
Abstract
Malignant ascites is a common phenomenon in cancer patients. It poses a great challenge to the clinician, because of limited treatment options and strong impairment of the quality of life of the often palliative patients. The SECIMAS study investigated the feasibility of a re-challenge with four catumaxomab intraperitoneal infusions in patients who had already received a first cycle of four infusions in the phase III CASIMAS study, which compared catumaxomab with and without prednisolone premedication. The primary endpoint was the proportion of patients who received at least three catumaxomab infusions. Secondary endpoints included a composite safety score (CSS) summarising the worst grades for the main catumaxomab-related adverse events (pyrexia, nausea, vomiting and abdominal pain), safety, efficacy and the occurrence of anti-drug antibodies (ADAs). Eight of nine screened patients received a second catumaxomab cycle. Compliance with a catumaxomab re-challenge was high: all eight patients (100 %) received all four infusions. The median CSS was 3.0 versus 3.4 in CASIMAS. The tolerability profile of the second catumaxomab cycle was comparable to that of the first cycle. Median puncture-free survival (48 days) and overall survival (407 days) were longer than in CASIMAS (35 and 103 days, respectively), although median time to next puncture was shorter (60 vs. 97 days). Of six patients sampled, all were ADA positive at screening and remained ADA positive until the end of the study. The presence of ADAs did not affect catumaxomab’s safety or efficacy. The CSS and tolerability profile for catumaxomab in SECIMAS were comparable to those in CASIMAS. The majority of patients benefitted from a second cycle of catumaxomab. A re-challenge seems to be feasible and safe for selected patients with recurrent malignant ascites due to carcinoma after a first cycle of catumaxomab.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malignant ascites due to peritoneal carcinomatosis is a common complication of several cancers, including ovarian and gastrointestinal [1–4], and poses a great challenge to the clinician. It is usually associated with advanced disease and a poor prognosis, with median overall survival (OS) of approximately 3–9 months [1, 5–7]. Common symptoms, such as fatigue, dyspnoea and abdominal pain, bloating and swelling [1, 8], result in considerable discomfort and poor quality of life (QoL) [1, 4, 9, 10]. Currently, only limited guidelines for the treatment of malignant ascites are available [10] and the majority of treatment approaches, such as paracentesis, intraperitoneal chemotherapy, diuretics and shunt systems [10, 11], have only short-term effects or no effects. There is thus a need for more effective therapies.
The trifunctional monoclonal antibody catumaxomab [anti-epithelial cell adhesion molecule (EpCAM) × anti-CD3; Removab®, Fresenius Biotech GmbH, Munich, Germany] was approved by the European Medicines Agency in April 2009 for the intraperitoneal treatment of malignant ascites in patients with EpCAM-positive carcinomas where standard therapy is not available or no longer feasible [12]. EpCAM provides a good target for antibody therapy as it is overexpressed by most (87–100 %) epithelial cancers (e.g. ovarian, gastric and colorectal) [13–16], and EpCAM-positive tumour cells are found in the majority (70–100 %) of malignant effusions [17, 18]. Catumaxomab has two different antigen-binding sites that bind to epithelial tumour cells via EpCAM and to T cells via CD3 [19, 20] and an intact Fc domain that binds to type I, IIa and III Fcγ-receptor-positive accessory cells, e.g. natural killer cells, macrophages [21]. Its anti-tumour activity results from T cell-mediated lysis, antibody-dependent cell-mediated cytotoxicity and phagocytosis [21–23].
Catumaxomab has demonstrated efficacy in the treatment of patients with recurrent malignant ascites due to a number of epithelial cancers in a pivotal phase II/III study [24] and in a second phase IIIb study (CASIMAS) [25]. In the pivotal study, catumaxomab was associated with significantly longer puncture-free survival (PuFS) and time to next puncture (TTPu). It statistically significantly improved ascites-related symptoms compared with paracentesis alone [24]. The safety profile was acceptable and reflected its mode of action [24]. The safety and efficacy of catumaxomab were confirmed in the second phase IIIb study (CASIMAS), which compared catumaxomab with and without prednisolone premedication [25].
