong VW, Parney IF, Roa WH, Petruk KC (2001) Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res 61:1162–1170
Puduvalli VK, Sampath D, Bruner JM, Nangia J, Xu R, Kyritsis AP (2005) TRAIL-induced apoptosis in gliomas is enhanced by Akt-inhibition and is independent of JNK activation. Apoptosis 10:233–243
Panner A, James CD, Berger MS, Pieper RO (2005) mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multiforme cells. Mol Cell Biol 25:8809–8823
O’Neill J, Manion M, Schwartz P, Hockenbery DM (2004) Promises and challenges of targeting Bcl-2 anti-apoptotic proteins for cancer therapy. Biochim Biophys Acta 1705:43–51
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87:99–163
Zoratti M, Szabò I (1995) The mitochondrial permeability transition. Biochim Biophys Acta 1241:139–176
Reifenberger J, Ring GU, Gies U, Cobbers L, Oberstrass J, An HX, Niederacher D, Wechsler W, Reifenberger G (1996) Analysis of p53 mutation and epidermal growth factor receptor amplification in recurrent gliomas with malignant progression. J Neuropathol Exp Neurol 55:822–831
Gaiser T, Becker MR, Meyer J, Habel A, Siegelin MD (2009) p53-mediated inhibition of angiogenesis in diffuse low-grade astrocytomas. Neurochem Int 54:458–463
Weller M, Malipiero U, Aguzzi A, Reed JC, Fontana A (1995) Protooncogene bcl-2 gene transfer abrogates Fas/APO-1 antibody-mediated apoptosis of human malignant glioma cells and confers resistance to chemotherapeutic drugs and therapeutic irradiation. J Clin Invest 95:2633–2643
Krajewski S, Krajewska M, Ehrmann J, Sikorska M, Lach B, Chatten J, Reed JC (1997) Immunohistochemical analysis of Bcl-2, Bcl-X, Mcl-1, and Bax in tumors of central and peripheral nervous system origin. Am J Pathol 150:805–814
Nakasu S, Nakasu Y, Nioka H, Nakajima M, Handa J (1994) bcl-2 protein expression in tumors of the central nervous system. Acta Neuropathol 88:520–526
Strik H, Deininger M, Streffer J, Grote E, Wickboldt J, Dichgans J, Weller M, Meyermann R (1999) BCL-2 family protein expression in initial and recurrent glioblastomas: modulation by radiochemotherapy. J Neurol Neurosurg Psychiatry 67:763–768
Streffer JR, Rimner A, Rieger J, Naumann U, Rodemann HP, Weller M (2002) BCL-2 family proteins modulate radiosensitivity in human malignant glioma cells. J Neurooncol 56:43–49
Jiang Z, Zheng X, Rich KM (2003) Down-regulation of Bcl-2 and Bcl-xL expression with bispecific antisense treatment in glioblastoma cell lines induce cell death. J Neurochem 84:273–281
Zhu CJ, Li YB, Wong MC (2003) Expression of antisense bcl-2 cDNA abolishes tumorigenicity and enhances chemosensitivity of human malignant glioma cells. J Neurosci Res 74:60–66
Guensberg P, Wacheck V, Lucas T, Monia B, Pehamberger H, Eichler HG, Jansen B (2002) Bcl-xL antisense oligonucleotides chemosensitize human glioblastoma cells. Chemotherapy 48:189–195
Stegh AH, Kim H, Bachoo RM, Forloney KL, Zhang J, Schulze H, Park K, Hannon GJ, Yuan J, Louis DN, DePinho RA, Chin L (2007) BCL-2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev 21:98–111
Wagenknecht B, Glaser T, Naumann U, Kügler S, Isenmann S, Bähr M, Korneluk R, Liston P, Weller M (1999) Expression and biological activity of X-linked inhibitor of apoptosis (XIAP) in human malignant glioma. Cell Death Differ 6:370–376
Chen Z, Naito M, Hori S, Mashima T, Yamori T, Tsuruo T (1999) A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem Biophys Res Commun 264:847–854
Fulda S, Wick W, Weller M, Debatin KM (2002) Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med 8:808–815
Giagkousiklidis S, Vogler M, Westhoff MA, Kasperczyk H, Debatin KM, Fulda S (2005) Sensitization for gamma-irradiation-induced apoptosis by second mitochondria-derived activator of caspase. Cancer Res 65:10502–10513
Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG (2004) A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science 305:1471–1474
Letai A (2006) Growth factor withdrawal and apoptosis: the middle game. Mol Cell 21:728–730
Broniscer A, Gajjar A (2004) Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist 9:197–206
Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313:144–147
Fleming TP, Saxena A, Clark WC, Robertson JT, Oldfield EH, Aaronson SA, Ali IU (1992) Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res 52:4550–4553
Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, Nistér M (1992) Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 52:3213–3219
Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, Kieran MW, Luster AD, Segal RA (2003) A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A 100:13513–13518
Knobbe CB, Reifenberger G (2003) Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol 13:507–518
Knobbe CB, Trampe-Kieslich A, Reifenberger G (2005) Genetic alteration and expression of the phosphoinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropathol Appl Neurobiol 31:486–490
Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O’Fallon JR, Schaefer PL, Scheithauer BW, James CD, Buckner JC, Jenkins RB (2001) PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst 93:1246–1256
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87:99–163
Cartron PF, Gallenne T, Bougras G, Gautier F, Manero F, Vusio P, Meflah K, Vallette FM, Juin P (2004) The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell 16:807–818
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111:331–342
Breckenridge DG, Xue D (2004) Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol 16:647–652
Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X (2000) Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol 2:754–761
Lutter M, Perkins GA, Wang X (2001) The pro-apoptotic Bcl-2 family member tBid localizes to mitochondrial contact sites. BMC Cell Biol 2:22
Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, Chen J, Rabinowich H, Amoscato AA, Yin XM (2004) Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell 15:3061–3072
Garcia Fernandez M, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, Dobrucki J, Cossarizza A (2002) Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ 13:449–455
Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG (2005) Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1:223–232
Chelli B, Lena A, Vanacore R, Da Pozzo E, Costa B, Rossi L, Salvetti A, Scatena F, Ceruti S, Abbracchio MP, Gremigni V, Martini C (2004) Peripheral benzodiazepine receptor ligands: mitochondrial transmembrane potential depolarization and apoptosis induction in rat C6 glioma cells. Biochem Pharmacol 68:125–134
Miettinen H, Kononen J, Haapasalo H, Helén P, Sallinen P, Harjuntausta T, Helin H, Alho H (1995) Expression of peripheral-type benzodiazepine receptor and diazepam binding inhibitor in human astrocytomas: relationship to cell proliferation. Cancer Res 55:2691–2695
Lena A, Rechichi M, Salvetti A, Bartoli B, Vecchio D, Scarcelli V, Amoroso R, Benvenuti L, Gagliardi R, Gremigni V, Rossi L (2009) Drugs targeting the mitochondrial pore act as citotoxic and cytostatic agents in temozolomide-resistant glioma cells. J Transl Med 7:13
Moreno-Sánchez R, Rodríguez-Enríquez S, Saavedra E, Marín-Hernández A, Gallardo-Pérez JC (2009) The bioenergetics of cancer: is glycolysis the main ATP supplier in all tumor cells? Biofactors 35:209–225
Shaw RJ (2006) Glucose metabolism and cancer. Curr Opin Cell Biol 18:598–608
Cuezva JM, Ortega AD, Willers I, Sánchez-Cenizo L, Aldea M, Sánchez-Aragó M (2009) The tumor suppressor function of mitochondria: translation into the clinics. Biochim Biophys Acta 1792:1145–1158
Fulda S (2009) Tumor resistance to apoptosis. Int J Cancer 124:511–515
Kang MH, Reynolds CP (2009) Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 15:1126–1132
Berridge MV, Herst PM, Lawen A (2009) Targeting mitochondrial permeability in cancer drug development. Mol Nutr Food Res 53:76–86
Gogvadze V, Zhivotovsky B (2007) Alteration of mitochondrial function and cell sensitization to death. J Bioenerg Biomembr 39:23–30
Kiebish MA, Seyfried TN (2005) Absence of pathogenic mitochondrial DNA mutations in mouse brain tumors. BMC Cancer 5:102
Deighton RF, Mcgregor R, Kemp J, Mcculloch J, Whittle IR (2010) Glioma pathophysiology: insights emerging from proteomics. Brain Pathol (in press)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ordys, B.B., Launay, S., Deighton, R.F. et al. The Role of Mitochondria in Glioma Pathophysiology. Mol Neurobiol 42, 64–75 (2010). https://doi.org/10.1007/s12035-010-8133-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-010-8133-5