
Abstract
The methylotrophic yeast Pichia pastoris is garnering interest as a chassis cell factory for the manufacture of recombinant proteins because it effectively satisfies the requirements of both laboratory and industrial set up. The optimisation of P. pastoris cultivation is still necessary due to strain- and product-specific problems such as promoter strength, methanol utilisation type, and culturing conditions to realize the high yields of heterologous protein(s) of interest. Techniques integrating genetic and process engineering have been used to overcome these problems. Insight into the Pichia as an expression system utilizing MUT pathway and the development of methanol free systems are highlighted in this systematic review. Recent developments in the improved production of proteins in P. pastoris by (i) diverse genetic engineering such as codon optimization and gene dosage; (ii) cultivating tactics including co-expression of chaperones; (iii) advances in the use of the 2A peptide system, and (iv) CRISPR/Cas technologies are widely discussed. We believe that by combining these strategies, P. pastoris will become a formidable platform for the production of high value therapeutic proteins.
Graphical Abstract

Similar content being viewed by others

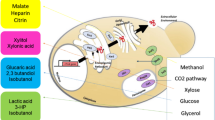

Avoid common mistakes on your manuscript.
Introduction
Introduced as methylotrophic yeast in the 1960s, Pichia pastoris now reassigned as Komagataella phaffii (K. phaffii) has been proven as a valuable expression system for a growing list of therapeutic proteins and industrial enzymes. This species was initially used for the production of single-cell protein as an animal feed additive, leveraging methanol as a carbon and energy source but the rising cost of oil made it economically unviable. Approximately ten years later Philips Petroleum developed P. pastoris as a heterologous gene expression system along with the Salt Institute Biotechnology/Industry Associates, Inc. (SIBIA) [1, 2]. Two decades earlier, this system was licensed to Invitrogen to be made available for researchers worldwide and The Food and Drug Administration (FDA) considered P. pastoris as Generally Recognised as Safe (GRAS) [3].
The publication of a high-quality genome sequence of CBS7435 (parental strain) in the year 2011 served as a breakthrough and the first strain developed for protein expression is GS115 [4]. High cell culture density, ease of genetic manipulations, ability to secrete recombinant proteins, protein processing, eukaryotic post-translational modifications, and stable genetic constructs [5,6,7] makes K. phaffii (often referred to as P. pastoris) a versatile and most favored eukaryotic expression system. It also lacks the well-known drawbacks present in bacterial expression systems such as the formation of inclusion bodies and the presence of endotoxins or mammalian systems such as high cultivation, handling costs, and reduced levels of protein expression [8]. The basic characteristics of different host systems are listed in Table 1. The most studied yeast expression system platform for recombinant protein production is the Baker’s yeast Saccharomyces cerevisiae owing to its advantages such as secretory expression, glycosylation, and eukaryotic post-translational modifications but the expressed proteins are often hyper glycosylated which in turn affects the protein immunogenicity [9]. The major advantage of P. pastoris gaining momentum over Saccharomyces cerevisiae as an expression platform is attaining high cell density culture due to the aerobic mode of respiration, expression of protein under tightly regulated methanol inducible AOX1 promoter, both the intracellular and extracellular expression of recombinant proteins by secretory expression (alpha mating factor—MATα) pathway, and metabolic engineering of yeast glycosylation pathway(Glycoengineered strains) to mimic the mammalian system [3, 10, 11]. Recombinant proteins can be produced in either a constitutive or induced manner depending on the type of promoter used and are secreted to the culture supernatant with less than 10% of endogenous proteins making it easier for various purification strategies in this host system [12].
Strong methanol inducible promoters from the alcohol oxidase genes (alcohol oxidase 1, AOX1, and to a lesser extent, alcohol oxidase 2, AOX2), as well as from the glyceraldehyde 3-phosphate genes, have helped P. pastoris succeed in producing recombinant proteins. Despite the effectiveness, strict control, and recombinant protein productivity attained when PAOX1 is used to drive transgene expression, this manufacturing system has some issues related to the use of methanol. Methanol usage results in high oxygen demand for catabolism because it is harmful to cells and causes oxidative stress [13]. Apart from this, recombinant protein production is hindered at two different levels, one is protein synthesis and the other is protein folding and secretion. Considerable attempts have been made to better understand the physiology and cell biology of P. pastoris [2, 14, 15], protein folding and intracellular trafficking [5, 16], metabolic engineering concerning glycosylation [17], and process monitoring [14] to increase recombinant protein production in recent years. However, it is important to emphasize and summarize the use of methanol-independent promoters as a substitute (a full list of vectors with various promoters that are commercially available are listed out under “Promoters” section), developments on strain engineering, and methods to overcome the challenges in the production of recombinant proteins with respect to copy number, codon optimization, culturing conditions, use of 2A peptide system, and CRISPR techniques and their contribution to Pichia’s effectiveness to produce recombinant proteins for commercial use.
Methodology
For this review, we conducted an extensive search of literature survey on various reliable databases such as PubMed, Google Scholar, Wiley Online Library, Elsevier, and Scopus. Full-length articles about the subject of interest—Bio therapeutics in P. pastoris expression system, published between 2010 and 2023, written in the English language were taken into consideration for this systematic review. The keywords used in various combinations to search the articles in the above-mentioned databases were K. phaffii, P. pastoris, recombinant proteins, CRISPR/Cas, 2A peptide system, promoters, and chaperones. Since a large amount of data is available online, certain inclusion and exclusion criteria were used to refine the screening process as described in Fig. 1. The inclusion criteria included proteins expressed for therapeutic applications using P. pastoris, investigation on various promoters, and strategies to overcome recombinant protein production. Conference abstracts, duplicated studies, and non-peer-reviewed articles were excluded from the study. 95 out of 200 articles were included for this study purpose.

Methodology for the search of literature
Pichia pastoris as a Lower Eukaryotic System
Expression in P. pastoris follows a distinct workflow different from the other expression systems such as bacteria and higher eukaryotic systems (Fig. 2). The target gene is cloned into the P. pastoris vectors such as pPICzαa or pPIC9K and tandem inserts of the gene of interest are also possible. The linearization of the construct gives rise to 5′ and 3′ regions of homology and upon transformation; it integrates into the genome of the appropriate strain used through homologous recombination [18]. Reports are suggesting that the integration of multiple copies enhances the expression levels of the recombinant protein [19]. This is followed by a screening of various constructs based on the integration event, antibiotics selection, and Mut phenotype (MutS/Mut+). This gives rise to a few highest expressers for scale up in a shake flask or fermenter very much depending on the scale of the study.

Schematic illustration of the distinct workflow for the expression of recombinant proteins in the lower eukaryotic organism P. pastoris. Linearized DNA (pPIC9KVector with the gene of interest—GOI) (a) after confirmation through sequencing is transformed into electrocompetent GS115 cells and gets integrated via homologous recombination, (b) The transformed colonies are selected using cell auxotrophy, (c) and then grown on a 96 well plate supplemented with yeast-peptone-dextrose broth, (d), further selected based on increasing concentrations of antibiotics (Geneticin), (e) Antibiotic selection is followed by determining Mut phenotype (on a scoring template) and integration into the host genome (gene-specific and AOX-specific PCR), (f) Small-scale expression studies are carried out in 96 well plates, (g) and in 10–30 ml culture volume in shake flasks, (h) Upon analysis of the protein of interest by SDS PAGE electrophoresis, (i) and functional assays, (j) the best expression clone is identified and taken further for bioreactors
MUT Pathway
The understanding of the MUT is essential when it comes to the expression of recombinant proteins in P. pastoris as it metabolizes methanol as its carbon and energy source. This methanol metabolism pathway takes place both in the peroxisome and cytosol of yeast. Briefly, the alcohol oxidase enzyme involves in the oxidation of methanol into formaldehyde and H2O2. Under the assimilatory pathway, the resulting formaldehyde is processed into dihydroxyacetone (DHA) and glyceraldehyde 3 phosphate (GAP) by the peroxisomal dihydroxyacetone synthase (DAS) in peroxisome and ends in the cytosol with the generation of fructose 1,6-biphosphate whereas H2O2 breaks down into water and oxygen (activity of catalase). Under the dissimilatory pathway, the formaldehyde is oxidized to carbon dioxide by the enzymatic route involving formaldehyde dehydrogenase (FLD), formyl glutathione hydrolase (FGH), and formate dehydrogenase (FDH) with the release of NADH (Fig. 3) [20, 21]. The widely used strain for heterologous protein production is GS115 and it carries two alcohol oxidase genes AOX1 and AOX2. The majority of alcohol oxidase is produced by the AOX1 gene and comprises about 30% of the total proteins produced in yeast. Knocking down of the AOX1 gene results in reduced or slow growth of cells on methanol and this particular phenotype is referred to as MutS roughly translating to methanol utilization (MUT) slow and the presence of AOX1 and AOX2 intact results in the fast growth of cells on methanol and this particular phenotype is referred as Mut+ (methanol utilizing plus). When both the genes are knocked down, the strains won’t be able to grow on methanol and are referred to as Mut− (methanol utilization minus). These phenotypes give rise to the strain diversity in P. pastoris.

