
Abstract
Despite the great importance of human membrane proteins involved in detoxification mechanisms, their wide use for biochemical approaches is still hampered by several technical difficulties considering eukaryotic protein expression in order to obtain the large amounts of protein required for functional and/or structural studies. Lactococcus lactis has emerged recently as an alternative heterologous expression system to Escherichia coli for proteins that are difficult to express. The aim of this work was to check its ability to express mammalian membrane proteins involved in liver detoxification, i.e., CYP3A4 and two isoforms of MGST1 (rat and human). Genes were cloned using two different strategies, i.e., classical or Gateway-compatible cloning, and we checked the possible influence of two affinity tags (6×-His-tag and Strep-tag II). Interestingly, all proteins could be successfully expressed in L. lactis at higher yields than those previously obtained for these proteins with classical expression systems (E. coli, Saccharomyces cerevisiae) or those of other eukaryotic membrane proteins expressed in L. lactis. In addition, rMGST1 was fairly active after expression in L. lactis. This study highlights L. lactis as an attractive system for efficient expression of mammalian detoxification membrane proteins at levels compatible with further functional and structural studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Membrane proteins (MPs) are involved in many essential cellular and biological functions, and are thus important as potential drug targets. Eukaryotic integral MPs account for approximately 25 % of open reading frames that are identified in sequenced genomes [1]. However, the vast majority of MPs still have no assigned function, and only about four hundred unique high-resolution 3D structures of transmembrane proteins have been obtained so far. Most of these structures are from bacterial and archeal proteins, and only very few from eukaryotic systems [2, 3]. Since structural studies require large amounts of purified proteins, novel approaches for their over-production need to be developed in order to increase our knowledge of the functions and structures of eukaryotic MPs. Several teams have extensively explored various systems for MP expression [4, 5]. Different expression hosts, such as bacteria (Escherichia coli), yeasts (Pichia pastoris and Saccharomyces cerevisiae), or higher eukaryotic cells (mammalian and insect cells) are often used for MP production [6]. However, the production of MPs in a functional state and in sufficient yields for structural analysis often remains challenging [7].
In the last decade, the bacterial expression system Lactococcus lactis has emerged as a good alternative to E. coli for expression of MPs, in particular for eukaryotic MPs [8–10]. In contrast to E. coli, this Gram-positive bacterium displays three interesting features for expression and further studies of MPs: it has a small genome [8], it does not form inclusion bodies [9], and it has only one membrane. These two last characteristics allow functional studies of recombinant proteins on whole bacterium, without purification, refolding, and reconstitution processes. However, in the case of the active site of the protein of interest is present at its cytosolic side, studies could be performed only when substrates, ligands, or inhibitors can pass through the cell membrane to interact directly with the MP of interest [8, 9]. Moreover, a tightly controlled system (NICE, NIsin-Controlled gene Expression; [11]) based on the use of sub-inhibitory amounts of the antimicrobial compound nisin, has already been successfully used for expression of prokaryotic and eukaryotic MPs in L. lactis [12–14]; for review, see [15]). Indeed, this system has been recently used for functional characterization of MPs from diverse origins (plants, bacteria, or mammals) and functional families (ABC transporters, mitochondrial carriers, and others; [15]).
Detoxification is a vital metabolic and defense capability exhibited by living organisms against potentially toxic xenobiotics. This process involves complex metabolizing enzymatic systems, consisting in two phases before the efflux of the xenobiotic metabolites (the so-called phase III): the phase I of functionalization, which uses oxygen to form a reactive metabolite [16], and the phase II of conjugation, which results in addition of a water-soluble group to the reactive metabolite [17]. The functionalization phase is generally the first level of enzymatic defense against exogenous compounds, and is mainly realized by cytochrome P450 enzymes (CYPs). This superfamily of membrane-bound hemoproteins is found in most organisms, and is responsible for the biotransformation of endobiotic and xenobiotic lipophilic/amphiphilic substrates in order to increase their solubility for a facilitated urinary and biliary excretion [18, 19]. CYP3A4 (EC 1.14.13.97) is the most abundant P450 isoform in human liver, i.e., 30 % of total CYP protein [20] and belongs to the microsomal class of P450 enzymes [21]. It is predominantly found on the cytoplasmic side of endoplasmic reticulum (ER; [22]). It is responsible for metabolizing about 50 % of drugs currently available on the market [23, 24]. Its first crystal structure was obtained 10 years ago [25, 26] for a soluble protein deleted of its anchoring transmembrane helix. Nevertheless no structures are currently available for this protein in its native form with its single transmembrane helix. Moreover, it seems that the N-terminus placed upstream the helix is involved in oligomerization of the protein in homo-/hetero-trimer [27].
Phase I detoxification is generally followed by a phase of conjugation, resulting in the transformation of the xenobiotic into a more water-soluble compound for facilitated excretion through bile and urine. Glutathione S-transferases (GSTs) are one important family of enzymes involved in this phase II of metabolic cell detoxification. They conjugate the tripeptide glutathione (GSH) to electrophilic compounds, including numerous carcinogenic, mutagenic, and pharmacologically active molecules [28]. They display diverse substrate specificities, and include both cytosolic and membrane-bound proteins. Microsomal glutathione S-transferase 1 (MGST1) (EC 2.1.5.18) is one of the most studied members of the family of membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG; [29]). This enzyme is highly expressed in liver, constituting about 3 % of rat ER MPs [30]. The protein was first purified from rat [31], then from human [32] livers. MGST1 exhibits both GSH-transferase and GSH-peroxidase activities [33]. Only one 3.2 Å electron microscopy structure resolved on 2D crystals of rat MGST1 bound with GSH has been reported and revealed that rMGST1 is composed of 4 α-helices organized in a homotrimeric functional conformation [34]. This is the first atomic-level structural information available for any MAPEG protein.
