
Abstract
RNA methylation, which was identified back in 1970s, has gained remarkable interest in recent years as it was shown to be a reversible modification involved in many cellular processes like mRNA and miRNA processing, mRNA localisation, translation suppression, or activation. These, in turn, affect important bioprocesses such as tissue development, sex determination, and DNA damage response. Important group of proteins are responsible for adding, recognizing, and removing the methyl group to and from the RNA molecules, which are referred as writers, readers, and erasers, respectively. If any of the processes is not strictly controlled, this can cause abnormalities in gene expression, which result in diseases including cancers such as lung, pancreas, glioblastoma, and breast cancer. Mechanisms of RNA methylation and its role in various cancer types and diagnostic methods for RNA methylation are discussed in this article.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Methylation, by definition, is the addition of a methyl (-CH3) group to a molecule. Methylation is observed on nucleic acids (DNA, RNA) and several proteins including histones, and is recognized as a key process underlying epigenetic regulation of histone architecture and gene expression in eukaryotic cells [1].
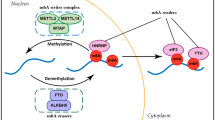
The addition of methyl groups is controlled at several different levels in cells and is carried out by a family of enzymes called methyltransferases (writers). Equally important and coupled with methylation is demethylation, which is the removal of the methyl group. Demethylation is necessary for epigenetic reprogramming of genes and this makes the methylation process reversible and dynamic. Demethylation is achieved by another enzyme family called demethylases (erasers). In addition to writers and erasers, there is another important group of proteins that recognize these modifications, bind to them, and carry out different biological functions (readers) [2].
DNA and histone methylation processes are very extensively studied subjects. DNA methylation, especially on promoter regions (CpG islands), is known to suppress gene transcription [3], whereas effect of histone methylation primarily depends on the level of methylation and position of the methylated residue on the histone protein [4]. In both cases, methylation results in a strict epigenetic control of gene expression in eukaryotic cells. In addition to DNA and histone methylation, another level of epigenetic regulation has become a hot topic in biological sciences in the last decade, which is RNA methylation. RNA methylation involves the addition of the methyl group either to the 5th carbon of cytosine nucleotide (5-mC) or to the nitrogen on the 6th carbon of adenine nucleotide (m6A) [5]. RNA methylation modifications have been found for decades of years, which occur at different RNA types of numerous species [6]. Growing evidence suggests a major regulatory role of RNA methylation modifications in mRNA processing (splicing, polyadenylation, etc.), localisation, miRNA processing, tRNA stabilization, and translation suppression or activation. These regulations have a big impact on tissue development, circadian rhythm, DNA damage response, sex determination, and diseases including tumorigenesis [7, 8]. MODOMICS database is a web-based database which contains the information related to the RNA modification pathways. Until now, 171 RNA modifications have been described and 72 of them include methyl groups [9].
5-Methylcytosine (5-mC) RNA modification
Even though cytosine methylation (5-mC) as an epigenetic mark in DNA has been studied widely, the location, exact mechanism of formation, and possible cellular functions of the same modified nucleotide in different cellular RNAs still remain to be investigated [10, 11]. Recent progress in the field includes the characterization of several enzymes belonging to the RNA (cytosine-5) methyltransferase (RNCMT) family of proteins in various organisms, such as NSun2, TRDMT1, and DNMT2. These have been shown to act as cytosine methyltransferases for tRNAs and rRNAs using a catalytic mechanism [10, 12].
Functional studies indicated 5-mC methylation’s involvement in structural and metabolic stabilization of tRNAs, and also its importance for tRNA translation suppression in vivo has been shown [11, 13]. However, as mentioned above, the exact mechanisms and functions of 5-mC modifications, especially in mRNAs, still need to be elucidated.
N6-methyladenosine (m6A) RNA modification
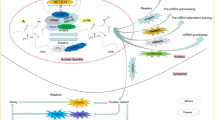
First identified in 1970s, m6A RNA methylation is one of the most abundant post-transcriptional modifications, making up approximately 50% of total methylated ribonucleotides in total cellular RNA content [14]. It has gained prodigious interest in recent years as being a predominant internal modification of especially mRNA molecules in higher eukaryotes. Through its impact on protein binding, m6A influences every step in the lifespan of an mRNA molecule, from splicing, localization, polyadenylation to translation and decay. A large protein complex named as the m6A methyltransferase complex is responsible for the addition of the methyl group to the nitrogen on 6th carbon of the aromatic ring of an adenosine residue (Fig. 1).