Methanol utilization pathway in methylotrophic yeasts
Promoters
For the expression of recombinant proteins, much importance is given to the design of the expression system which comprises promoters, selection markers, signal sequences, and host strains. Promoters, generally located upstream of a gene or at the 5′ end of the transcription initiation site are the most important part of the recombinant protein expression and it is classified into two groups. They are tightly regulated inducible promoters and unregulated constitutive promoters not affected by environmental or other chemical factors. Different promoters under inducible and constitutive conditions are listed in Fig. 4.

Constitutive and inducible promoter systems available in P. pastoris for the expression of recombinant proteins (accession codes—7MB0, 3VM5 shown as an example)
Inducible Promoters
An inducible promoter is a regulated promoter that expresses its associated genes only in response to specific chemical and physical factors such as alcohol, steroid, antibiotics, oxygen level, temperature. Alcohol Oxidase (AOX1), a ~ 960 bp fragment corresponding to the alcohol oxidase I gene regulates the metabolism of methanol and is a widely used promoter in the expression of recombinant proteins in P. pastoris. It is one of the major components in the MUT pathway and uses methanol as a sole carbon source for the induction of expression of recombinant proteins. This promoter is strongly repressed during the growth phase of Pichia pastors in glucose or glycerol and upon depletion of these sources, depression of the promoter takes place but is fully induced upon the addition of methanol. One out of many proteins produced using this promoter is Thermus thermophilus’s glucose isomerase (xylA) with a yield of 137 U/g DCW [22]. Dihydroxyacetone synthase (DAS) is similar to the AOX1 promoter and relies on methanol for carbon and energy source. This promoter is also part of the MUT pathway [23, 24].
Glutathione-dependent Formaldehyde Dehydrogenase 1 (FLD1), a ~ 1200 bp fragment was found to be an alternative for AOX1 and also as a marker for the selection of multi-copy expression strains [3]. It uses methanol as a carbon source (with ammonium sulfate as a nitrogen source) and methylamine as a nitrogen source (with glucose as a carbon source), an inexpensive nontoxic nitrogen source for the induction of protein expression. Isocitrate lyase ICL1, a ~ 1563 bp fragment found to have a 64% identity between P. pastoris Icl and Saccharomyces cerevisiae Icl. The promoter is repressed by glucose and induced in the absence of glucose or by the addition of ethanol [25]. Dextranase gene (dexA) from Penicillium minioluteum under the control of the ICL1 promoter in P. pastoris is considered a good alternative for the expression of heterologous proteins [26].
Putative Sodium Coupled Phosphate Symporter (PHO89), a ~ 1044 bp fragment exhibited stronger transcriptional activity with higher specific productivity. Expression of a bacterial lipase gene upon phosphate starvation in different modes of fermentation was shown to be regulated using the PHO89 promoter [25]. Thiamine Biosynthesis (THI11), a ~ 1000 bp fragment gene encodes a protein involved in the synthesis of thiamine precursor, and this promoter in P. pastoris is repressed by thiamin. Fed-batch cultivation of human serum albumin under this promoter showed high transcript levels at a low specific growth rate and also interesting regulatory properties on the availability of thiamine in a growth medium [27].
Alcohol Dehydrogenase (ADH1), a ~ 1000 bp fragment encoding the alcohol dehydrogenase gene is required for the reduction of acetaldehyde to ethanol. This particular promoter is repressed on glucose and methanol while induced on glycerol ad ethanol for the production of recombinant proteins in P. pastoris. Five different synthetic promoters derived from ADH1 by addition and deletion of regulatory sites within the promoter were found to increase the product range of extracellular xylanase between 165 and 200% when compared to the native promoter [28]. Glycerol Kinase (GUT1) is similar to ADH1 and gets repressed on methanol and induced on glucose, glycerol, and ethanol. Enolase (ENO1) gets repressed on glucose, methanol, and ethanol and induced on glycerol. These three promoters cannot be compared with widely used AOX1 due to the lack of absolute expression values. Peroxisomal Matrix (PEX8), a moderately expressing promoter directs the peroxisomal matrix protein formation which is essential for peroxisomal biogenesis.
Constitutive Promoters
A constitutive promoter is an unregulated promoter that allows the transcription of its associated genes continuously and is not affected by environmental or developmental factors. Glyceraldehyde-3-P Dehydrogenase (GAP), a 477 bp fragment is a key enzyme in the glycolysis pathway. Like AOX1 in inducible promoters, this is widely used for constitutive expression of recombinant proteins using glucose and to a lesser extent glycerol [29]. Since methanol is not used for induction, this promoter makes it straightforward without any carbon source change during the growth and induction of various heterologous proteins [30]. Rab Family GTPase Yeast Protein (YPT1), a 618 bp fragment encodes GTPase essential in secretion and is involved in moderate expression levels of recombinant proteins. This provides a low but constitutive expression of glucose, methanol, or mannitol as a carbon source.
Phosphoglycerate Kinase (PGK1), a 1251 bp gene shows high identity to homologous proteins from other yeasts and codes for the protein 3-phosphoglycerate kinase. This promoter is shown to be a potential alternative for the constitutive expression of glucose and less on glycerol and methanol [31]. Sorbitol Dehydrogenase (SDH), a 211 bp fragment is an important enzyme in carbohydrate metabolism. This promoter was evaluated for expression of two heterologous proteins, human serum albumin and erythrina trypsin inhibitor under repressive as well as non-repressive carbon sources [32].
Translation Elongation Factor (TEF1), a gene with an open reading frame of 1380 bp with the potential to encode 450 amino acids was tested in comparison to the well-studied GAP promoter. It is found to have a strong promoter activity in high glucose and carbon-limited conditions and produces recombinant heterologous proteins at levels similar to or greater than the GAP promoter [33]. Glycosyl Phosphatidyl inositol (GPI)-anchored protein (GCW14), an 822 bp promoter region was identified and characterized the regulatory sequences involved in this particular promoter and showed that it enables stronger expression than GAP and TEF1 when enhanced green fluorescent protein was used as a reporter constitutively [34].
Glucose (G1) and (G6), genes encoding for high-affinity glucose transporter and putative aldehyde dehydrogenase were identified as novel regulated promoters that do not use methanol as an inducer. These promoters get repressed on glycerol and induced upon glucose limitation. G1 was well suited for the protein production processes when compared to G6 and Gap promoter [25].
To address the limited number of promoters available, two distinct methodologies were used to identify novel promoters that aid in recombinant protein expression. The first strategy, heterologous microarray hybridization, was used to hybridize Pichia cDNA with S. cerevisiae cDNA microarrays after it was isolated being grown on separate carbon sources at two distinct pH values. The transcriptome data mining revealed 15 genes with high expression levels, indicating the presence of strong promoters. The second technique, the logical selection of promoters from different yeasts based on literature mining, resulted in the identification of nine probable strong promoter sequences. 80% of the promoter sequences discovered from the transcriptome data demonstrated their promoter activity on all carbon sources typically used for P. pastoris growth. A total of 24 potential promoter sequences were examined for their promoter activities for both intracellular and extracellular protein expression. The promoter sequences that were rationally chosen had a very low success rate. Many of these discovered promoters, including two chaperones (HSP82 and KAR2) and three ribosomal (RPL1, RPS2, and RPS31) promoters, exhibited behavior that was growth dependent [3]. The Lonza XS Pichia 2.0 glucose-regulated promoter system has also been engineered to circumvent the restrictions associated with the toxic effects of methanol, which can affect purity and productivity at high growth rates [35]. Based on these findings, a broad toolbox for promoter engineering in P. pastoris may be set up based on the rational design and a better understanding of the target promoter control mechanisms.
Vectors and Selection Markers
Vectors otherwise referred to as plasmids used in recombinant DNA technology usually possess an origin of replication, a multi-cloning site, and a selectable marker. They do have a bi-functional system that lets them propagate or replicate in E. coli and express the gene of interest in P. pastoris. They are characterized by the presence of selection markers such as auxotrophic markers or genes conferring resistance. The auxotrophic markers include HIS4, ARG4, ADE1, URA3, URA5, GUT1, and ADE2 to name a few while the antibiotic-resistant genes include Zeocin, Geneticin, Kanamycin, Blasticidin S, Hygromycin and so on. The different vectors used in the production of recombinant proteins both intracellular and extracellular with their characteristics and features using P. pastoris are listed in Table 2. Apart from the vectors presented in the table, academic researchers have been successful in modifying the commercially available vectors (with prior permission from the respective supplier) with different promoter sequences mentioned in “Promoters” section of this review.