Due to its murine nature, catumaxomab is highly immunogenic and is known to induce anti-drug antibodies (ADAs) [26, 27]. However, the safety and efficacy of catumaxomab may not be compromised by the occurrence of ADAs [26, 27]. In the pivotal study, catumaxomab had a similar safety and efficacy profile in the seven patients who were ADA positive prior to the fourth infusion as in the ADA-negative patients [24, 26]. A single case report showed that a second cycle of catumaxomab was effective despite the presence of ADAs and had acceptable tolerability [27]. The aim of the SECIMAS study was to investigate the feasibility of a re-challenge with four intraperitoneal infusions of catumaxomab in patients who had already been treated with a first complete cycle of four intraperitoneal infusions in the CASIMAS study.
Patients and methods
Patients
Male or female patients aged ≥18 years could be included in the study if they had a Karnofsky index ≥60 %, body mass index (BMI) 17–40 kg/m2, malignant ascites requiring a first therapeutic ascites puncture ≥60 days following the last catumaxomab intraperitoneal infusion in the CASIMAS study and for whom standard therapy was not available or no longer feasible. Exclusion criteria included: documented acute or chronic infection; concomitant treatment with investigational products other than catumaxomab, anti-cancer therapy, chemotherapy or radiotherapy; previous treatment with entirely murine monoclonal antibodies other than catumaxomab; known or suspected hypersensitivity to catumaxomab or similar antibodies; inadequate respiratory, renal (creatinine > 1.5 × upper limit of normal (ULN) or glomerular filtration rate <75 % of lower limit of normal) or hepatic (aspartate aminotransferase (AST), alanine aminotransferase (ALT) > 5 × ULN; bilirubin > 1.5 × ULN) function; platelets <80,000 cells/mm3; absolute neutrophil count <1,500 cells/mm3 and albumin <3 g/dL or total protein <6 g/dL.
Study design
This was a single-arm, open-label phase I/II study to investigate the feasibility and safety of a second cycle of catumaxomab in patients with malignant ascites due to carcinoma requiring their first therapeutic puncture after treatment with catumaxomab in the CASIMAS study. Up to 30 evaluable patients from both arms of the CASIMAS were planned to be enrolled. As in the first cycle of treatment in the CASIMAS study, patients received four, 3-h, constant rate, intraperitoneal infusions of catumaxomab via an indwelling intraperitoneal catheter at a dose of 10, 20, 50 and 150 μg on days 0, 3, 7 and 10, respectively. Paracetamol 1,000 mg i.v. was given 30 min before each infusion to prevent pyrexia and pain. In addition, to enable optimal distribution of catumaxomab in the peritoneal cavity, 500 mL of 0.9 % sodium chloride solution was administered intraperitoneally before each infusion. The study consisted of a 3-day screening period, an 11-day treatment phase, which could be extended to a maximum of 20 days, and two follow-up visits. Study visits were screening (visit 1): within ≤3 days before the first catumaxomab infusion; treatment visits (2–5): on each infusion day; follow-up (visit 6): 8 days (±2 days) after the last dose of catumaxomab; and end of study (visit 7): 28 days (±4 days) after the last dose of catumaxomab. After the end-of-study visit, post-study follow-up assessments were conducted by telephone every 2 months until either death or 6 months after the last patient started catumaxomab treatment, whichever occurred first.
The study was conducted according to the principles of the International Conference on Harmonization Guideline for Good Clinical Practice, the Declaration of Helsinki and local legal and regulatory requirements. The study protocol and other appropriate documents were approved by the relevant independent ethics committees and regulatory authorities. Patients provided written informed consent prior to study entry.