Structure of an N6-methyladenosine molecule
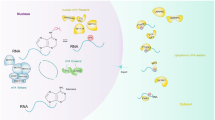
The complex contains a core structure composed of METTL3 and METTL14 proteins, which have methyltransferase activity [15]. Another protein, Wilms’ tumor 1-associating protein (WTAP), is also included in the complex. It interacts with METTL3 and METTL14, and is required for their localization into nuclear speckles enriched with pre-mRNA processing factors and for catalytic activity of the m6A methyltransferase in vivo [16]. In addition to METTL3, METTL14, and WTAP, RBM15, KIAA1429, and possibly ZC3H13 and CBLL1 are also found in the m6A methyltransferase complex [14] (Fig. 2).

Representation of proteins found in the m6A methyltransferase complex
Despite the identification of the m6A RNA modification and the components of the methyltransferase complex, there were no identified m6A RNA demethylases in higher eukaryotic cells, until 2010. Then, obesity-associated protein (FTO) and ALKBH5 were discovered to be mammalian demethylases that oxidatively reverse m6A in mRNA in vitro and in vivo [17, 18]. This discovery indicated that m6A RNA modification is reversible and dynamically regulated, suggesting it has regulatory roles in eukaryotic cells [19]. Therefore, reversible RNA methylation, analogous to reversible DNA and histone modifications, may affect gene expression and cell fate decisions by modulating multiple RNA-related cellular pathways, which potentially provides rapid responses to various cellular and environmental signals, including energy and nutrient availability in mammals [19].
Once the RNAs are modified with m6A methylation, some cytoplasmic and nuclear proteins, referred as the ‘reader’ proteins, can recognize the modified site and bind onto the RNA to carry out specific functions. YT521-B homology (YTH) domain-containing proteins including YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3 have been identified as readers of m6A marks on mRNA [14]. Binding of different YTH-domain family proteins to distinct subsets of m6A sites, results in production of different effects on gene expression. For example, binding of YTHDF2 results in the localization of bound mRNA to mRNA decay sites [20]. This results in reduced stability and increased turnover of m6A-modified mRNA. YTHDF1, on the other hand, binds to the modified mRNA and actively promotes protein synthesis by interacting with the translation initiation factor eIF3 [21]. YTHDF3 was shown to be able to interact cooperatively with YTHDF1 and YTHDF2. When bound to YTHDF1 and YTHDF2, it accelerates mRNA decay and translation initiation processes, respectively [22].
Another mechanism involves m6A methylation as a translational regulation during stress responses. When the stress responses are triggered in a cell, m6A methylation of the 5′-UTR of stress response genes leads to cap-independent translation [14, 23]. A transcript with a single 5′ UTR m6A residue directly binds to the eIF3, which is sufficient to recruit the ribosomal 43S complex to initiate translation in the absence of the cap-binding factor eIF4E [23]. During this, YTHDF2 is re-localized to the nucleus to prevent the removal of m6A modification by FTO [14].
Apart from functions in mRNA regulation, m6A methylation is also involved in the processing of pri-miRNAs. hnRNPA2B1 recruits the microRNA microprocessor complex protein DGCR8 to RNA sites that are modified by m6A methylation and promote primary pri-miRNA processing into mature miRNA [14, 24].
Biophysical studies indicated that m6A modification can also have a direct effect on RNA structure. Unmodified adenosine residues can form bonds with their adjacent bases in RNA structures. But when the residue is modified with m6A methylation, this destabilizes the RNA duplex, so bases adjacent to m6A sites tend to be more single stranded. As a result, RNA-binding motifs that were once buried in the hairpin structure is revealed and the accessibility of RNA-binding proteins such as hnRNPC is improved. This, in turn, mediates an indirect effect within the cell. This phenomenon is termed as the ‘m6A switch’ [25] (Fig. 3).

Representation of proteins found in the m6A methyltransferase complex
RNA methylation and cancer
The main hallmark of cancer is abnormal regulation of the gene expression. m6A is important for cellular differentiation control and pluripotency. Recent studies highlighted that not only abnormal gene expression and DNA methylation has an effect on cancer development, but also RNA methylation is important for cancer self-renewal and cell fate. It is also involved in cancer stem cell (CSC) pluripotency, extensive cell proliferation, metastasis, and potentially in tumor immunity, which makes it a new and promising therapeutic avenue for investigation [26].
METTL3, METTL14, NSun2, FTO, ALKBH5, YTHDF2 are noted as the abnormally methylated molecules in different types of cancers [27]. One important affected group of cells are hematopoietic stem cells. Hematopoietic stem cells have diverse differentiation pathways to reach their final differentiated state. Increased amount of m6A alters the normal differentiation pathway and this causes abnormalities in the progenitor cell state [26].