Expression Strain Development
The methylotrophic yeast strains extensively used for the production of recombinant proteins were all derived from NRRL-Y 11430 strain. They are briefly classified into auxotrophic strains, protease deficient strains, and glycoengineered strains based on the mutations on aox1,his4, arg4, pep4, prb1, etc. The most frequently used strains for heterologous protein productions are auxotrophic strain GS115 (his4), prototrophic strain X33(WT), aox knockout strains KM71 (his4 arg4 aox1::ARG4), KM71H (arg4 aox1::ARG4). The presence of these genes in the vector also facilitates a better screening protocol in place. The strain diversity also includes protease-deficient strains such as SMD1163 (his4 pep4 prb1) SMD1168 (his4 pep4::URA3 ura3) and SMD1168H (pep4) and ade2 auxotrophic PichiaPink™ strain. The strains derived from P. pastoris CBS7435 are not covered by any patent protection and are widely used for commercial purposes and a huge number of different strains are developed according to the need of the researcher. One such example is CBS7435 MutS strain provided by the Graz Pichia pool being a marker-free as it was developed using Flp/FRT recombinase system technology. Apart from these strains, glycoengineered strains using GlycoSwitch® technology were made available for facilitating homogenous, humanized glycosylation patterns in recombinant proteins. A detailed review of the host strains is available online on the topic of Protein expression in P. pastoris by Ahmed et al. [26]. The engineering of the CBS7435 strain to produce cholesterol instead of ergosterol was studied to functionally express human membrane proteins [36]. A recent study on 45 different strains of P. pastoris suggests that cumulative oxygen transfer might be used as a screening criterion to facilitate the pre-selection of high-producing strains [37]. The genetic toolsets required to generate P. pastoris strains secreting recombinant proteins have advanced dramatically in recent decades. As a result, the bottleneck in bioprocess development is no longer the creation of strains, but the identification of high-performing clones. Recently a microscale cultivation strategy has been shown to increase the efficiency of high throughput screening of recombinant clones by optimizing the architecture of 96 deep-well plates, shaking throw diameter, shaking frequency, culture volume/well, and media composition [38]. Through fluorescence-activated cell sorting, a switchable secrete and capture technology has demonstrated the ability to efficiently isolate high-producer clones of Fab fragments from millions of cells [39]. In a rather new study, the Mattanovich team created an autotroph P. pastoris strain that can grow on CO2. The peroxisomal methanol-assimilation route of Pichia was engineered into a CO2-fixation pathway reminiscent of the Calvin-Benson-Bassham cycle by the addition of eight heterologous genes and the deletion of three native genes; the resulting strain demonstrated the ability to grow continuously with CO2 as a unique carbon source [40]. Ito et al. showed that a combination of genome-wide screening of effective factors for gene disruption, their accumulation in one strain, and an Adaptive laboratory evolution(ALE) for recovering the reduced cell growth of gene-knockout strains is an effective strategy for enhancing the secretion of heterologous proteins by the unconventional yeast P. pastoris [41]. These new techniques, in conjunction with the well-studied host strains, may offer a time-effective strategy for primary screening, allowing for the accelerated selection of high-producing P. pastoris strains.
Strategies of Recombinant Protein Production and Its Challenges
The well-studied and easy-to-use P. pastoris expression has its challenges and some degree of optimization is essential for better production of recombinant proteins. It is evident from the vast literature that the expression of proteins in Pichia is highly target specific and various strategies are applied for significant achievements.
Gene Codon Optimization and Copy Number
Codon optimization has become a go-to tool for improved expression of a gene in P. pastoris and the usual strategy is replacing rare codons to match the codon usage bias [42,43,44]. Numerous studies suggest a substantial shift in the understanding of the function of codon bias and how it affects genes in native and expression hosts [45]. A version of α amylase (codon optimized) from Bacillus showed significantly higher expression levels and showed an activity of 8100/ml (2.31 fold higher) compared to the wild-type version of the same gene [43]. A new tool is developed called CPO (Codon Pair Optimisation) to provide a simple and efficient codon pair optimization for synthetic gene design in P. pastoris. They show that gene design based on codon pair bias significantly improved the protein expression levels and might be an alternate strategy to codon usage bias [46]. Karaoglan and Erden-Karaoglan proposed a model for the expression of the A.niger protein endo-polygalacturonase (Pgl), whose sequence was subjected to codon optimization, evaluating its performance under the control of two promoters (PAOX and PADH2). The best productivity was obtained in shaking flasks using the codon-optimized PGL employing the PADH2, with a productivity of 42.33 U/mL (fourfold increase) [47].
Gene copy number analysis has been carried out for the past 20 years and a direct correlation between the copy number and expression levels has been shown for intracellular expression [19, 48]. But the direct correlation is not necessarily valid when it comes to secretions of that particular protein. Multiple copy integration through a single crossover event accounts for only 1% of all transformants from a single transformation and screening of this is labor intensive and is possible through antibiotic selection and not by auxotrophic markers. A recent study suggests that human DNA topoisomerase I was successfully expressed with single and multicopy inserts (via in vivo strategy using pPIC3.5K) and the multicopy transformant was found to express the highest expression levels of total protein and also exhibited the highest enzyme activity [48]. But this was possible with the use of an intracellular expression vector system and the same is not apparent for secretory proteins. Reports are suggesting that multicopy integration also leads to genetic instability by excision of the integrated gene through the loop-out method owing to the highly recombinogenic nature of P. pastoris. Additionally, a spike in the number of copies of foreign genes may change Pichia's regular metabolism, which would have a detrimental effect on the multiple-copy recombinant yeast's regular cell physiology. This calls for testing transformants with increasing gene copy numbers to determine the ideal gene copy number for maximal protein synthesis. Extensive research is needed to use the multi-copy hypothesis for the production of secretory proteins which is indeed an essential trait for the ease of downstream processing.
Culturing Conditions
Various optimization strategies including media composition and culture conditions are in place to produce recombinant proteins in P. pastoris. Essentially all P. pastoris strains grow on a defined medium with supplements following the phenotype of the strains. The wild-type strain X33 grows on minimal media whereas the widely used GS115 grows only on minimal media supplemented with histidine. However, Yeast-Peptone-Dextrose (YPD), Buffered Glycerol complex media (BMGY), Buffered methanol complex media (BMMY), and Basal salts media are extensively used for screening, expression, and fermentation studies. To attain high cell density growth and better expression of heterologous proteins, the medium consists of biotin and ammonium hydroxide as the nitrogen source, glycerol, and methanol as the carbon and energy source along with basal salt medium(BSM), and trace elements such as zinc chloride, ferrous sulfate heptahydrate [38]. Methanol as an inducer for the expression of recombinant proteins is the principal carbon source and monitoring of methanol percentage during biomass production and induction is crucial for better optimization of productivity and toxicity management. The utilization of methanol very much depends on the Mut form of the strains used (Mut+, MutS, Mut−). An immense literature survey points out that the minimum concentration of methanol for the induction of heterologous proteins is 0.5% and the maximum is 2–2.5% for the production of fully expressed proteins. Any concentration of about 5% is considered toxic and results in the accumulation of formaldehyde and hydrogen peroxide and indeed death of the cells [10]. An extensively used alternative for glycerol with methanol is sorbitol and unlike glycerol, it does not induce or repress AOX promoters hence using sorbitol could reduce cell growth rate and increase recombinant protein production. It also can reduce intermediate metabolites, decrease toxicity production, and positively affect cell growth and energy supply for recombinant protein production [49,50,51,52]. In a different investigation, the production of three heterologous cellulases—an exoglucanase from Trichoderma reesei, β-glucosidase, and endoglucanase enzymes from Aspergillus niger—enabled the resulting strain to thrive on cellobiose and carboxymethyl cellulose [53].
P. pastoris is usually grown in an optimum temperature range of 28–30 °C and growth above 32 °C is considered detrimental to protein production. A reduction in temperature from 30 to 20 °C during induction has shown a tremendous change in protein production due to decreased folding stress and lower proteolytic activity. Two more critical factors in the production of heterologous proteins in P. pastoris are the incubation time and agitation speed. Production time is long in Pichia close to 100 h and the most preferred time point is 72–96 h of incubation time [54, 55]. In the shake flask culture, the agitation speed maintained is usually between 250 and 300 rpm and results in better aeration with high productivity titers.
Because the generation of high-titer recombinant proteins is heavily dependent on the target protein, incorporating cell engineering methodologies, as well as co-substrate feeding and auxiliary carbon sources, may provide a new approach for P. pastoris culture strategies. Some of the recently produced recombinant proteins expressed in P. pastoris are listed in Table 3 with their different culturing conditions.
Protein Synthesis and Its Secretion
With the help of codon optimization and various culturing conditions, a huge number of proteins can be produced with this expression system at high levels whereas complex, multimeric human proteins pose a great challenge. The expression of complex proteins often leads to the overproduction of misfolded proteins thereby triggering cellular stress in the host cells. The recombinant proteins intended for secretion undergo translocation into the endoplasmic reticulum lumen as a nascent peptide. This nascent peptide undergoes folding, disulfide bond formation, and other post-translational modifications which are unique to the expressed recombinant protein. Only the properly folded proteins are allowed to exit ER to the Golgi apparatus where post-translational modifications such as glycosylation occur.