Despite the great importance of human MPs involved in detoxification mechanisms, their wide use for biochemical studies is still hampered by several technical problems for expressing these proteins at sufficient levels. Here, we describe the expression of the three mammalian MPs, CYP3A4, human MGST1, and rat MGST1, in L. lactis using the NICE system since this heterologous expression system presents several advantages over E. coli and yeasts which allow successful production and characterization of eukaryotic MPs.
Materials and Methods
Chemicals
All chemical were purchased from Sigma-Aldrich, unless otherwise indicated.
Bacterial Strains and Growth Conditions
Bacterial strains described in this study are listed in Table 1. E. coli DH5α were cultured at 37 °C under shaking (150 rpm) in LB Broth. L. lactis subsp. cremoris NZ9000 and NZ9700 were grown at 30 °C in M17 medium (Oxoid, UK), supplemented either with 0.5 % (w/v) glucose (M17G0.5 medium) without shaking for DNA isolation or with 1 % glucose (M17G1) with gentle shaking (90 rpm) for expression of recombinant proteins. The following antibiotics were used for plasmid maintenance: chloramphenicol (Chl; 10 µg/mL, Roth) for L. lactis, ampicillin (Amp; 100 µg/mL), and Kanamycin (Kan; 50 µg/mL) for E. coli. The supernatant from overnight cultures of the nisin-producing strain L. lactis NZ9700 was filter-sterilized, and used in this study as a source of the inducer nisin.
Oligonucleotides
The different primers used in this study to amplify the cDNAs introduced into the Gateway entry vector or into pNZ8148 are listed in Tables 2, 3, and 4.
DNA Techniques
Plasmid DNA extraction and ligation reactions were performed as previously described [35]. Restriction endonucleases (NEB, USA), PCR amplifications (High-Fidelity DNA Polymerase, Finnzymes, Finland), and site-directed mutagenesis (QuikChange Multi Site-Directed Mutagenesis Kit, Stratagene) were performed according to the manufacturers’ instructions.
Cloning into Gateway Entry Vectors
Plasmids used in this study are listed in Table 1. The human mgst1 cDNA (P10620/NM_145764.1 variant 1d) was obtained from Source BioScience (Nottingham, UK) and rat mgst1 cDNA (P08011/NM_134349.3) from Ralf Morgenstern; the cyp3a4 cDNA was amplified from the pYedDP60-cyp3a4 plasmid (gift from D. Pompon; P08684/NM_017460.3 isoform 1). All cDNAs were first amplified in one step by PCR with or without affinity tags (6×-His-tag or Strep-tag II), using the corresponding primers (Tables 2, 3, and 4). The different PCR fragments were then inserted into the entry vector pDONR221 by a BP reaction according to the manufacturer’s recommendation (Invitrogen). Afterward, the NcoI site present at position 428 of the cDNA encoding hMGST1 and at position 23 for CYP3A4 were silently mutated to facilitate the further cloning into pNZ8148. Afterward, the cDNA was transferred by LR reaction (Invitrogen) into pBS-RfA as already described [13], and generated the ‘‘shuttle’’ vector pBS-RfA-cDNA.
Cloning cDNAs from Gateway Entry Vectors into pNZ8148NKle
The nisin-inducible vector pNZ8148 (Table 1) is an improved version of pNZ8048 (see the Mobitec web site: http://www.mobitec.de/de/products/bio/04_vector_sys/index.php?nisin.html). The different cDNAs were excised from the pBS-RfA-cDNA by digestion with EcoRV and ligated into pNZ8148NKle as already described [13]. Afterward, L. lactis strain NZ9000 was transformed with the recombinant plasmids [36], and the presence of the insert in the right orientation was confirmed using restriction analyses, PCR amplification, and subsequent sequencing [35].
Direct Cloning of cDNAs into pNZ8148NK Vector
The cDNAs were amplified by PCR using the corresponding primers (Tables 2, 3, and 4) in order to introduce new NcoI and KpnI restriction sites, with the exception of CYP3A4 where the primer CYP-NcoIm fwd also allowed mutating the NcoI site present at nucleotide +24 of cyp3a4 cDNA. Afterward, PCR fragments were purified using the Nucleospin Extract II kit (Macherey-Nagel, Germany), and then digested with the endonucleases, NcoI and KpnI. Subsequently, the digested fragments were ligated into pNZ8148NK previously digested with the same endonucleases. The ligation reactions were purified, eluted in water, and then used to transform NZ9000 strain by electroporation [36]. Chloramphenicol-resistant clones were selected on M17GChl agar Petri dishes after 1–2 days at 30 °C. Presence of the cDNA and the correct sequence of the clones were confirmed by both endonuclease digestion and sequencing analysis.
Finally, a glycerol stock (15 % (v/v)) was prepared for each recombinant bacterium containing the correct clones, and kept at −80 °C.
Expression of Recombinant Proteins and Crude Bacterial Protein Purification
Precultures (25 mL of M17G1Chl) were inoculated with concentrated glycerol stock of recombinant bacteria (for details, see [35]) and incubated overnight at 30 °C with gentle shaking (90 rpm). These precultures were used to inoculate 1 L of M17G1Chl in Schott bottles. Cultures were incubated at 30 °C with gentle shaking until the OD600 reached 0.8, after which transcription from the nisA promoter was switched on by the addition of 0.005 vol culture supernatant of the nisin A-producing strain NZ9700 [13] and incubation was carried on for four additional hours. Bacteria were harvested by centrifugation at 5000×g for 15 min, at 4 °C. The bacterial pellets were resuspended in either in CYP3A4 (50 mM Tris HCl pH 7.5, 1 mM EDTA [37, 38]) or MGST1 (15 mM Tris–HCl pH 8.0, 0.25 M sucrose, 0.1 mM EDTA, 1 mM GSH [39]) bacterial resuspension buffer, and kept at −20 °C overnight after a second centrifugation.