METTL3 mRNA and protein levels were shown to be increased in acute myeloid leukemia (AML) cells compared to normal human hematopoietic stem/progenitor cells (HSPCs) and other types of tumor cells [27]. Depletion of METTL3 in AML cell lines induces apoptosis and cell differentiation, which in turn delays leukemia progression in vivo [27]. Like METTL3, another key component of the m6A methyltransferase complex, METTL14 is also highly expressed in normal HSPCs and also in a group of AML cells and is downregulated during myeloid differentiation [14, 28]. Silencing of METTL14 in HSPCs and AML cells promotes terminal myeloid differentiation and inhibit AML cell survival and proliferation [28]. On the other hand, FTO, which is a m6A demethylase, was found to be highly expressed in some types of AML. This results in repression of a suppressor of cytokine signaling box-2 (ASB2) and the retinoic acid receptor alpha (RARA) due to decreased mRNA stability upon FTO-mediated decrease of m6A RNA methylation levels [14, 29].
It was shown that reduced mRNA m6A levels are critical for the maintenance of glioblastoma stem-like cell (GSC) growth, self-renewal, and tumor development. Knockdown of METTL3 or METTL14 reduces mRNA m6A levels of their target gene A disintegrin and metallopeptidase domain 19 (ADAM19) promotes ADAM19 expression, which in turn promote GSC self-renewal, growth, and tumorigenesis [30, 31]. Also, inhibition of FTO suppresses tumor progression and prolongs lifespan of GSC-grafted mice [31]. Additionally, ALKBH5 is highly expressed in GSCs, which demethylates the forkhead box protein M1 (FOXM1) nascent transcripts. The nuclear RNA-binding protein HuR is recruited to these unmethylated transcripts, which results in stabilization of the mRNA [14, 32]. Stabilized FOXM1 mRNA causes elevated levels of FOXM1 protein that functions as a key cell-cycle molecule required for G1/S and G2/M transition and M-phase progression in cells [14, 33].
In breast cancer, it was shown that the exposure of the cells to hypoxia could stimulate hypoxia-induced factor HIF-1α- and HIF-2α-dependent expression of ALKBH5. ALKBH5 then induces stabilization of NANOG mRNA through m6A demethylation [34]. NANOG is a pluripotency factor and is required for the maintenance of cancer stem cells, including breast cancer stem cells (BCSC). So, this way, increased m6A demethylation correlates with the promotion of BCSC phenotype [34]. Additionally, aberrant mammalian hepatitis B X-interacting protein (HBXIP) expression was shown to upregulate the expression of METTL3 through the suppression of let-7 g miRNA in breast cancers. Elevated METTL3 then increases the expression of HBXIP forming a positive feedback loop, leading to accelerated cell proliferation in breast cancer [35]. In hepatocellular carcinoma (HCC), on the other hand, overexpression of METTL3 represses suppressor of cytokine signaling 2 (SOCS2) expression through an m6A-YTHDF2-dependent mechanism and promotes tumor growth [36]. However, another subunit of the m6A methyltransferase, METTL14, promotes expression of miR126, which is a metastasis suppressor molecule, and causes suppression of metastasis in HCC cells [14].
The reader protein YTHDF2 is shown to be upregulated in human pancreatic cancer cells compared to healthy cells in both mRNA and protein levels [37]. By interacting with Akt/GSK3b/CyclinD1 pathway, YTHDF2 promotes cell proliferation while it can suppress the migration and adhesion capability of cancer cells by inhibiting EMT, likely via the downregulation of YAS gene. This way, YTHDF2 is said to orchestrate EMT/proliferation dichotomy in pancreatic cancer cells [14, 37].
Moreover, it was shown that NSun2 methylates the pre-miR125b and reduces the levels of mature miR-125b [38]. This causes augmentation of its target gene products such as GRB2-associated-binding protein 2 (Gab2) and results in the promotion of cancer cell migration [14, 38].
Methods of detecting RNA methylation
Several methods have been developed for detection of DNA methylation, which are based on bisulfite modification, chemical modification of non-methylated cytosines, like methylation specific PCR, bisulfite sequencing, MS-RFLP, pyrosequencing, traditional PCR-based amplification fragment length polymorphism (AFLP), restriction fragment length polymorphism (RFLP), LUminometric Methylation Assay, array, or NGS-based methods.
The identification of the RNA methylation events on RNA structure and metabolism needs combination with novel, high-throughput methods. For RNA 5-mC detection, some methods that depend on the conversion of non-methylated cytosine residues to uracil after bisulfite treatment are established and are widely used [39]. However, different from the 5-mC, m6A RNA methylation has no such chemical conversion [40]. Lack of sensitive methods for the detection of RNA modifications and their static and non-reversible nature led to restrained scientific interest in this field.