Endpoints
The primary endpoint was the proportion of patients who were able to receive a second cycle of at least three intraperitoneal infusions of catumaxomab. The main secondary endpoint was a composite safety score, consisting of a score calculated from the incidence and intensity of the most frequent catumaxomab-related adverse events (Medical Dictionary for Regulatory Activities preferred terms pyrexia, nausea, vomiting and abdominal pain) occurring between the first infusion and 28 days after the last infusion. Adverse events were graded using the Common Terminology Criteria for Adverse Events (CTCAE) v3.0. The worst grade for each of the four adverse events was added together for each patient to obtain the composite safety score. A higher score indicates a higher and/or more severe occurrence of these catumaxomab-related adverse events (possible score range 0–20). In addition, patient intra-individual comparisons were made by comparing the score after the first cycle in the CASIMAS study with the score after the second cycle. The intra-individual difference between the composite safety scores after the second cycle and first cycle in the CASIMAS study were calculated and analysed descriptively. Other secondary endpoints were safety, efficacy (PuFS, TTPu, number of ascites punctures and OS), the development of ADAs, quality of life (QoL) assessed with the EQ-5D-3L questionnaire and visual analogue scale (VAS) [28] and ascites-related symptoms assessed with the Functional Assessment of Chronic Illness Therapy Ascites Index (FACIT-AI) [29]. PuFS was defined as the time from the day of catheter removal to the first need for therapeutic ascites puncture or death, whichever occurred first. TTPu was defined as the time from the day of catheter removal to the first need for therapeutic ascites puncture. OS was defined as the time from randomization in the CASIMAS study until death of the patient in the SECIMAS study. The number of ascites punctures was measured from the day of catheter removal until death or 6 months after the last patient started treatment, whichever occurred first. Study results were compared with those from the CASIMAS study.
Statistics
Statistical analyses were performed with the SAS software package version 9.1 or higher at an alpha level of 0.05. All statistical tests were two sided. The analyses were of an exploratory nature with no adjustments being made for multiplicity. There was no confirmative hypothesis testing. No sample size calculation was performed as it was not necessary for exploratory studies. Results were analysed descriptively.
Results
Patients
Nine female patients who completed treatment in the CASIMAS study met the inclusion criteria and were screened for enrolment in the SECIMAS study. Of these patients, eight were re-challenged with catumaxomab and were included in the analysis. In the CASIMAS study, four of these patients had received catumaxomab plus prednisolone and the other four patients had received catumaxomab alone. Patient characteristics are shown in Table 1. As in the CASIMAS study, ovarian cancer was the most common tumour type. Compared with the overall population in the CASIMAS study, most patients in the SECIMAS study had a higher Karnofsky index at screening, indicating a highly selected patient population. All patients had undergone primary debulking surgery. Seven patients (87.5 %) had received chemotherapy (five who had received carboplatin and paclitaxel); two patients (25.0 %) had received radiotherapy; and three patients (37.5 %) had received hormone therapy prior to catumaxomab treatment.
Primary endpoint
All eight patients received at least three catumaxomab infusions; thus, the proportion of patients who met the primary endpoint was 100 %. In addition, all eight patients (100 %) received all four infusions compared with 76 % of patients in the CASIMAS study.
Secondary endpoints
The mean composite safety score was 3.0, comparable to that (3.4) for the first cycle of catumaxomab the SECIMAS patients received in the CASIMAS study and to that (3.8) for the 107 patients who received catumaxomab alone in the CASIMAS study (Table 2). All eight patients experienced a total of 84 adverse events and a total of 68 catumaxomab-related adverse events (Table 3). The most common adverse events were fatigue (six patients; 75 %), nausea (four patients; 50 %), abdominal pain (four patients; 50 %) and pyrexia (three patients; 37.5 %). The majority of the most frequently reported adverse events resolved within 7 days. The most common catumaxomab-related adverse events were fatigue (five patients; 62.5 %), nausea (four patients; 50.0 %), abdominal pain (three patients; 37.5 %) and pyrexia (three patients; 37.5 %). Nine serious adverse events were reported in three patients (38 %), being abdominal discomfort (one patient; 12.5 %), ascites (one patient; 12.5 %), general physical health deterioration (one patient; 12.5 %) and nausea (two patients; 25 %), vomiting (two patients; 25 %) and subileus (two patients; 25 %), respectively. No hypersensitivity or allergic reactions were observed after catumaxomab administration, and no patients discontinued the study due to an adverse event. One patient experienced a fatal adverse event (general physical health deterioration), which was not considered to be related to catumaxomab, and died on day 29, 17 days after the last catumaxomab infusion. Compared with patients who received catumaxomab alone in the CASIMAS study, there was a similar incidence of adverse events and catumaxomab-related adverse events and a lower incidence of grade ≥3 and serious adverse events (overall and catumaxomab related), cytokine-release-related symptoms and adverse events leading to discontinuation.