The bacteria were lysed by twofold passages through a One Shot cell disruptor (Constant Cell Disruption Systems, Northants, UK) at 35,000 p.s.i. (2.3 kbars), and the lysates clarified by centrifugation at 10,000×g for 10 min, at 4 °C. The supernatant containing total proteins was transferred into Ti45 tubes for further ultracentrifugation at 150,000×g for 90 min, at 4 °C; the supernatant containing soluble fractions was kept for further analysis and membrane fractions present in the pellet were resuspended in 1–2 mL of protein resuspension buffers (50 mM Tris–HCl pH 7.5/1 mM EDTA/20 % glycerol for CYP3A4 [37, 38] or 10 mM KPi pH 7.0, 0.1 mM EDTA, 1 mM GSH, 20 % glycerol [39] for MGST1). Bacteria containing the empty vector pNZ8148 were systematically grown in parallel and used as a negative control (EV).
SDS-Polyacrylamide Gel Electrophoresis and Western Blotting of Expressed Proteins
Protein content of membrane fractions were estimated using the Bio-Rad protein assay (Bio-Rad, Hercules, CA [40]). SDS-PAGE was performed on soluble and membrane fractions with 12 or 15 % polyacrylamide gels for CYP3A4 and MGST1, respectively, followed by Western blotting and ECL detection. Afterward, Western blots were performed with antibodies specific to the protein to be detected (ab71493 for human and rat MGST1; Abcam, Cambridge, UK; CYP3A4 antibody from I. de Waziers, Paris V) or to the affinity tag used for the construction (His Probe TM-HRP, Thermo Scientific Pierce, USA or Strep-Tactin HRP conjugate, IBA, Germany). Liver microsomes from rat (prepared as previously described by [37]) and human (XTreme 200, pooled human liver microsomes, XenoTech) containing defined amounts of CYP3A4 and MGST1, the Strep-tag Protein ladder (MW from 16 to 100 kDa; 0.4 µg/protein; IBA, Germany) and a His-tagged protein (1 µg) were used as positive detection controls and to estimate the amount of recombinant protein produced. Protein amounts were quantified by the software Quantity One (Bio-Rad), after exclusion of the dye front.
Glutathione Transferase Activity of rMGST1
GST activity was measured on 200 µg/mL of membrane fractions isolated from recombinant bacteria of the various constructions of rMGST1. Indeed, L. lactis NZ9000 strains can neither synthetize nor import GSH on its own [41]. The activity was measured using 1 mM 1-chloro-2,4-dinitrobenzene (ε 340 =9.6 mM-1/cm) and 5 mM GSH as substrates in 0.1 M potassium phosphate buffer (pH 6.5) containing 0.1 % Triton X-100 (required for membrane solubilization) as previously described by [42]. The absorbance changes were measured at 340 nm in a gently stirred 2 mL spectrophotometric cuvette. All enzymatic measurements were performed at 30 °C. Microsomes of rat liver prepared according to [37] were used as a positive control. For a given cuvette containing the reaction medium, measurement accuracy of the enzymatic activity is essentially dependent on the accuracy of the determination of the slope of optical absorbance variations: under our standard conditions, this has been routinely observed at being ±2 nmol/min/mg total MP. This value gave a rough estimation of the confidence interval within which the measured activity values were found for these pilot functional tests, allowing to compare them as a first approach.
Results
Sequence Search
First, a sequence search was performed for these proteins on KEGG database in order to check whether L. lactis possesses homologues of the proteins of interest. In one hand, L. lactis does not possess P450 cytochromes, but presents hemoproteins including cytochromes bd oxidases (cydA (llmg_1864) and cydB (llmg_1863); [43]) in the membrane. On the other hand, L. lactis displays glutathione pathways [44] but the enzymes involved display no homology with either hMGST1 or rMGST1.
Engineering of Recombinant L. lactis Strains
In order to generate recombinant L. lactis strains, we cloned cDNA encoding for the proteins of interest into pNZ8148 vector. This vector possesses the nisin-inducible promoter with the obligatory NcoI site for translational fusions [12], but it may not be propagated into E. coli because of instability [45, 46] and of the partial shut off of the nisin promoter, which can be problematic in case of expression of toxic heterologous proteins. This is the reason why, in addition to the classical strategy [8], other methods have been developed to facilitate the cloning of genes into L. lactis vectors, such as the LIC-VBEx [47], as well as the Gateway-compatible strategy [13]. In our study, we have chosen to compare expression yields for proteins after cloning with either Gateway-compatible or classical strategies, either in the absence or presence of affinity tags.
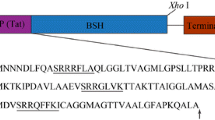
First, genes were cloned by the Gateway-compatible strategy which has allowed successful expression of MPs from diverse origins in L. lactis [13, 14]. The Gateway technology is one of the most efficient systems routinely used for cloning DNA fragments into a wide number of expression systems, from prokaryotic or eukaryotic origins. It facilitates gene cloning by using homologous recombination between att sites instead of the classical restriction enzymes and ligase [48]. All genes were first cloned into pDONR221, and subsequently into the shuttle vector pBS-RfA prior to insertion into the L. lactis expression vector, pNZ8148 [13]. All these steps resulted in the insertion of ten additional amino acids (ITSLYKKAGS; at the N-terminal end) between the ATG of the NcoI site and the ATG of the gene. In order to test the impact of this additional N-terminal, Gateway-specific, sequence on the production level of the proteins expressed, all genes were also cloned with the classical strategy after digestion with restriction enzymes (i.e., NcoI and KpnI; see “Methods” section) and ligation into the pNZ8148 vector. Moreover, for further detection and purification of recombinant proteins, and in order to test the impact of the nature of affinity tag on protein expression, the three proteins were also tagged at the C-terminus end with different affinity tags among the wide variety available [49], i.e., 6×-His-tag and Strep-tag II. These two affinity tags have already been used for successful expression of membrane proteins in Gram-positive bacteria [13, 50]. The 6×-His-tag is one of the most widely used purification tags since its small size minimizes interference with folding and structure of target proteins. There are many examples of proteins crystallized with intact His-tags having little or no impact on the structure of the tagged protein [51]. Strep-tag II (WSHPQFEK) is a small tag with a strong and specific interaction with Strep-Tactin, an optimized and recombinant form of streptavidin [52, 53].