Dot-blot and high-performance liquid chromatography coupled to triple–quadrupole mass spectrometry (LC–MS/MS) can be a useful tool for information of m6A modification. But these techniques cannot be appropriate for the identification of localization of these modified sites [40]. m6A-seq or MeRIP-seq are important tools for the identification of m6A because of their accuracy and reproducibility [41]. Another detection technique for RNA methylation is m6A individual-nucleotide-resolution crosslinking and immunoprecipitation (miCLIP) [42]. Also, NGS-based protocols were developed for the detection of different RNA modifications [43].
Conclusion
Identification of important roles of m6A in human cancers makes targets editing of m6A for an effective treatment strategy. The identification of the increase of m6A RNA methylation in circulating tumor cells (CTCs) in lung cancer patients should be used as a key for identification of the metastasis mechanism of cancers in the future. Also, m6A RNA methylation could be a brand new therapeutic target in AML or glioblastoma.
References
Moore LD, Le T, Fan G. DNA methylation its basic function. Neuropsychopharmacology 2013;38(1):23–38.
Torres IO, Fujimori DG. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr Opin Struct Biol. 2015;35:68–75.
Curradi M, et al. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol Cell Biol. 2002;22(9):3157–73.
Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343–57.
EPIGENTEK. RNA methylation analysis made easy. 2010 http://dx.doi.org/10.1093/nar/gkp1117.
Liu J, Jia G. Methylation modifications in eukaryotic messenger RNA. J Genet Genomics. 2014;41(1):21–33.
Roignant JY, Soller M. m(6)A in mRNA: an ancient mechanism for fine-tuning gene expression. Trends Genet. 2017;33(6):380–90.
Deng X, et al. Role of N(6)-methyladenosine modification in cancer. Curr Opin Genet Dev. 2018;48:1–7.
Boccaletto P, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303-d307.
Squires JE, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40(11):5023–33.
Motorin Y, Lyko F, Helm M. 5-Methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res. 2010;38(5):1415–30.
Amort T, et al. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 2017. 18(1).
Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 2006;34(2):721–33.
Pan Y, et al. Multiple functions of m6A RNA methylation in cancer. J Hematol Oncol 2018;11(1):48.
Zhou KI, Pan T. Structures of the m6A methyltransferase complex: two subunits with distinct but coordinated roles. Mol Cell. 2016;63(2):183–5.
Ping XL, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
Zheng G, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Jia G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29(2):108–15.
Wang X, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Wang X, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Shi H, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Meyer KD, et al. 5′ UTR m(6)A promotes cap-independent translation. Cell. 2015;163(4):999–1010.
Alarcon CR, et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–308.
Liu N, et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Jaffrey SR, Kharas MG. Emerging links between m6A and misregulated mRNA methylation in cancer. Genome Med. 2017. 9.
Vu LP, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Weng H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22(2):191–205.e9.
Li Z, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. 2017;31(1):127–41.
Mochizuki S, Okada Y. ADAMs in cancer cell proliferation progression. Cancer Sci. 2007;98(5):621–8.
Cui Q, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–34.
Zhang S, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591–606.e6.
Li Y, Zhang S, Huang S. FoxM1: a potential drug target for glioma. Future Oncol. 2012;8(3):223–6.
Zhang C, et al., Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA 2016. 113(14):E2047-56
Cai X, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7 g. Cancer Lett. 2018;415:11–9.
Chen M, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70.
Chen J, et al. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16(23):2259–71.
Yuan S, et al. Methylation by NSun2 represses the levels and function of microRNA 125b. Mol Cell Biol. 2014;34(19):3630–41.
Pollex T, Hanna K, Schaefer M. Detection of cytosine methylation in RNA using bisulfite sequencing. Cold Spring Harb Protoc 2010. 2010(10): p. pdb.prot5505.
Peer E, Rechavi G, Dominissini D. Epitranscriptomics: regulation of mRNA metabolism through modifications. Curr Opin Chem Biol. 2017;41:93–8.
Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-sEq. Nature. 2012;485(7397):201–6.
Linder B, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–72.
Schwartz S, Motorin Y. Next-generation sequencing technologies for detection of modified nucleotides in RNAs. RNA Biol. 2017;14(9):1124–37.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that they have NO affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tuncel, G., Kalkan, R. Importance of m N6-methyladenosine (m6A) RNA modification in cancer. Med Oncol 36, 36 (2019). https://doi.org/10.1007/s12032-019-1260-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-019-1260-6