Liver function tests showed transient increases in AST, ALT, gamma-glutamyltransferase and alkaline phosphatase. In the majority of cases, these changes were without clinical signs and symptoms and were generally fully reversible. The median white blood cell count and neutrophil count remained within normal limits. There was a transient decrease in the peripheral lymphocyte count during treatment, but this returned to normal at follow-up.
In terms of efficacy (Table 4), median PuFS (48 days) and median OS (407 days) were longer than in the CASIMAS study for the overall population (35 and 103 days, respectively), although median TTPu (60 days) was shorter than in CASIMAS study (97 days). OS after the second cycle of catumaxomab was also longer than in a comparable subpopulation of 64 patients from the CASIMAS study (201 days) who had received all four catumaxomab infusions in the first treatment cycle and had not received a therapeutic ascites puncture for ≥60 days but were not enrolled in SECIMAS. A comparison of PuFS and TTPu between SECIMAS and CASIMAS for individual patients is shown in Table 5. After the second catumaxomab cycle in SECIMAS, TTPu was still ≥4 weeks in four of eight patients (50.0 %) and the other four patients (50.0 %) did not require a further therapeutic puncture before death (Table 5). The median number of ascites punctures per patient was 0.5 (range 0–13; mean ± standard deviation 2.3 ± 4.46) compared with 0 in CASIMAS (range 0–24; mean 2.4 ± 6.12; n = 211).
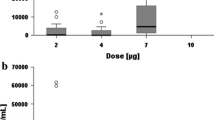
Ascites and plasma samples for determination of ADA concentrations were obtained from six patients (75.0 %). All six patients were ADA positive in ascites and plasma at screening and remained ADA positive until the end of the study. In both ascites and plasma, the formation of ADAs occurred in a similar pattern, but ADA concentrations were consistently higher in plasma compared with ascites. Individual plasma ADA concentrations started to increase between the third and fourth catumaxomab infusion and increased further during follow-up until day 28 after the last infusion. Individual ADAs concentrations over time were higher in plasma (range 0.002–101.41 mg/mL) than in ascites (range 0.001–0.77 mg/mL). During the treatment period, the highest ADA concentrations occurred at the end of the treatment period in ascites (range 0.007–0.77 mg/mL) and plasma (range 0.576–27.44 mg/mL). In the CASIMAS study, all patients were ADA negative at screening; plasma was ADA positive in up to 15 % of patients during the treatment phase; and at days 8 and 28 follow-up visits, 100 % of the evaluated samples were ADA positive. In ascites, up to 2 % of the evaluated samples were ADA positive during treatment and 67 % were ADA positive at the puncture visit. In general, median ADA concentrations in ascites and plasma were higher during the second cycle of catumaxomab in SECIMAS (range 0.008–11.67 mg/mL and 0.029–0.247 mg/mL, respectively) compared with the first cycle in CASIMAS (range 0–0 mg/mL and 0–0.00003 mg/mL, respectively).