Some recombinant proteins might fail to undergo proper post-translational modification for unknown reasons and that is when unfolded protein response (UPR) pathway and ER-associated degradation (ERAD) pathway come into play [65]. Overexpression of chaperones or helper proteins such as BiP/Kar2p [64, 66, 67], Pdi1 [44], Ero1p, and a transcriptional regulator HAC1 was found to be an alternate strategy to overcome secretory bottlenecks. A crucial technique for raising the yield of many recombinant proteins is the overexpression of the transcription factor Hac1. Huang et al. investigated the effect of HAC1p on a raw starch hydrolyzing-amylase (Gs4j-amyA) to improve the heterologous synthesis of the enzyme in P. pastoris. They also looked at the promoter and copy number variations used for HAC1 overexpression. In this case, a strain with basal expression of 305 U/mL and 12 copies of the GS4J-AMYA gene driven by PAOX1 was used. Amylase activity rose to 2200 U/mL upon the inclusion of six copies of PAOX1-driven HAC1 [68, 69]. Yu et al. reported the distinct impacts of the co-overexpression of nine proteins under PAOX1 regulation on PGAP-driven k-carrageenase production, including seven chaperones (Pdi: protein disulfide isomerase; Ire1: endoplasmic reticulum stress transducer; ero1: endoplasmic reticulum oxidoreductase; Kar2: immunoglobulin-binding protein; Aha1: activator of Hsp90 ATPase; Ypt6; GTPase; Prx1: thioredoxin-linked peroxidase) and two transcription factors Yap1 and Rpn4 (proteasome subunit transcription factors) [70, 71].
The production of transcription factors like Yap1 and Hac1 is another method that enhances protein folding and secretion. Although it was anticipated that their expression in combination with other folding facilitator proteins like Pdi1 and Kar2 would increase the production of the target recombinant protein, research by Sun et al. and Duan et al. revealed that the productivity of the recombinant protein was either maintained or decreased [72, 73]. As a result, many researchers have implemented multiple modifications at the same time, combining the co-expression of chaperones and/or foldases with other genetic manipulation tools such as optimization of codon usage, gene copy number, co-expression/modulation of transcription factors, and variation in culture conditions to improve heterologous protein productivity [47, 66, 67, 74, 75]. These results show that no combinatorial approach offers equivalent benefits for the secretion of all recombinant proteins, and all the above-mentioned parameters need to be tested individually for the specific protein of interest.
2A Peptide System
Co-expression of several genes in eukaryotic cells can be accomplished by introducing separate plasmids into the host cell, utilizing a plasmid with multiple promoters, creating proteolytic sites, internal ribosome entry sites (IRES), or self-processing 2A sequences between genes. 2A sequences are short 18–20 aminoacid peptides derived primarily from viral polyproteins that enable a ribosome-skipping event that allows numerous distinct proteins to be synthesized from a single open reading frame. When used in metabolic engineering and synthetic gene circuits, 2A peptides enable co-regulated and consistent expression of several genes in eukaryotic cells. The core sequence motif of a 2A peptide is DXEXNPG↓P (where X refers to any amino acid and ↓ refers to the site of cleavage between the C terminal glycine of 2A and N terminal proline of the downstream protein) [76]. The 2A peptides extensively used for the co-expression of multiple proteins are P2A (porcine teschovirus-1), F2A (foot and mouth disease virus 18), E2A (equine rhinitis A virus), and T2A (thosea asigna virus). Out of these 4 2A peptide sequences, the most widely used are the P2A system [77]. Recently, the F2A system was applied to construct a polycistronic system to produce fusaruside by co-expressing biosynthetic genes in engineered P. pastoris due to its capacity for simultaneous and efficient expression of multiple genes. The yield obtained using this system after 120 h of induction was 5× higher than that of the yield obtained after 10 days of induction [78].
By using a P2A peptide sequence instead of a 3CD (viral protease precursor protein) to terminate translation between individual capsid proteins and comparing this to protease-dependent production of enterovirus virus-like particle (VLP), Sherry et al. investigated the potential for protease-independent production VLPs in P. pastoris. They demonstrated that one of the VLPs tagged with P2A maintained their native antigenicity and are thermostable and also corroborate with VLPs produced in other expression systems [79]. Another study on producing thermophilic cellulases using a polycistronic construct established successful transcription of the genes and recombinant proteins were detected by enzymatic assay and fluorescent microscopy [80]. To the best of our knowledge, the construct with the most genes expressed in a coordinated manner is the work by Geier et al., in which nine genes were successfully expressed from a single polycistronic transcript via T2A peptides in the yeast P. pastoris [81]. Self-cleaving 2A peptides enable the formation of polycistronic sequences for gene co-expression in eukaryotes, making them the finest genetic tool for reducing the number of transcription units in pathways design. With a large decrease in the number of promoters and terminators participating in the synthetic network, it may be possible to incorporate an entire pathway or a full synthetic gene circuit into a single transcription unit [77]. Although there haven’t been many reports of 2A-based multicistronic vectors up to now due to the lack of a complete understanding and characterization of how 2A peptides function in various expression systems and especially P. pastoris, there has been a resurgence of interest in this particular DNA sequence in recent years. This strategy might be employed to engineer P. pastoris in the synchronous production of recombinant proteins.
CRISPR/Cas Techniques
The effectiveness and precision of genome editing have improved recently with the development of CRISPR/Cas technology (Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9). Many advances have been made by utilizing the advantages of CRISPR/Cas in the bioengineering of non-conventional yeasts such as P. pastoris. To achieve effective and accurate genome editing in P. pastoris, Weninger et al. systematically improved the CRISPR/Cas9 expression system. This included but was not limited to, Cas9 coding sequences, gRNA sequences, gRNA structures (such as those with ribozyme sequences), and promoters for the expression of Cas9 and gRNAs. Only 6 constructs out of 95 combinations were discovered to be functional for genome editing, demonstrating the need for additional optimization [82]. Multiplex integration of heterologous genes is another crucial synthetic biology approach for creating P. pastoris as cell factories for heterologous proteins in addition to gene disruption. Later research improved the system's targeted integration of donor DNAs by introducing two simultaneous double-strand breaks, followed by donor DNA replacement in the intermittent region. This is especially true in the P. pastoris ku70 knockout strain, where the homologous recombination DNA repair process is enhanced by lowering non-homologous end joining (NHEJ), whereas broken DNA strand repair by NHEJ predominates in wildtype strains [83, 84]. This has recently been aided by the improved P. pastoris genome sequence, which includes over 500 corrected locations, corrected annotations, and comparative analysis of the most commonly used P. pastoris strain genomes and transcriptomes [85,86,87]. For double- and triple-locus co-integration, three high-efficiency gRNA targets (PAOX1UP, PTEF1UP, and PFLD1UP) were chosen. These were integrated simultaneously in a KU70-deficient strain of P. pastoris, with integration efficiencies ranging from 57.7 to 70% and 12.5 to 32.1% for double- and triple-loci, respectively [88].
For precise genomic changes in P. pastoris and ease of use, Gassler et al. have created a CRISPR/Cas9-based kit for gene insertions, deletions, and substitutions. The CRISPi kit from Addgene is a ready-to-use plasmid kit based on Golden PiCS modular cloning for CRISPR/Cas9 mediated genome editing in P. pastoris [89]. Another recent study reported multiplexed genome integration of three glycosylation-related genes (gnt1, mns1, and mnn2) using orthogonal tRNA-sgRNA cassettes expressed via the tRNA promoter. Using this method, rapid, multiplexed engineering of a complicated phenotype was feasible, resulting in humanized product glycosylation in two successive engineering phases [90]. These methods can now be quickly applied to a wide range of applications, such as the introduction of mammalian chaperones to improve the folding of complex molecules, the creation of new protease-deficient strains to increase the yields of full-length products, the reduction and redirection of vacuolar and endoplasmic reticulum-associated protein degradation pathways, the enhancement of lipid synthesis and vesicular machinery [91,92,93,94]. This approach enables quick, marker-free genome engineering in P. pastoris, allowing for novel strain and metabolic engineering applications.
Conclusion
Pichia pastoris has established itself as a successful platform for producing biotechnologically and commercially appealing products, owing to its ability to produce a wide range of functionally active heterologous proteins, ranging from microbial proteins to complex eukaryotic proteins with numerous applications. As each protein requires the optimization of different parameters, we have elaborated on different strategies employed in the production of recombinant proteins as this process is sometimes based on trial and error experiments. Despite AOX1 being the widely used promoters, on-going study on the availability of methanol independent promoters might pave way in circumventing the disadvantages of using methanol. On the other hand, considerable discussion is given regarding the role of molecular and chemical chaperones for a better understanding of the production and secretion of proteins, as well as how these chaperones assist in reducing the degradation of proteins. Since most of the reviews talk about metabolic engineering, we focused on genetic engineering approaches such as CRISPR/Cas technology and 2A peptide system which has been a recent trend and not much has been reported. We have extensively reviewed research articles pertaining to the above said techniques in the last 3–4 years and carefully analyzed how they can be used as one of the strategies to improve protein production in the versatile cell factory P. pastoris. We believe that the moment and technology are likely more appropriate than ever to have a significant impact on bio manufacturing in the upcoming decade due to the growing demand for cost-effective recombinant therapeutic protein production.