The use of two cloning strategies (classical and Gateway compatible) and the presence or absence of affinity tags led us to obtain 18 different expression clones (listed in Table 5), which were used to test the expression of the two detoxification enzymes, CYP3A4, and (rat and human) MGST1, in L. lactis.
NICE System to Produce the Detoxification Enzymes CYP3A and MGST1
After cloning all the constructs in pNZ8148, we tested the expression of CYP3A4 and MGST1 in NZ9000, a MG1363-derived strain using the NICE system (Table 1). Over the last 10 years, the NICE system has been extensively used in L. lactis to produce homologous and heterologous proteins [8, 15]. Bacteria containing the empty pNZ8148 vector were systematically used as negative controls (Figs. 1, 2, 3, ‘‘EV’’) to validate the specificity of the detected signals.

Expression of human CYP3A4 protein in L. lactis. Western blot analysis of membrane fractions with CYP3A4-specific antibody (a), anti-His antibody (b), and with anti-Strep antibody (c). cDNA was either cloned with the classical or the Gateway-compatible strategy (G) and in the presence or absence of affinity tags (H or S, see Table 1). Total proteins (25 µg for a and c, 20 µg for b) were separated in a 12 % SDS-PAGE, and analyzed by Western blot performed using either a protein-specific antibody (a; 1/5000e), or a HRP conjugate specific to His-tag (b; 1/2000e) or specific to the Strep-tag II (c; 1/2500e). In all panels, EV means crude membrane proteins derived from bacteria containing the empty pNZ8148 vector; defined amounts of positive control proteins (P+) were loaded to estimate the expression levels of the recombinant proteins, either human liver microsomes (a), His-tagged protein (b), or Strep-tag II ladder (c)

Expression of human and rat MGST1 proteins in L. lactis. Western blot analysis of membrane fractions with MGST1-specific antibody after expression of rMGST1 (a) or hMGST1 (b). cDNAs were either cloned with the classical or the Gateway-compatible strategy (G) and in the presence or absence of affinity tags (H or S, see Table 1). Total membrane proteins (25 µg for a and b) were separated in a 15 % SDS-PAGE and analyzed by Western blot performed using a protein-specific antibody (1/75e). In all panels, EV means crude membrane proteins derived from bacteria containing the empty pNZ8148 vector; defined amounts of positive control proteins (P+) were loaded to estimate the expression levels of the recombinant proteins, rat (a) or human (b) liver microsomes

Enzymatic activity of rMGST1 heterologously expressed in L. lactis. Total GSH-transferase activity of the various constructions was measured on crude membrane fractions isolated from recombinant bacteria expressing rMGST1, as described in “Materials and Methods” section. It has been obtained after subtraction of the non-enzymatic part corresponding to the spontaneous conjugation between CDNB and GSH. Accuracy of the measurement of enzymatic activity is ±2 nmol/min/mg total MP (error bars)
The nisin used for induction was produced in our laboratory, stored at −80 °C and after calibration, the same stock was used for all experiments. Production of the different constructs was achieved by adding 0.005 volume nisin to a culture at OD600 ≤ 0.8. In addition, induction was performed at 30 °C for 4 h since production levels were two to three-times higher than for an overnight induction at 20 °C [13]. Disruption of L. lactis cells allowed us to extract about 50 mg of crude MPs per liter culture.
CYP3A4 is Expressed at High Levels in the Membrane of L. lactis
After cloning the native form of CYP3A4 cDNA into pNZ8148, we tested the expression of the different versions in L. lactis. After induction with nisin, L. lactis cells were disrupted, and membrane proteins were analyzed by Western blot with antibodies specific either to the protein (Fig. 1a) or to the affinity tag used (Fig. 1b, c).
CYP3A4 protein was recovered only in the membrane fractions, where it accounts for around 5–6 % of total membrane proteins detected (Fig. 1 a–c). Western blot analysis of soluble fractions with the same antibodies did not show any bands (data not shown). Only versions obtained with the Gateway-compatible strategy (i.e., CG, CHG, and CSG constructs) could be detected on the Western blot analysis using either protein-specific antibody (Fig. 1a) or affinity tag-specific antibodies (Fig. 1b, c). There is no difference observed between these 3 different Gateway constructs in terms of expression levels. These results showed that, for CYP3A4, the presence of the additional N-terminal sequence positively affects the production and/or detection of the protein.
Human and Rat MGST1 are Expressed at Similar Levels in the Membrane of L. lactis
All the versions of human and rat MGST1 were cloned into pNZ8148, and then tested for expression in L. lactis. Indeed, rMGST1 has already been successfully produced in E. coli, but hMGST1 was mainly produced in inclusion bodies [54]. These two proteins display a primary sequence identity of 83 % [55]. Expression of both MGST1 isoforms in L. lactis was evaluated by immunoblotting of the different constructs extracted from membrane fractions, and using antibodies specific either to the protein (Fig. 2a, b) or to the affinity tag used (Fig. S1, S2).