QoL showed little change during the study. The EQ-5D-3L questionnaire results showed that the proportion of patients with no problems remained unchanged for two dimensions (usual activities and anxiety/depression) and decreased for the remaining dimensions (mobility, self-care and pain/discomfort) during the treatment phase (from screening to day 10). In the follow-up period from day 8 to day 28, the proportion of patients with no problems remained stable for two dimensions (usual activities and mobility), increased for two dimensions (self-care and anxiety/depression) and decreased for one dimension (pain/discomfort). In general, the mean (±SD) EQ-5D-3L VAS score remained unchanged between screening (61.2 ± 18.54), the end of treatment (63.0 ± 15.33) and follow-up day 28 (60.8 ± 18.55). Ascites symptoms were also relatively stable during the study: the median FACIT-AI scores for individual ascites-related symptoms from screening to follow-up day 28 showed little change over time. In addition, the FACIT-AI total score, which summarises all 13 symptoms, remained generally unchanged between screening (mean 30.80 ± 3.271; median 32.0, range 27.0–35.0) and follow-up day 28 (mean 31.35 ± 3.591; median 31.5, range 27.1–37.0).
Discussion
The SECIMAS study investigated the safety and feasibility of a re-challenge with four intraperitoneal catumaxomab infusions for patients who benefitted from a first catumaxomab treatment cycle in the CASIMAS study but still presented with recurrent ascites. There were two major points to consider when administering a second catumaxomab treatment cycle due to the immunogenicity of the antibody and the anticipated high ADA response. Firstly, there might be a risk of allergic reactions due to prior exposure to catumaxomab. Secondly, efficacy might be impaired due to ADA-mediated neutralisation of catumaxomab before binding to the tumour cells could occur.
Generally, it has to be noted that the patients enrolled in the SECIMAS study and were able to receive a second cycle of catumaxomab were highly selected patients from the CASIMAS study. They needed to have a puncture-free interval of at least 60 days after having received a full catumaxomab cycle in the CASIMAS study and still had to be in a good general health condition despite their already advanced stage of disease after the first treatment cycle. This kind of selection was in indeed confirmed by the fact that only 8 patients could be included in the SECIMAS.
The results of this study show that compliance with a second cycle of catumaxomab was very high: all eight patients received all four infusions, so the primary endpoint (the proportion of patients who were able to receive a second cycle of at least three intraperitoneal catumaxomab infusions) was 100 %. In the CASIMAS study, 76 % of patients received all four catumaxomab infusions, while in the pivotal phase III study, the proportion was 83 %.
The mean composite safety score was 3.0, which is comparable to that (3.4) for the first cycle of catumaxomab, the patients received in the CASIMAS study and to that (3.8) for patients who received catumaxomab alone in the CASIMAS study. This small difference in the composite safety score could lead to the interpretation that a second cycle of catumaxomab does not show cumulative side effects. This would be in concordance with the clinical experience and study data that do not show cumulative effects in the first cycle. This is even though one cycle consists of four repetitive doses. The side effects of the second cycle of catumaxomab were comparable to other treatments in the selected patient population, generally in line with the well-known safety profile of the drug and similar to that of the first treatment cycle in the CASIMAS study [25]. The main catumaxomab-related adverse events were fatigue, nausea, abdominal pain and pyrexia. Although all six patients who were sampled were ADA positive in ascites and plasma at screening and remained ADA positive until the end of the study, the presence of ADAs did not affect the safety of catumaxomab and no hypersensitivity reactions or type I allergic reactions were noted, despite the high immunogenicity of the drug.
In addition, the efficacy of catumaxomab did not appear to be impaired by the presence of ADAs. The efficacy results show that patients can benefit from a second cycle of catumaxomab despite having advanced disease. Despite the fact that our results could be subject to selection bias, it was noted that median PuFS (48 days) and median OS (407 days) were longer than in the CASIMAS study (35 and 103 days, respectively). Median TTPu was still longer than 4 weeks, which is clinically relevant in this patient population with advanced disease. The median number of ascites punctures per patient (0.5; range 0–13) was similar to that in the CASIMAS study (0; range 0–24), and 50 % of the patients did not require a further therapeutic puncture before death, indicating a comparable clinical benefit for the second cycle of catumaxomab.