Data availability
Not applicable.
Abbreviations
- ADH:
-
Alcohol dehydrogenase
- ALE:
-
Adaptive laboratory evolution
- AOX1, AOX2:
-
Alcohol Oxidase 1 and 2
- BMGY:
-
Buffered glycerol complex media
- BMMY:
-
Buffered methanol complex media
- CPO:
-
Codon pair optimization
- CRISPR/Cas9:
-
Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9
- CUP1:
-
Metallothionein protein 1
- DAK:
-
Dihydroxyacetone kinase
- DAS:
-
Dihydroxyacetone synthase
- DHA:
-
Dihydroxyacetone
- DHAP:
-
Dihydroxyacetone phosphate
- ENO1:
-
Enolase 1
- ER:
-
Endoplasmic reticulum
- ERAD:
-
ER-associated degradation
- F1,6BP:
-
Fructose 1,6 bisphosphate
- F6P:
-
Fructose 6 phosphate
- FBA1,2:
-
Fructose bisphosphate aldolase
- FBP:
-
Fructose 1,6-bisphosphatase
- FDA:
-
Food and Drug Administration
- FDH:
-
Formate dehydrogenase
- FGH:
-
Formyl glutathione hydrolase
- FLD:
-
Formaldehyde dehydrogenase
- Flp/FRT:
-
Flippase/Flippase recognition target
- G1, G6:
-
Glucose 1 and 6
- GAP:
-
Glyceradehyde 3 phosphate
- GCW14:
-
Glycosyl Phosphatidyl inositol anchored protein
- GOI:
-
Gene of Interest
- GRAS:
-
Generally Recognised as Safe
- gRNA:
-
Guide RNA
- GSH:
-
Glutathione
- GUT1:
-
Glycerol Kinase 1
- HAC1:
-
Homologous to ATF/CREB 1
- HIS4:
-
Histidine 4
- HSP82:
-
Heat shock protein 82
- ICL1:
-
Isocitrate Lyase
- IRES:
-
Internal ribosome entry sites
- KAR2:
-
Karyogamy 2
- MATα:
-
Alpha mating factor
- NADH:
-
Nicotinamide adenine dinucleotide hydrogen
- NHEJ:
-
Nonhomologous end joining
- PEX8:
-
Peroxisomal matrix 8
- PGK1:
-
Phosphoglycerate kinase
- PH089:
-
Putative Sodium Coupled Phosphate Symporter
- PHO1:
-
Phosphate transporter 1
- PYR:
-
Pyruvate
- RCT:
-
Research Corporation Technologies
- SDH:
-
Sorbitol dehydrogenase
- SDS PAGE:
-
Sodium dodecyl-sulfate polyacrylamide gel electrophoresis
- SIBIA:
-
Salt Institute Biotechnology/Industry Associates, Inc
- TCA:
-
Tricaboxylic cycle
- TEF1:
-
Translational elongation factor 1
- THI11:
-
Thiamine Biosynthesis
- TPI:
-
Triosephosphate isomerase
- TT:
-
Transcription termination
- UPP:
-
Uracil phosphoribosyltransferase
- UPR:
-
Unfolded protein response
- VLP:
-
Viral like particles
- YPT1:
-
Rab Family GTPase yeast protein
References
Barone, G. D., Emmerstorfer-Augustin, A., Biundo, A., Pisano, I., Coccetti, P., Mapelli, V., & Camattari, A. (2023). Industrial production of proteins with Pichia pastoris—Komagataella phaffii. Biomolecules, 13(3), 441. https://doi.org/10.3390/biom13030441
Bernauer, L., Radkohl, A., Lehmayer, L. G. K., & Emmerstorfer-Augustin, A. (2021). Komagataella phaffii as emerging model organism in fundamental research. Frontiers in Microbiology, 11(January), 1–16. https://doi.org/10.3389/fmicb.2020.607028
Juturu, V., & Wu, J. C. (2018). Heterologous protein expression in Pichia pastoris: Latest research progress and applications. ChemBioChem, 19(1), 7–21. https://doi.org/10.1002/cbic.201700460
Küberl, A., Schneider, J., Thallinger, G. G., Anderl, I., Wibberg, D., Hajek, T., Jaenicke, S., Brinkrolf, K., Goesmann, A., Szczepanowski, R., Pühler, A., Schwab, H., Glieder, A., & Pichler, H. (2011). High-quality genome sequence of Pichia pastoris CBS7435. Journal of Biotechnology, 154(4), 312–320. https://doi.org/10.1016/J.JBIOTEC.2011.04.014
Raschmanová, H., Weninger, A., Knejzlík, Z., Melzoch, K., & Kovar, K. (2021). Engineering of the unfolded protein response pathway in Pichia pastoris: Enhancing production of secreted recombinant proteins. Applied Microbiology and Biotechnology, 105(11), 4397–4414. https://doi.org/10.1007/s00253-021-11336-5
Ata, Ö., Ergün, B. G., Fickers, P., Heistinger, L., Mattanovich, Di., Rebnegger, C., & Gasser, B. (2021). What makes Komagataella phaffii non-conventional? FEMS Yeast Research, 21(8), 1–15. https://doi.org/10.1093/femsyr/foab059
Heistinger, L., Gasser, B., & Mattanovich, D. (2020). Microbe profile: Komagataella phaffii: A methanol devouring biotech yeast formerly known as Pichia pastoris. Microbiology (United Kingdom), 166(7), 614–616. https://doi.org/10.1099/mic.0.000958
O’Flaherty, R., Bergin, A., Flampouri, E., Mota, L. M., Obaidi, I., Quigley, A., Xie, Y., & Butler, M. (2020). Mammalian cell culture for production of recombinant proteins: A review of the critical steps in their biomanufacturing. Biotechnology Advances, 43(May), 107552. https://doi.org/10.1016/j.biotechadv.2020.107552
Tran, A. M., Nguyen, T. T., Nguyen, C. T., Huynh-Thi, X. M., Nguyen, C. T., Trinh, M. T., Tran, L.-T., Cartwright, S. P., Bill, R. M., & Tran-Van, H. (2017). Pichia pastoris versus Saccharomyces cerevisiae: A case study on the recombinant production of human granulocyte-macrophage colony-stimulating factor. BMC Research Notes, 10(1), 6–13. https://doi.org/10.1186/s13104-017-2471-6
Karbalaei, M., Rezaee, S. A., & Farsiani, H. (2020). Pichia pastoris: A highly successful expression system for optimal synthesis of heterologous proteins. Journal of Cellular Physiology, 235(9), 5867–5881. https://doi.org/10.1002/jcp.29583
De Wachter, C., Van Landuyt, L., & Callewaert, N. (2021). Engineering of yeast glycoprotein expression. Advances in Biochemical Engineering/Biotechnology, 175, 93–135. https://doi.org/10.1007/10_2018_69
Zhu, T., Sun, H., Wang, M., & Li, Y. (2019). Pichia pastoris as a versatile cell factory for the production of industrial enzymes and chemicals: Current status and future perspectives. Biotechnology Journal, 14(6), 1–13. https://doi.org/10.1002/biot.201800694
Jia, L., Li, T., Wu, Y., Wu, C., Li, H., & Huang, A. (2021). Enhanced human lysozyme production by Pichia pastoris via periodic glycerol and dissolved oxygen concentrations control. Applied Microbiology and Biotechnology, 105(3), 1041–1050. https://doi.org/10.1007/s00253-021-11100-9
Yang, Z., & Zhang, Z. (2018). Engineering strategies for enhanced production of protein and bio-products in Pichia pastoris: A review. Biotechnology Advances, 36(1), 182–195. https://doi.org/10.1016/j.biotechadv.2017.11.002
Gao, J., Jiang, L., & Lian, J. (2021). Development of synthetic biology tools to engineer Pichia pastoris as a chassis for the production of natural products. Synthetic and Systems Biotechnology, 6(2), 110–119. https://doi.org/10.1016/j.synbio.2021.04.005
Puxbaum, V., Mattanovich, D., & Gasser, B. (2015). Quo vadis? The challenges of recombinant protein folding and secretion in Pichia pastoris. Applied Microbiology and Biotechnology, 99(7), 2925–2938. https://doi.org/10.1007/s00253-015-6470-z
Schwarzhans, J. P., Luttermann, T., Geier, M., Kalinowski, J., & Friehs, K. (2017). Towards systems metabolic engineering in Pichia pastoris. Biotechnology Advances, 35(6), 681–710. https://doi.org/10.1016/j.biotechadv.2017.07.009
Royle, K. E., & Polizzi, K. (2017). A streamlined cloning workflow minimising the time-to-strain pipeline for Pichia pastoris. Scientific Reports, 7(1), 1–10. https://doi.org/10.1038/s41598-017-16172-0
Aw, R., & Polizzi, K. M. (2013). Can too many copies spoil the broth? Microbial Cell Factories, 12(1), 1–9. https://doi.org/10.1186/1475-2859-12-128
Gonçalves, A. M., Pedro, A. Q., Maia, C., Sousa, F., Queiroz, J. A., & Passarinha, L. A. (2013). Pichia pastoris: A recombinant microfactory for antibodies and human membrane proteins. Journal of Microbiology and Biotechnology, 23(5), 587–601. https://doi.org/10.4014/jmb.1210.10063
Yu, Y. Fan., Yang, J., Zhao, F., Lin, Y., & Han, S. (2022). Comparative transcriptome and metabolome analyses reveal the methanol dissimilation pathway of Pichia pastoris. BMC Genomics, 23(1), 1–14. https://doi.org/10.1186/s12864-022-08592-8
Ata, Ö., Boy, E., Güneş, H., & Çalik, P. (2015). Codon optimization of xylA gene for recombinant glucose isomerase production in Pichia pastoris and fed-batch feeding strategies to fine-tune bioreactor performance. Bioprocess and Biosystems Engineering, 38(5), 889–903. https://doi.org/10.1007/s00449-014-1333-z