MGST1 protein was only recovered in the membrane fractions and detected within an electrophoretic band at 17 kDa where it accounts for around 2–3 % of total MPs (Fig. 2a, b). As expected, no signal was found in supernatant fractions (not shown). Using the anti-MGST1 antibody, we were only able to detect two versions for rMGST1 (i.e., rM and, at a lower extent, rMSG; Fig. 2a) and, for hMGST1 constructs, Western blot analysis showed bands for hM, hMG, hMSG, and hMHG at the expected size with various intensities; the protein hMGST1 labeled with an affinity tag could only be revealed by Western blot analysis when the construction was performed through the Gateway-compatible strategy (hMHG and hMSG, Fig. 2b). In contrast, we were unable to detect hMGST1 and rMGST1 proteins labeled with the two affinity tags using the respective tag-specific antibodies (Fig. S1 and S2); this could be probably due to a problem of accessibility to the tags. These data showed that the presence of the additional 10 amino-acid N-terminal, Gateway-specific, sequence could affect the production and/or the detection of MGST1.
rMGST1 is Functional After Expression in L. lactis
In order to check whether the protein MGST1 exhibits its physiologic GSH-transferase activity [31] when expressed in L. lactis, we performed experiments based on the spectroscopic detection of the conjugation of the GSH moiety onto the dye CDNB (2,4-chlorodinitrobenzene). Whereas L. lactis cells carrying the empty vector, considered as the negative control, did not present any significant GSH-transferase activity, we could measure the GSH-transferase activity using membrane fractions isolated from L. lactis cells expressing the various versions of rMGST1 (Fig. 3). For the construct rM, we estimated the corresponding specific enzymatic activity to almost 400 nmol/min/mg of rMGST1. These data indicate that the recombinant rMGST1 protein expressed in L. lactis is able to exhibit its GSH-transferase activity, and, at least for some of the constructs, is thus produced in a fairly folded and active form.
Discussion
In this work, we showed how the L. lactis system can be used for the successful expression of mammal MPs playing central roles in liver detoxification against xenobiotics, illustrated in this work by the expression of CYP3A4 (human) and MGST1 (human and rat).
Lactococcus lactis has emerged in the last years as an alternative system for expression and study of MPs, in particular for the proteins most problematic to express [8, 9, 13, 15]. We successfully expressed in L. lactis the two enzymes, CYP3A4 and MGST1 (human and rat isoforms), at around 5–6 and 2–3 % of total MPs, respectively. These expression yields are relatively high compared to those already obtained for other mammal MPs in L. lactis. Indeed, these proteins were expressed, using the NICE system, with expression yields all ranging from 0.1 to 1 % of total MPs [15–57], although higher yields (up to 5 %) have been reported for proteins of eukaryotic, not mammal, origin. It is noteworthy that the only human MP produced in a functional form in L. lactis, Erd2, was expressed at a level below 0.1 % [8]. To our knowledge, our study is the first one reporting the expression of human MPs with high expression yields, i.e., above 1 %, using this heterologous expression system.
In order to check the influence of the cloning strategy, genes encoding CYP3A4 and MGST1 have been cloned into pNZ8148 using two different strategies, i.e., either the classical or the Gateway-compatible strategy [13]. When using the Gateway-compatible strategy, 30 additional nucleotides were added at the 5′ extremity of both genes, corresponding to 10 amino acids at the N-terminus of the protein. Remarkably, about one residue out of four is hydrophobic (in bold in ITSLYKKAGS), in alternation with charged and polar residues. These additional amino acids had already a slight positive impact on the production of plant peripheric proteins expressed using the Gateway-compatible strategy and on the activity of the protein produced [13]. In our present study, this slight impact has been observed for both rMGST1 and hMGST1, but only in presence of affinity tags and, unexpectedly, the CYP3A4 protein could only be detected when the gene was cloned using the Gateway-compatible strategy. The different impact of the N-terminal Gateway-specific sequence on expression levels of CYP3A4 and MGST1 could be explained by overall structural differences between these two proteins, since CYP3A4 possesses only one transmembrane (TM) domain, whereas MGST1 is an intrinsic MP with 4 transmembrane helices per sub-unit [34]. This unexpected positive effect on CYP3A4 production could be due to the fact that this 10 amino acid peptide is likely structured as an amphipathic α-helix (Fig. 4) at the membrane interface, with its hydrophobic face bound to membrane, thus stabilizing the anchoring of the whole protein. It can thus be speculated that the added Gateway sequence could display a more marked stabilizing effect on a one TM helix-containing MP, such as CYP3A4, than on an intrinsic MP (with 4 TM helices), such as the MGST1 monomer. In the particular case of CYP3A4, we could not reject the fact that this additional sequence could influence also translation since the protein could be detect only after Gateway cloning, in contrast to previous studies [13].

Schematic representation of the additional sequence linked to the Gateway-compatible strategy. Upper part each amino acid of the Gateway sequence is represented by a symbol depending on its nature: hydrophilic residues as circles, hydrophobic as diamonds, and potentially positively charged as pentagons; the representations have been performed with the PHDhtm software used for the prediction and analysis of the transmembrane (TM) domains [66]. The arrow represents the direction of the hydro-lipophilic moment. Lower part the nucleotide sequence of the additional sequence linked to the Gateway-compatible strategy and its translation into amino acids (performed with Pretty Seq)
In this work, we showed that CYP3A4 can be successfully expressed in L. lactis using the NICE system. This key metabolizing protein, as other cytochromes P450, has already been expressed in other classical heterologous systems, such as E. coli and S. cerevisiae. In E. coli, human CYP3A4 has been obtained in membranes at a level of 1–1.5 % of total MPs [58], but this yield required sequence modifications (for review see [59]), codon usage optimization [58], and addition of leader peptide sequences to avoid the formation of inclusion bodies [60]; these two last points are the major drawbacks of E. coli over L. lactis [8, 15]. In S. cerevisiae, the protein represented only around 0.5 % of total MPs [61]. In our study, we were able to produce human CYP3A4 at 5–6 % of total MPs, a much higher level compared to those obtained for the protein produced in E. coli and S. cerevisiae. This opens the road to further functional and structural characterizations of the membrane anchored CYP3A4.