The development of ADAs after the administration of murine antibodies is well known, and their presence has not been associated with any major safety issues [31–33]. ADAs may in fact be associated with positive humoral effects and prolonged survival [33–35]. In a post hoc analysis of the pivotal phase II/III study in patients with malignant ascites, there was a strong correlation between the humoral response to catumaxomab and clinical outcome: patients who developed ADAs had a significant improvement in clinical outcome in terms of PuFS, TTPu and OS compared with ADA-negative patients in the overall population and in the ovarian, non-ovarian and gastric cancer subpopulations.
QoL, assessed with the EQ-5D-3L questionnaire and visual analogue scale, remained relatively unchanged throughout the study and reflect the patients’ advanced stage of disease. In addition, the FACIT-AI scores showed that patients’ ascites symptoms remained fairly stable during treatment and follow-up.
The results of this study confirm the previous clinical experience in one patient who was treated with a second cycle of catumaxomab [27]. This case study showed that despite elevated ADA concentrations, a second catumaxomab cycle was still effective providing relief from ascites-related symptoms and prolonged PuFS [27]. This preserved efficacy was also reflected in the efficient elimination of tumour cells and a rapidly increasing number of CD45+ immune cells in the peritoneal compartment [27]. It may be that the efficacy of a second cycle of catumaxomab is due to an immunological booster effect. This might be indicated by the finding that humoral immune responses against human epidermal growth factor receptor 2 (HER2) (anti-HER2 immunoglobulin G) were detected in two patients who received a second treatment cycle after recurring malignant ascites [30].
In conclusion, compliance with a second cycle of catumaxomab was very high; the composite safety score and tolerability profile for catumaxomab in SECIMAS were comparable to those in CASIMAS. The presence of ADAs did not seem to affect either the safety or efficacy of catumaxomab. The majority of patients benefitted from a second cycle of catumaxomab despite further disease progression. A re-challenge seems to be safe and feasible for selected patients with recurrent malignant ascites due to carcinoma after a first cycle of catumaxomab.
Limitations
This study reports the first experience of a re-challenge with a second cycle of catumaxomab under controlled study conditions. Even though we think that this first report consists of valuable data, it has to be stated that our study is subject to certain limitations. Obviously, we can only draw conclusions on the effect of a second cycle of catumaxomab in female patients, since no male participants have been included in this study. Also this first experience is limited to patients with ovarian, breast and urachal cancer; no conclusions can be drawn regarding other entities. Also the small number of patients poses the treat of selection bias and limits the evidence level of our data. Nevertheless, we feel that these first results are encouraging to warrant future research with a greater number of patients.
Abbreviations
- ADA:
-
Anti-drug antibody
- CTCAE:
-
Common terminology criteria for adverse events
- EpCAM:
-
Epithelial cell adhesion molecule
- OS:
-
Overall survival
- PuFS:
-
Puncture-free survival
- TTPu:
-
Time to next puncture
- VAS:
-
Visual analogue scale
References
Ayantunde AA, Parsons S. Pattern and prognostic factors in patients with malignant ascites: a retrospective study. Ann Oncol. 2007;18:945–9.
Chung M, Kozuch P. Treatment of malignant ascites. Curr Treat Options Oncol. 2008;9:215–33.
Tamsma J. The pathogenesis of malignant ascites. Cancer Treat Res. 2007;134:109–18.
Woopen H, Sehouli J. Current and future options in the treatment of malignant ascites in ovarian cancer. Anticancer Res. 2009;29:3353–9.
Cui S, Ba M, Tang Y, Liu J, Wu Y, Wang B, Zhang X, Tang H, Zhong S. B ultrasound-guided hyperthermic intraperitoneal perfusion chemotherapy for the treatment of malignant ascites. Oncol Rep. 2012;28:1325–31.