Takagi, S., Tsutsumi, N., Terui, Y., Kong, X. Y., Yurimoto, H., & Sakai, Y. (2019). Engineering the expression system for Komagataella phaffii (Pichia pastoris): An attempt to develop a methanol-free expression system. FEMS Yeast Research, 19(6), 1–10. https://doi.org/10.1093/femsyr/foz059
Vogl, T., Sturmberger, L., Kickenweiz, T., Wasmayer, R., Schmid, C., Hatzl, A., Gerstmann, M. A., Pitzer, J., Wagner, M., Thallinger, G. G., Geier, M., & Glieder, A. (2016). A toolbox of diverse promoters related to methanol utilization: Functionally verified parts for heterologous pathway expression in Pichia pastoris. ACS Synthetic Biology, 5(2), 172–186. https://doi.org/10.1021/acssynbio.5b00199
Prielhofer, R., Maurer, M., Klein, J., Wenger, J., Kiziak, C., & Gasser, B. (2013). Induction without methanol: Novel regulated promoters enable high-level expression in Pichia pastoris. Microbial Cell Factories, 12, 5. https://doi.org/10.1186/1475-2859-12-5
Ahmad, M., Hirz, M., Pichler, H., & Schwab, H. (2014). Protein expression in Pichia pastoris: Recent achievements and perspectives for heterologous protein production. Applied Microbiology and Biotechnology, 98(12), 5301–5317. https://doi.org/10.1007/s00253-014-5732-5
Landes, N., Gasser, B., Vorauer-Uhl, K., Lhota, G., Mattanovich, D., & Maurer, M. (2016). The vitamin-sensitive promoter PTHI11 enables pre-defined autonomous induction of recombinant protein production in Pichia pastoris. Biotechnology and Bioengineering, 113(12), 2633–2643. https://doi.org/10.1002/bit.26041
Karao, M., & Mehmet, İ. (2019). Identification of major ADH genes in ethanol metabolism of Pichia pastoris. Microbial Cell Factories, 37(2), 227–236. https://doi.org/10.1002/yea.3443
García-Ortega, X., Cámara, E., Ferrer, P., Albiol, J., Montesinos-Seguí, J. L., & Valero, F. (2019). Rational development of bioprocess engineering strategies for recombinant protein production in Pichia pastoris (Komagataella phaffii) using the methanol-free GAP promoter. Where do we stand? New Biotechnology, 53(June), 24–34. https://doi.org/10.1016/j.nbt.2019.06.002
Qin, X., Qian, J., Yao, G., Zhuang, Y., Zhang, S., & Chu, J. (2011). gap promoter library for fine-tuning of gene expression in Pichia pastoris. Applied and Environmental in Microbiology, 77(11), 3600–3608. https://doi.org/10.1128/AEM.02843-10
Arruda, A., Reis, V. C. B., Batista, V. D. F., Daher, B. S., Piva, L. C., De Marco, J. L., de Noraes, L. M. P., & Torres, F. A. G. (2016). A constitutive expression system for Pichia pastoris based on the PGK1 promoter. Biotechnology Letters, 38(3), 509–517. https://doi.org/10.1007/s10529-015-2002-2
Periyasamy, S., Govindappa, N., Sreenivas, S., & Sastry, K. (2013). Isolation, characterization and evaluation of the Pichia pastoris sorbitol dehydrogenase promoter for expression of heterologous proteins. Protein Expression and Purification, 92(1), 128–133. https://doi.org/10.1016/j.pep.2013.09.008
Vogl, T., & Glieder, A. (2013). Regulation of Pichia pastoris promoters and its consequences for protein production. New Biotechnology, 30(4), 385–404. https://doi.org/10.1016/j.nbt.2012.11.010
Liang, S., Zou, C., & Lin, Y. (2013). Identification and characterization of P GCW14: A novel, strong constitutive promoter of Pichia pastoris. Biotechnological Letters. https://doi.org/10.1007/s10529-013-1265-8
Tools for successful biologics—XS® Pichia Expression System. 2020. LONZA Pharma and Biotech Technical Bulletin, pp 1–4. https://www.lonza.com/-/media/Lonza/knowledge/Licensing/Pichia-Technical-Note-Update_0121_TSB_XS-TLI.pdf
Hirz, M., Richter, G., Leitner, E., Wriessnegger, T., & Pichler, H. (2013). A novel cholesterol-producing Pichia pastoris strain is an ideal host for functional expression of human Na, K-ATPase α3β1 isoform. Applied Microbiology and Biotechnology. https://doi.org/10.1007/s00253-013-5156-7
Wollborn, D., Munkler, L. P., Horstmann, R., Germer, A., Blank, L. M., & Büchs, J. (2022). Predicting high recombinant protein producer strains of Pichia pastoris MutS using the oxygen transfer rate as an indicator of metabolic burden. Scientific Reports, 12(1), 1–13. https://doi.org/10.1038/s41598-022-15086-w
Kaushik, N., Lamminmäki, U., Khanna, N., & Batra, G. (2020). Enhanced cell density cultivation and rapid expression-screening of recombinant Pichia pastoris clones in microscale. Scientific Reports, 10(1), 1–11. https://doi.org/10.1038/s41598-020-63995-5
Gätjen, D., Wieczorek, M., Listek, M., Tomszak, F., Nölle, V., Hanack, K., & Droste, M. (2022). A switchable secrete-and-capture system enables efficient selection of Pichia pastoris clones producing high yields of Fab fragments. Journal of Immunological Methods, 511(November), 113383. https://doi.org/10.1016/j.jim.2022.113383
Gassler, T., Sauer, M., Gasser, B., Egermeier, M., Troyer, C., Causon, T., Hann, S., Mattanovich, D., & Steiger, M. G. (2020). The industrial yeast Pichia pastoris is converted from a heterotroph into an autotroph capable of growth on CO2. Nature Biotechnology, 38(2), 210–216. https://doi.org/10.1038/s41587-019-0363-0
Ito, Y., Ishigami, M., Terai, G., Nakamura, Y., Hashiba, N., Nishi, T., Nakazawa, H., Hasunuma, T., Asai, K., Umetsu, M., Ishii, J., & Kondo, A. (2022). A streamlined strain engineering workflow with genome-wide screening detects enhanced protein secretion in Komagataella phaffii. Communications Biology, 5(1), 1–12. https://doi.org/10.1038/s42003-022-03475-w
Liu, Y., Wu, C., Wang, J., Mo, W., & Yu, M. (2013). Codon optimization, expression, purification, and Functional characterization of recombinant human IL-25 in Pichia pastoris. Applied Microbiology and Biotechnology, 97(24), 10349–10358. https://doi.org/10.1007/s00253-013-5264-4
Wang, J. R., Li, Y. Y., Liu, D. N., Liu, J. S., Li, P., Chen, L. Z., & Xu, S. D. (2015). Codon optimization significantly improves the expression level of α-Amylase gene from Bacillus licheniformis in Pichia pastoris. BioMed Research International. https://doi.org/10.1155/2015/248680
He, H., Wu, S., Mei, M., Ning, J., Li, C., Ma, L., Zhang, G., & Yi, L. (2020). A combinational strategy for effective heterologous production of functional human lysozyme in Pichia pastoris. Frontiers in Bioengineering and Biotechnology, 8(March), 1–12. https://doi.org/10.3389/fbioe.2020.00118
Che, Z., Cao, X., Chen, G., & Liang, Z. (2020). An effective combination of codon optimization, gene dosage, and process optimization for high-level production of fibrinolytic enzyme in Komagataella phaffii (Pichia pastoris). BMC Biotechnology, 20(1), 1–13. https://doi.org/10.1186/s12896-020-00654-7
Huang, Y., Lin, T., Lu, L., Cai, F., Lin, J., Jiang, Y., & Lin, Y. (2021). Codon pair optimization (CPO): A software tool for synthetic gene design based on codon pair bias to improve the expression of recombinant proteins in Pichia pastoris. Microbial Cell Factories, 20(1), 1–10. https://doi.org/10.1186/s12934-021-01696-y
Karaoğlan, M., & Erden-Karaoğlan, F. (2020). Effect of codon optimization and promoter choice on recombinant endo-polygalacturonase production in Pichia pastoris. Enzyme and Microbial Technology. https://doi.org/10.1016/j.enzmictec.2020.109589
Fadzil, N. A., Lim, S. K., Chew, A. L., & Khoo, B. Y. (2022). Multiple gene copy number increases total protein expression and enzyme activity of DNA topoisomerase I in Pichia pastoris. World Academy of Sciences Journal. https://doi.org/10.3892/wasj.2022.167
Berrios, J., Flores, M. O., Díaz-Barrera, A., Altamirano, C., Martínez, I., & Cabrera, Z. (2017). A comparative study of glycerol and sorbitol as co-substrates in methanol-induced cultures of Pichia pastoris: Temperature effect and scale-up simulation. Journal of Industrial Microbiology and Biotechnology, 44(3), 407–411. https://doi.org/10.1007/s10295-016-1895-7
Azadi, S., Mahboubi, A., Naghdi, N., Solaimanian, R., & Mortazavi, S. A. (2017). Evaluation of Sorbitol-Methanol Co-Feeding strategy on production of recombinant human growth hormone in Pichia pastoris. Iranian Journal of Pharmaceutical Research, 16(4), 1555–1564.