The two isoforms rMGST1 and hMGST1 display 83 % of sequence identity; however, all structural and functional studies so far mainly focused on rMGST1, purified either from rat liver, or heterologously expressed in E. coli, or in mammalian COS cells [34, 54]. Otherwise, there are only very few reports of expression of hMGST1, one in E. coli but with high amounts of proteins found in inclusion bodies [54], and one in mammalian cells [62]. Apparently, the other MAPEG proteins are also difficult to express since very few studies have been reported, and only in P. pastoris [63, 64]. When expressed in E. coli, rMGST1 represented around 1 % of bacterial MPs, and about 1 mg of pure protein could be obtained from 1 L of bacterial culture [39]. It is noticeable that we were able to produce MGST1, either from human and rat origins, in L. lactis at 2–3 % of total MPs, which represents clearly higher yields compared to E. coli expression. These yields are similar to native yields for both proteins in their physiological environment, liver microsomes. Indeed, L. lactis presents the major advantage not to form inclusion bodies, therefore leading to higher MP production efficiency.
Glutathione transferase enzymatic activity of the various expressed versions of rMGST1 have been measured, and were all found in the range 0.001–0.009 µmol/min/mg total MP (Fig. 3). When specific protein quantification was possible to be assayed, we could calculate a corresponding specific enzymatic activity of around 0.4 µmol/min/mg of rMGST1 (rM). This activity is somewhat lower than, but comparable to the values previously reported for rMGST1, obtained either from purified microsomes (2 µmol/min/mg of rMGST1; [33]) or from heterologous expression in E. coli (2.9 µmol/min/mg of rMGST1; [54]). The low functional activity that we unexpectedly observed for the human isoform might be explained by a more stringent requirement for the surrounding lipids than the rat isoform, in line with the different effect of NEM on rat and human MGST1 in microsomes, respectively, activating or not [33]. But such a functional difference between rat and human isoforms could also be related to different sensitivities to the various tags used and/or to the various experimental conditions. Our study shows that L. lactis expression system can produce quite fairly folded and functional rMGST1 and is the second one for functional expression of rodent MPs [65] in L. lactis, and the first one for a functional mammalian homotrimer, i.e., rMGST1.
References
Wallin, E., & von Heijne, G. (1998). Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Science, 7, 1029–1038.
von Heijne, G. (2007). The membrane protein universe: What’s out there and why bother? Journal of Internal Medicine, 261, 543–557.
White, S. H. (2009). Biophysical dissection of membrane proteins. Nature, 459, 344–346.
Lundstrom, K. (2006). Structural genomics for membrane proteins. Cellular and Molecular Life Sciences, 63, 2597–2607.
Bill, R. M., Henderson, P. J., Iwata, S., Kunji, E. R., Michel, H., Neutze, R., et al. (2011). Overcoming barriers to membrane protein structure determination. Nature Biotechnology, 29, 335–340.
Grisshammer, R., & Tate, C. G. (1995). Overexpression of integral membrane proteins for structural studies. Quarterly Reviews of Biophysics, 28, 315–422.
Lacapere, J. J., Pebay-Peyroula, E., Neumann, J. M., & Etchebest, C. (2007). Determining membrane protein structures: Still a challenge! Trends in Biochemical Sciences, 32, 259–270.
Kunji, E. R., Slotboom, D. J., & Poolman, B. (2003). Lactococcus lactis as host for overproduction of functional membrane proteins. Biochimica et Biophysica Acta, 1610, 97–108.
Kunji, E. R., Chan, K. W., Slotboom, D. J., Floyd, S., O’Connor, R., & Monné, M. (2005). Eukaryotic membrane protein overproduction in Lactococcus lactis. Current Opinion in Biotechnology, 16, 546–551.
Junge, F., Schneider, B., Reckel, S., Schwarz, D., Dötsch, V., & Bernhard, F. (2008). Large-scale production of functional membrane proteins. Cellular and Molecular Life Sciences, 65, 1729–1755.
Pontes, D. S., de Azevedo, M. S., Chatel, J. M., Langella, P., Azevedo, V., & Miyoshi, A. (2011). Lactococcus lactis as a live vector: Heterologous protein production and DNA delivery systems. Protein Expression and Purification, 79, 165–175.
Zhou, X. X., Li, W. F., Ma, G. X., & Pan, Y. J. (2006). The nisin-controlled gene expression system: Construction, application and improvements. Biotechnology Advances, 24, 285–295.
Frelet-Barrand, A., Boutigny, S., Moyet, L., Deniaud, A., Seigneurin-Berny, D., Salvi, D., et al. (2010). Lactococcus lactis, an alternative system for functional expression of peripheral and intrinsic Arabidopsis membrane proteins. PLoS ONE, 5, e8746.
Bernaudat, F., Frelet-Barrand, A., Pochon, N., Dementin, S., Hivin, P., Boutigny, S., et al. (2011). Heterologous expression of membrane proteins: Choosing the appropriate host. PLoS ONE, 6, e29191.
Bakari, S., André, F., Seigneurin-Berny, D., Delaforge, M., Rolland, N., & Frelet-Barrand, A. (2014). Lactococcus lactis, recent developments in functional expression of membrane proteins. In I. Mus-Veteau (Ed.), Membrane proteins production for structural analysis (pp. 107–132). New York: Springer.
Estabrook, R. W. (1996). The remarkable P450s: A historical overview of these versatile hemeprotein catalysts. FASEB Journal, 10, 202–204.
Testa, B. (2008). Biotransformation reactions and their enzymes. In C. G. Wermuth (Ed.), The practice of medicinal chemistry (3rd ed., pp. 655–673). Amsterdam: Elsevier.
Sono, M., Roach, M. P., Coulter, E. D., & Dawson, J. H. (1996). Heme-containing oxygenases. Chemical Reviews, 96, 2841–2888.
Bernhardt, R. (2006). Cytochromes P450 as versatile biocatalysts. Journal of Biotechnology, 124, 128–145.