Takeyoshi I, Makita F, Iwazaki S, Ishikawa H, Kakinuma S, Sato Y, Ohya T, Nakagami K, Tomizawa N, Izumi M, Kobayashi I, Tanahashi Y, Kobayashi J, Kamoshita N, Kawate S, Sunose Y, Sakamoto I, Yoshinari D, Yamada T, Okabe T. Weekly paclitaxel in combination with doxifluridine for peritoneally disseminated gastric cancer with malignant ascites. Anticancer Res. 2011;31:4625–30.
Valle M, Van der Speeten K, Garofalo A. Laparoscopic hyperthermic intraperitoneal preoperative chemotherapy (HIPEC) in the management of refractory malignant ascites: a multi-institutional retrospective analysis in 52 patients. Surg Oncol. 2009;100:331–4.
Adam RA, Adam YG. Malignant ascites: past, present, and future. J Am Coll Surg. 2004;198:999–1011.
Becker G, Galandi D, Blum HE. Malignant ascites: systematic review and guideline for treatment. Eur J Cancer. 2006;42:589–97.
Cavazzoni E, Bugiantella W, Graziosi L, Franceschini MS, Donini A. Malignant ascites: pathophysiology and treatment. Int J Clin Oncol. 2013;18:1–9.
Barni S, Cabiddu M, Ghilardi M, Petrelli F. A novel perspective for an orphan problem: old and new drugs for the medical management of malignant ascites. Crit Rev Oncol Hematol. 2011;79:144–53.
Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010;36:458–67.
Went P, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, Dirnhofer S. Frequent EpCAM protein expression in human carcinomas. Hum Pathol. 2004;35:122–8.
Went P, Vasei M, Bubendorf L, Terracciano L, Tornillo L, Riede U, Kononen J, Simon R, Sauter G, Baeuerle PA. Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Br J Cancer. 2006;94:128–35.
Spizzo G, Fong D, Wurm M, Ensinger C, Obrist P, Hofer C, Mazzoleni G, Gastl G, Went P. EpCAM expression in primary tumour tissues and metastases: an immunohistochemical analysis. Clin Pathol. 2011;64:415–20.
Patriarca C, Macchi RM, Marschner AK, Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev. 2012;38:68–75.
Davidson B, Risberg B, Kristensen G, Kvalheim G, Emilsen E, Bjåmer A, Berner A. Detection of cancer cells in effusions from patients diagnosed with gynaecological malignancies: evaluation of five epithelial markers. Virchows Arch. 1999;435:43–9.
Passebosc-Faure K, Li G, Lambert C, Cottier M, Gentil-Perret A, Fournel P, Pérol M, Genin C. Evaluation of a panel of molecular markers for the diagnosis of malignant serous effusions. Clin Cancer Res. 2005;11:6862–7.
Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98:2526–34.
Ruf P, Gires O, Jäger M, Fellinger K, Atz J, Lindhofer H. Characterisation of the new EpCAM-specific antibody HO-3: implications for trifunctional antibody immunotherapy of cancer. Br J Cancer. 2007;97:315–21.
Zeidler R, Mysliwietz J, Csánady M, Walz A, Ziegler I, Schmitt B, Wollenberg B, Lindhofer H. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer. 2000;83:261–6.
Jäger M, Schoberth A, Ruf P, Hess J, Hennig M, Schmalfeldt B, Wimberger P, Ströhlein M, Theissen B, Heiss MM, Lindhofer H. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3). Cancer Res. 2012;72:24–32.
Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubal S, Schmitt B, Lindhofer H. Simultaneous activation of T-cells and accessory cells by a new class of intact bispecific antibody results in efficient tumour cell killing. J Immunol. 1999;163:1246–52.
Heiss MM, Murawa P, Koralewski P, Kutarska E, Kolesnik OO, Ivanchenko VV, Dudnichenko AS, Aleknaviciene B, Razbadauskas A, Gore M, Ganea-Motan E, Ciuleanu T, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: results of a prospective randomized phase II/III trial. Int J Cancer. 2010;127:2209–21.