Zepeda, A. B., Pessoa, A., & Farías, J. G. (2018). Carbon metabolism influenced for promoters and temperature used in the heterologous protein production using Pichia pastoris yeast. Brazilian Journal of Microbiology, 49, 119–127. https://doi.org/10.1016/j.bjm.2018.03.010
Chen, L., Mohsin, A., Chu, J., Zhuang, Y., Liu, Y., & Guo, M. (2017). Enhanced protein production by sorbitol co-feeding with methanol in recombinant Pichia pastoris strains. Biotechnology and Bioprocess Engineering, 22(6), 767–773. https://doi.org/10.1007/s12257-017-0011-9
Kickenweiz, T., Glieder, A., & Wu, J. C. (2018). Construction of a cellulose-metabolizing Komagataella phaffii (Pichia pastoris) by co-expressing glucanases and β-glucosidase. Applied Microbiology and Biotechnology. https://doi.org/10.1007/s00253-017-8656-z
Soleimanpour, S., Farsiani, H., Mosavat, A., Ghazvini, K., Eydgahi, M. R. A., Sankian, M., Sadeghian, H., Meshkat, Z., & Rezaee, S. A. (2015). APC targeting enhances immunogenicity of a novel multistage Fc-fusion tuberculosis vaccine in mice. Applied Microbiology and Biotechnology, 99(24), 10467–10480. https://doi.org/10.1007/s00253-015-6952-z
Mohammadzadeh, R., Karbalaei, M., Soleimanpour, S., Mosavat, A., Rezaee, S. A., Ghazvini, K., & Farsiani, H. (2021). Practical methods for expression of recombinant protein in the Pichia pastoris system. Current Protocols, 1(6), 1–24. https://doi.org/10.1002/cpz1.155
Prabhu, A. A., Veeranki, V. D., & Dsilva, S. J. (2016). Improving the production of human interferon gamma (hIFN-γ) in Pichia pastoris cell factory: An approach of cell level. Process Biochemistry, 51(6), 709–718. https://doi.org/10.1016/j.procbio.2016.02.007
Li, J., Sun, C., Chen, L., Sun, L., Duan, L., Zheng, Q., & Hu, X. (2017). Optimization of the secretory expression of recombinant human C-reactive protein in Pichia pastoris. 3 Biotech, 7(5), 1–8. https://doi.org/10.1007/s13205-017-0917-0
Lee, J. S., Kim, J., Im, S. P., Kim, S. W., Jung, J. W., Lazarte, J. M. S., Lee, H.-O., Thompson, K., & Jung, T. S. (2018). Expression and characterization of monomeric variable lymphocyte receptor B specific to the glycoprotein of viral hemorrhagic septicemia virus (VHSV). Journal of Immunological Methods, 462(August), 48–53. https://doi.org/10.1016/j.jim.2018.08.006
Jiao, L., Zhou, Q., Su, Z., Xu, L., & Yan, Y. (2018). High-level extracellular production of Rhizopus oryzae lipase in Pichia pastoris via a strategy combining optimization of gene-copy number with co-expression of ERAD-related proteins. Protein Expression and Purification, 147(February), 1–12. https://doi.org/10.1016/j.pep.2018.02.005
Eissazadeh, S., Moeini, H., Dezfouli, M. G., Heidary, S., Nelofer, R., & Abdullah, M. P. (2017). Production of recombinant human epidermal growth factor in Pichia pastoris. Brazilian Journal of Microbiology, 48(2), 286–293. https://doi.org/10.1016/j.bjm.2016.10.017
Dagar, V. K., & Khasa, Y. P. (2018). Combined effect of gene dosage and process optimization strategies on high-level production of recombinant human interleukin-3 (hIL-3) in Pichia pastoris fed-batch culture. International Journal of Biological Macromolecules, 108, 999–1009. https://doi.org/10.1016/j.ijbiomac.2017.11.008
Vieira, S. M., da Rocha, S. L. G., Neves-Ferreira, A. G. C., Almeida, R. V., & Perales, J. (2017). Heterologous expression of the antimyotoxic protein DM64 in Pichia pastoris. PLoS Neglected Tropical Diseases, 11(7), 1–20. https://doi.org/10.1371/journal.pntd.0005829
Navone, L., Vogl, T., Luangthongkam, P., Blinco, J. A., Luna-Flores, C. H., Chen, X., von Hellens, J., Mahler, S., & Speight, R. (2021). Disulfide bond engineering of AppA phytase for increased thermostability requires co-expression of protein disulfide isomerase in Pichia pastoris. Biotechnology for Biofuels, 14(1), 1–14. https://doi.org/10.1186/s13068-021-01936-8
Deng, J., Li, J., Ma, M., Zhao, P., Ming, F., Lu, Z., Shi, J., Fan, Q., Liang, Q., Jia, J., Li, J., Zhang, S., & Zhang, L. (2020). Co-expressing GroEL-GroES, Ssa1-Sis1 and Bip-PDI chaperones for enhanced intracellular production and partial-wall breaking improved stability of porcine growth hormone. Microbial Cell Factories, 19(1), 1–17. https://doi.org/10.1186/s12934-020-01304-5
Zahrl, R. J., Mattanovich, D., & Gasser, B. (2018). The impact of ERAD on recombinant protein secretion in Pichia pastoris (Syn komagataella spp.). Microbiology (United Kingdom), 164(4), 453–463. https://doi.org/10.1099/mic.0.000630
Sallada, N. D., Harkins, L. E., & Berger, B. W. (2019). Effect of gene copy number and chaperone coexpression on recombinant hydrophobin HFBI biosurfactant production in Pichia pastoris. Biotechnology and Bioengineering, 116(8), 2029–2040. https://doi.org/10.1002/bit.26982
Roth, G., Vanz, A. L., Lünsdorf, H., Nimtz, M., & Rinas, U. (2018). Fate of the UPR marker protein Kar2/Bip and autophagic processes in fed-batch cultures of secretory insulin precursor producing Pichia pastoris. Microbial Cell Factories, 17(1), 1–11. https://doi.org/10.1186/s12934-018-0970-3
Yu, S., Miao, L., Huang, H., Li, Y., & Zhu, T. (2020). High-level production of glucose oxidase in Pichia pastoris: Effects of Hac1p overexpression on cell physiology and enzyme expression. Enzyme and Microbial Technology, 141(1), 109671. https://doi.org/10.1016/j.enzmictec.2020.109671
Huang, M., Gao, Y., Zhou, X., Zhang, Y., & Cai, M. (2017). Regulating unfolded protein response activator HAC1p for production of thermostable raw-starch hydrolyzing Α-amylase in Pichia pastoris. Bioprocess and Biosystems Engineering. https://doi.org/10.1007/s00449-016-1701-y
Yu, Y., Liu, Z., Chen, M., Yang, M., Li, L., & Mou, H. (2020). Enhancing the expression of recombinant κ-carrageenase in Pichia pastoris using dual promoters, co-expressing chaperones and transcription factors. Biocatalysis and Biotransformation, 38(2), 104–113. https://doi.org/10.1080/10242422.2019.1655001
Shirozu, R., Yashiroda, H., & Murata, S. (2015). Identification of minimum Rpn4-responsive elements in genes related to proteasome functions. FEBS Letters, 589(8), 933–940. https://doi.org/10.1016/j.febslet.2015.02.025
Sun, J., Jiang, J., Zhai, X., Zhu, S., Qu, Z., Yuan, W., Wang, Z., & Wei, C. (2019). Coexpression of Kex2 endoproteinase and Hac1 transcription factor to improve the secretory expression of bovine lactoferrin in Pichia pastoris. Biotechnology and Bioprocess Engineering, 24(6), 934–941. https://doi.org/10.1007/s12257-019-0176-5
Duan, G., Ding, L., Wei, D., Zhou, H., Chu, J., Zhang, S., & Qian, J. (2019). Screening endogenous signal peptides and protein folding factors to promote the secretory expression of heterologous proteins in Pichia pastoris. Journal of Biotechnology, 306, 193–202. https://doi.org/10.1016/j.jbiotec.2019.06.297
Ben Azoun, S., Belhaj, A. E., Göngrich, R., Gasser, B., & Kallel, H. (2016). Molecular optimization of rabies virus glycoprotein expression in Pichia pastoris. Microbial Biotechnology, 9(3), 355–368. https://doi.org/10.1111/1751-7915.12350