Thummel, K. E., & Wilkinson, G. R. (1998). In vitro and in vivo drug interactions involving human CYP3A. Annual Review of Pharmacology and Toxicology, 38, 389–430.
Hannemann, F., Bichet, A., Ewen, K. M., & Bernhardt, R. (2007). Cytochrome P450 systems—biological variations of electron transport chains. Biochimica et Biophysica Acta, 1770, 330–344.
Estabrook, R. W., Franklin, M. R., Cohen, B., Shigamatzu, A., & Hildebrandt, A. G. (1971). Biochemical and genetic factors influencing drug metabolism. Influence of hepatic microsomal mixed function oxidation reactions on cellular metabolic control. Metabolism, 20, 187–199.
Isin, E. M., & Guengerich, F. P. (2006). Kinetics and thermodynamics of ligand binding by cytochrome P450 3A4. Journal of Biological Chemistry, 281, 9127–9136.
Denisov, I. G., Grinkova, Y. V., McLean, M. A., & Sligar, S. G. (2007). The one-electron autoxidation of human cytochrome P450 3A4. Journal of Biological Chemistry, 282, 26865–26873.
Williams, P. A., Cosme, J., Vinkovic, D. M., Ward, A., Angove, H. C., Day, P. J., et al. (2004). Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science, 305, 683–686.
Yano, J. K., Wester, M. R., Schoch, G. A., Griffin, K. J., Stout, C. D., & Johnson, E. F. (2004). The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. Journal of Biological Chemistry, 279, 38091–38094.
Davydov, D. R., Davydova, N. Y., Sineva, E. V., & Halpert, J. R. (2015). Interactions among cytochromes P450 in microsomal membranes: Oligomerization of cytochromes P450 3A4, 3A5, and 2E1 and its functional consequences. Journal of Biological Chemistry, 290, 3850–3864.
Oakley, A. J., Harnnoi, T., Udomsinprasert, R., Jirajaroenrat, K., Ketterman, A. J., & Wilce, M. C. (2001). The crystal structures of glutathione S-transferases isozymes 1-3 and 1-4 from Anopheles dirus species B. Protein Science, 10, 2176–2185.
Jakobsson, P. J., Morgenstern, R., Mancini, J., Ford-Hutchinson, A., & Persson, B. (1999). Common structural features of MAPEG—a widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Science, 8, 689–692.
Morgenstern, R., Lundqvist, G., Andersson, G., Balk, L., & DePierre, J. W. (1984). The distribution of microsomal glutathione transferase among different organelles, different organs and different organisms. Biochemical Pharmacology, 33, 3609–3614.
Morgenstern, R., Guthenberg, C., & DePierre, J. W. (1982). Microsomal glutathione S-transferase. Purification, initial characterization and demonstration that it is not identical to the cytosolic glutathione S-transferases A, B and C. European Journal of Biochemistry, 128, 243–248.
McLellan, L. I., Wolf, C. R., & Hayes, J. D. (1989). Human microsomal glutathione S-transferase. Its involvement in the conjugation of hexachlorobuta-1,3-diene with glutathione. Biochemical Journal, 258, 87–93.
Morgenstern, R., & DePierre, J. W. (1983). Microsomal glutathione transferase purification in inactivated form and further characterization of the activation process, substrate specificity and amino acid composition. European Journal of Biochemistry, 134, 591–597.
Holm, P. J., Bhakat, P., Jegerschold, C., Gyobu, N., Mitsuoka, K., Fujiyoshi, Y., et al. (2006). Structural basis for detoxification and oxidative stress protection in membranes. Journal of Molecular Biology, 360, 934–945.
Frelet-Barrand, A., Boutigny, S., Kunji, E. R., & Rolland, N. (2010). Membrane protein expression in Lactococcus lactis. In I. Mus-Vuteau (Ed.), Methods in molecular biology: Heterologous expression of membrane proteins (Vol. 601, pp. 67–85). New York: Springer, Humana.
Holo, H., & Nes, I. F. (1989). High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Applied and Environment Microbiology, 55, 3119–3123.
Peyronneau, M. A., Delaforge, M., Riviere, R., Renaud, J. P., & Mansuy, D. (1994). High affinity of ergopeptides for cytochromes P450 3A. Importance of their peptide moiety for P450 recognition and hydroxylation of bromocriptine. European Journal of Biochemistry, 223, 947–956.
Guengerich, F. P., Parikh, A., Johnson, E. F., Richardson, T. H., von Wachenfeldt, C., Cosme, J., et al. (1997). Heterologous expression of human drug-metabolizing enzymes. Drug Metabolism and Disposition, 25, 1234–1241.
Morgenstern, R. (2005). Microsomal glutathione transferase 1. Methods in Enzymology, 401, 136–146.
Bradford, M. M. (1976). A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Li, Y., Hugenholtz, J., Sybesma, W., Abee, T., & Molenaar, D. (2005). Using Lactococcus lactis for glutathione overproduction. Applied Microbiology and Biotechnology, 67, 83–90.
Habig, W. H., Pabst, M. J., & Jakoby, W. B. (1974). Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. Journal of Biological Chemistry, 249, 7130–7139.
Baureder, M., & Hederstedt, L. (2013). Heme proteins in lactic acid bacteria. Advances in Microbial Physiology, 62, 1–43.
Pophaly, S. D., Singh, R., Pophaly, S. D., Kaushik, J. K., & Tomar, S. K. (2012). Current status and emerging role of glutathione in food grade lactic acid bacteria. Microbial Cell Factories, 11, 114.
Kok, J., van der Vossen, J. M., & Venema, G. (1984). Construction of plasmid cloning vectors for lactic streptococci which also replicate in Bacillus subtilis and Escherichia coli. Applied and Environment Microbiology, 48, 726–731.
de Vos, W. M., & Simons, G. F. M. (1994). Gene cloning and expression systems in Lactococci. In M. J. Gasson & W. M. de Vos (Eds.), Genetics and biotechnology of lactic acid bacteria (pp. 52–105). London: Blackie Academic and Professional.