Sehouli J, Wimberger P, Vergote I, Rosenberg P, Schneeweiss A, Bokemeyer C, Salat C, Scambia G, Berton Rigaud D, Lordick F. Catumaxomab versus catumaxomab plus prednisolone in patients with malignant ascites due to epithelial cancer: results from the CASIMAS study. Ann Oncol. 2012;23 (Suppl 9):ix539[Abstract 1683P].
Ott MG, Marmé F, Moldenhauer G, Lindhofer H, Hennig M, Spannagl R, Essing MM, Linke R, Seimetz D. Humoral response to catumaxomab correlates with clinical outcome: results of the pivotal phase II/III study in patients with malignant ascites. Int J Cancer. 2012;130:2195–203.
Pietzner K, Jäger M, Schoberth A, Oskay-Özcelik G, Kuhberg M, Lindhofer H, Sehouli J. First patient treated with a re-challenge of catumaxomab in recurrent malignant ascites: a case report. Med Oncol. 2012;29:1391–6.
The EuroQol Group. EuroQol—a new facility for the measurement of health-related quality of life. Health Policy. 1990;16:199–208.
Cella D, Neubauer N, Thomas J, Kutner J, Seiden MV. The FACIT-AI, a new tool for assessing symptoms associated with malignant ascites. Gynecol Oncol. 2013;128:187–90.
Ruf P, Foerster B, Jager M, Martinius H, Seimetz D, Lindhofer H. Humoral tumor-associated immune responses induced by catumaxomab in patients with malignant ascites. J Clin Oncol. 2011;29 (suppl: abstr 2575).
DeNardo GL, Bradt BM, Mirick GR, DeNardo SJ. Human antiglobulin response to foreign antibodies: therapeutic benefit? Cancer Immunol Immunother. 2003;52:309–16.
Marmé A, Strauss G, Bastert G, Grischke EM, Moldenhauer G. Intraperitoneal bispecific antibody (HEA125xOKT3) therapy inhibits malignant ascites production in advanced ovarian carcinoma. Int J Cancer. 2002;101:183–9.
Mirick GR, Bradt BM, Denardo SJ, Denardo GL. A review of human antiglobulin antibody (HAGA, HAMA, HACA, HAHA) responses to monoclonal antibodies. Not four letter words. Q J Nucl Med Mol Imaging. 2004;48:251–7.
Azinovic I, DeNardo GL, Lamborn KR, Mirick G, Goldstein D, Bradt BM, DeNardo SJ. Survival benefit associated with human anti-mouse antibody (HAMA) in patients with B-cell malignancies. Cancer Immunol Immunother. 2006;55:1451–8.
DeNardo GL, Mirick GR, Kroger LA, Bradt BM, Lamborn KR, DeNardo SJ. Characterization of human IgG antimouse antibody in patients with B-cell malignancies. Clin Cancer Res. 2003;9:4013S–21S.
Acknowledgments
The study was sponsored by Neovii Biotech GmbH and supported by the North-Eastern German Society of Gynecology (NOGGO). The authors would like to thank the freelance medical writer Kevin De-Voy (funded by Fresenius Biotech GmbH) for his writing support.
Conflict of interest
Holger Martinius and Hilke Friccius-Quecke are employees of Neovii Biotech GmbH (formerly Fresenius Biotech GmbH). Klaus Pietzner has received honoraria for scientific presentations. All other authors report no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Novelty and impact of the work Due to its murine nature, catumaxomab is highly immunogenic and induces anti-drug antibodies (ADAs). However, its safety and efficacy may not be compromised by ADAs. This study shows that compliance with a second cycle is high and that the presence of ADAs did not affect either the safety or efficacy of catumaxomab. A re-challenge seems to be safe and feasible for selected patients with recurrent malignant ascites due to carcinoma after a first cycle of catumaxomab.
Rights and permissions
About this article

Cite this article
Pietzner, K., Vergote, I., Santoro, A. et al. Re-challenge with catumaxomab in patients with malignant ascites: results from the SECIMAS study. Med Oncol 31, 308 (2014). https://doi.org/10.1007/s12032-014-0308-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-014-0308-x