Bustos, C., Quezada, J., Veas, R., Altamirano, C., Braun-Galleani, S., Fickers, P., & Berrios, J. (2022). Advances in cell engineering of the Komagataella phaffii platform for recombinant protein production. Metabolites. https://doi.org/10.3390/metabo12040346
Luke, G. A., & Ryan, M. D. (2018). Therapeutic applications of the ‘NPGP’ family of viral 2As. Reviews in Medical Virology, 28(6), 1–12. https://doi.org/10.1002/rmv.2001
Wang, X., & Marchisio, M. A. (2021). Synthetic polycistronic sequences in eukaryotes. Synthetic and Systems Biotechnology, 6(4), 254–261. https://doi.org/10.1016/j.synbio.2021.09.003
Tian, Y., Li, Y., Zhao, F., & Meng, C. (2019). Engineered Pichia pastoris production of fusaruside, a selective immunomodulator. BMC Biotechnology, 19(1), 1–9. https://doi.org/10.1186/s12896-019-0532-8
Sherry, L., Swanson, J. J., Grehan, K., Xu, H., Uchida, M., Jones, I. M., Stonehouse, N. J., & Rowlands, D. J. (2023). Protease-independent production of poliovirus virus-like particles in Pichia pastoris: Implications for efficient vaccine development and insights into capsid assembly. Microbiology Spectrum. https://doi.org/10.1128/spectrum.04300-22
Javanmard, A. S., Matin, M. M., & Bahrami, A. R. (2021). Polycistronic cellulase gene expression in Pichia pastoris. Biomass Conversion and Biorefinery. https://doi.org/10.1007/s13399-021-01765-7
Geier, M., Fauland, P., Vogl, T., & Glieder, A. (2015). Compact multi-enzyme pathways in P. pastoris. Chemical Communications, 51(9), 1643–1646. https://doi.org/10.1039/c4cc08502g
Weninger, A., Hatzl, A. M., Schmid, C., Vogl, T., & Glieder, A. (2016). Combinatorial optimization of CRISPR/Cas9 expression enables precision genome engineering in the methylotrophic yeast Pichia pastoris. Journal of Biotechnology, 235, 139–149. https://doi.org/10.1016/j.jbiotec.2016.03.027
Weninger, A., Fischer, J. E., Raschmanová, H., Kniely, C., Vogl, T., & Glieder, A. (2018). Expanding the CRISPR/Cas9 toolkit for Pichia pastoris with efficient donor integration and alternative resistance markers. Journal of Cellular Biochemistry, 119(4), 3183–3198. https://doi.org/10.1002/jcb.26474
Fischer, J. E., & Glieder, A. (2019). Current advances in engineering tools for Pichia pastoris. Current Opinion in Biotechnology, 59, 175–181. https://doi.org/10.1016/j.copbio.2019.06.002
Valli, M., Tatto, N. E., Peymann, A., Gruber, C., Landes, N., Ekker, H., Thallinger, G. G., Mattanovich, D., Gasser, B., & Graf, A. B. (2016). Curation of the genome annotation of Pichia pastoris (Komagataella phaffii) CBS7435 from gene level to protein function. FEMS Yeast Research, 16(6), 1–12. https://doi.org/10.1093/femsyr/fow051
Sturmberger, L., Chappell, T., Geier, M., Krainer, F., Day, K. J., Vide, U., Trstenjak, S., Schiefer, A., Richardson, T., Soriaga, L., Darnhofer, B., Birner-Gruenberger, R., Glick, B. S., Tolstorukov, I., Cregg, J., Madden, K., & Glieder, A. (2016). Refined Pichia pastoris reference genome sequence. Journal of Biotechnology, 235, 121–131. https://doi.org/10.1016/j.jbiotec.2016.04.023
Love, K. R., Shah, K. A., Whittaker, C. A., Wu, J., Bartlett, M. C., Ma, D., Leeson, R. L., Priest, M., Borowsky, J., Young, S. K., & Love, J. C. (2016). Comparative genomics and transcriptomics of Pichia pastoris. BMC Genomics, 17(1), 1–17. https://doi.org/10.1186/s12864-016-2876-y
Liu, Q., Shi, X., Song, L., Liu, H., Zhou, X., Wang, Q., Zhang, Y., & Cai, M. (2019). CRISPR-Cas9-mediated genomic multiloci integration in Pichia pastoris. Microbial Cell Factories, 18(1), 1–11. https://doi.org/10.1186/s12934-019-1194-x
Gassler, T., Heistinger, L., Mattanovich, D., Gasser, B., & Prielhofer, R. (2019). CRISPR/Cas9-mediated homology-directed genome editing in Pichia pastoris. Methods in Molecular Biology, 1923, 211–225. https://doi.org/10.1007/978-1-4939-9024-5_9
Dalvie, N. C., Leal, J., Whittaker, C. A., Yang, Y., Brady, J. R., Love, K. R., & Love, J. C. (2021). Host-informed expression of CRISPR guide RNA for genomic engineering in Komagataella phaffii. ACS Synthetic Biology, 9(1), 26–35. https://doi.org/10.1021/acssynbio.9b00372.Host-informed
Jiang, H., Horwitz, A. A., Wright, C., Tai, A., Znameroski, E. A., Tsegaye, Y., Warbington, H., Bower, B. S., Alves, C., Co, C., Jonnalagadda, K., Platt, D., Walter, J. M., Natarajan, V., Ubersax, J. A., Cherry, J. R., & Love, J. C. (2019). Challenging the workhorse: Comparative analysis of eukaryotic micro-organisms for expressing monoclonal antibodies. Biotechnology and Bioengineering, 116(6), 1449–1462. https://doi.org/10.1002/bit.26951
Marsalek, L., Puxbaum, V., Buchetics, M., Mattanovich, D., & Gasser, B. (2019). Disruption of vacuolar protein sorting components of the HOPS complex leads to enhanced secretion of recombinant proteins in Pichia pastoris. Microbial Cell Factories, 18(1), 1–16. https://doi.org/10.1186/s12934-019-1155-4
Li, P., Sun, H., Chen, Z., Li, Y., & Zhu, T. (2015). Construction of efficient xylose utilizing Pichia pastoris for industrial enzyme production. Microbial Cell Factories, 14(1), 1–10. https://doi.org/10.1186/s12934-015-0206-8
Love, K. R., Dalvie, N. C., & Love, J. C. (2018). The yeast stands alone: The future of protein biologic production. Current Opinion in Biotechnology, 53, 50–58. https://doi.org/10.1016/j.copbio.2017.12.010
Acknowledgements
The authors are thankful to Indian Council of Medical Research (ICMR), New Delhi, India for the Senior Research Fellowship. The authors are thankful to Indo French Centre for the Promotion of Advanced Research (IFCPAR/CEFIPRA) for providing Senior Research Fellowship to the lead author. The authors are thankful to the Vellore Institute of Technology, Vellore, India for providing the necessary facilities to carry out this work.
Funding
The lead author has received Senior Research Fellowship (5/3/8/9/ITR-F/2018) fund from Indian Council of Medical Research (ICMR), New Delhi, India and also Senior Research Fellowship fund from Indo French Centre for the Promotion of Advanced Research (IFCPAR/CEFIPRA). Corresponding author received grant (IFC/7126/Hemophilia) for the work from Indo French Centre for the Promotion of Advanced Research (IFCPAR/CEFIPRA).
Author information
Authors and Affiliations
Contributions
VEV and KV conceptualized the study. VEV carried out review of literature, conception of ideas, data collection, curation, manuscript writing, editing and artwork. KV supervised review of literature, conception of ideas, writing, editing, and correspondence. Both the authors have read the manuscript and accepted the content for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vijayakumar, V.E., Venkataraman, K. A Systematic Review of the Potential of Pichia pastoris (Komagataella phaffii) as an Alternative Host for Biologics Production. Mol Biotechnol 66, 1621–1639 (2024). https://doi.org/10.1007/s12033-023-00803-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-023-00803-1