Geertsma, E. R., & Poolman, B. (2007). High-throughput cloning and expression in recalcitrant bacteria. Nature Methods, 4, 705–707.
Hartley, J. L., Temple, G. F., & Brasch, M. A. (2000). DNA cloning using in vitro site-specific recombination. Genome Research, 10, 1788–1795.
Terpe, K. (2003). Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Applied Microbiology and Biotechnology, 60, 523–533.
Marreddy, R. K., Pinto, J. P., Wolters, J. C., Geertsma, E. R., Fusetti, F., Permentier, H. P., et al. (2011). The response of Lactococcus lactis to membrane protein production. PLoS ONE, 6, e24060.
Carson, M., Johnson, D. H., McDonald, H., Brouillette, C., & Delucas, L. J. (2007). His-tag impact on structure. Acta Crystallographica. Section D, Biological Crystallography, 63, 295–301.
Schmidt, T. G., & Skerra, A. (1994). One-step affinity purification of bacterially produced proteins by means of the “Strep tag” and immobilized recombinant core streptavidin. Journal of Chromatography A, 676, 337–345.
Schmidt, T. G., & Skerra, A. (2007). The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nature Protocols, 2, 1528–1535.
Weinander, R., Mosialou, E., DeJong, J., Tu, C. P. D., Dypbukt, J., Bergman, T., et al. (1995). Heterologous expression of rat liver microsomal glutathione transferase in simian COS cells and Escherichia coli. Biochemical Journal, 311, 861–866.
DeJong, J. L., Morgenstern, R., Jörnvall, H., DePierre, J. W., & Tu, C. P. D. (1988). Gene expression of rat and human microsomal glutathione S-transferases. Journal of Biological Chemistry, 263, 8430–8436.
Mifsud, J., Ravaud, S., Krammer, E. M., Chipot, C., Kunji, E. R., Pebay-Peyroula, E., et al. (2013). The substrate specificity of the human ADP/ATP carrier AAC1. Molecular Membrane Biology, 30, 160–168.
Janvilisri, T., Venter, H., Shahi, S., Reuter, G., Balakrishnan, L., & van Veen, H. W. (2003). Sterol transport by the human breast cancer resistance protein (ABCG2) expressed in Lactococcus lactis. Journal of Biological Chemistry, 278, 20645–20651.
Pan, Y., Abd-Rashid, B. A., Ismail, Z., Ismail, R., Mak, J. W., & Ong, C. E. (2011). Heterologous expression of human cytochromes P450 2D6 and CYP3A4 in Escherichia coli and their functional characterization. Protein Journal, 30, 581–591.
Zelasko, S., Palaria, A., & Das, A. (2013). Optimizations to achieve high-level expression of cytochrome P450 proteins using Escherichia coli expression systems. Protein Expression and Purification, 92, 77–87.
Guengerich, F. P., & Parikh, A. (1997). Expression of drug-metabolizing enzymes. Current Opinion in Biotechnology, 8, 623–628.
Peyronneau, M. A., Renaud, J. P., Truan, G., Urban, P., Pompon, D., & Mansuy, D. (1992). Optimization of yeast-expressed human liver cytochrome P450 3A4 catalytic activities by coexpressing NADPH-cytochrome P450 reductase and cytochrome b5. European Journal of Biochemistry, 207, 109–116.
Otieno, M. A., & Anders, M. W. (1997). Stable transfection of LLC-PK1 cells with human microsomal glutathione S-transferase gene increases haloalkene glutathione S-conjugate formation and cytotoxicity. Biochemical Biophysical Research Communications, 234, 481–484.
Wetterholm, A., Martinez Molina, D., Nordlund, P., Eshaghi, S., & Haeggström, J. Z. (2008). High-level expression, purification, and crystallization of recombinant rat leukotriene C(4) synthase from the yeast Pichia pastoris. Protein Expression and Purification, 60, 1–6.
Ahmad, S., Niegowski, D., Wetterholm, A., Haeggström, J. Z., Morgenstern, R., & Rinaldo-Matthis, A. (2013). Catalytic characterization of human microsomal glutathione S-transferase 2: Identification of rate-limiting steps. Biochemistry, 52, 1755–1764.
Herzig, S., Raemy, E., Montessuit, S., Veuthey, J. L., Zamboni, N., Westermann, B., et al. (2012). Identification and functional expression of the mitochondrial pyruvate carrier. Science, 337, 93–96.
Zidovetzki, R., Rost, B., Armstrong, D. L., & Pecht, I. (2003). Transmembrane domains in the functions of Fc receptors. Biophysical Chemistry, 100, 555–575.
Acknowledgments
We are grateful to Denis Pompon (LISBP, Toulouse, France) for his gift of the pYEDP60-CYP3A4 plasmid, Ralf Morgenstern (IEM, Karolinska Institute, Sweden) for cDNA encoding rMGST1 and for fruitful exchanges, to Isabelle de Waziers (Inserm U775, Paris V, France) for CYP3A4-specific antibody, and to François Parcy (LPCV, CEA Grenoble, France) for pBS-RfA plasmid. We thank Ludovic Robillard for technical assistance and Elizabeth Kish-Perrin for linguistic revisions of the manuscript. This work has been supported by the Region Ile de France (DIM SeNt, PhD fellowship to SB AAP2010-3-10T6).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Human and Animal Rights Statement
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Additional information
Sana Bakari and Mehdi Lembrouk have contributed equally to this work.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bakari, S., Lembrouk, M., Sourd, L. et al. Lactococcus lactis is an Efficient Expression System for Mammalian Membrane Proteins Involved in Liver Detoxification, CYP3A4, and MGST1. Mol Biotechnol 58, 299–310 (2016). https://doi.org/10.1007/s12033-016-9928-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-016-9928-z