
Abstract
Metformin, one of most widely prescribed oral hypoglycemic agents, has recently received increased attention because of its potential antitumorigenic effects that are thought to be independent of its hypoglycemic effects. Several potential mechanisms have been suggested for the ability of metformin to suppress cancer growth in vitro and vivo: (1) activation of LKB1/AMPK pathway, (2) induction of cell cycle arrest and/or apoptosis, (3) inhibition of protein synthesis, (4) reduction in circulating insulin levels, (5) inhibition of the unfolded protein response (UPR), (6) activation of the immune system, and (7) eradication of cancer stem cells. There is also a growing number of evidence, mostly in the form of retrospective clinical studies that suggest that metformin may be associated with a decreased risk of developing cancer and with a better response to chemotherapy. There are currently several ongoing randomized clinical trials that incorporate metformin as an adjuvant to classic chemotherapy and aim to evaluate its potential benefits in this setting. This review highlights basic aspects of the molecular biology of metformin and summarizes new advances in basic science as well as intriguing results from recent clinical studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metformin (N′,N′-dimethylbiguanide) is the most widely prescribed oral hypoglycemic agent. It is believed to exert its effect by reducing hepatic glucose production and by increasing insulin sensitivity as well as glucose use by peripheral tissues. Guanidine was the active ingredient of Galega officinalis (goat’s-rue or French lilac), which was used to alleviate polyuria in medieval Europe. In the 1920s, diabetes pathophysiology was traced to the pancreas, and in the 1950s metformin and phenformin, the two main biguanides, were introduced. Metformin for the treatment of diabetes was approved in the 1970s in Europe and in 1995 in the United States. Since then, metformin use has been gradually increasing with 25 million prescriptions filled in 2000 and more than 40 million in 2008 in the United States. Its use in diabetes has shown to increase overall survival and prevent macrovascular complications better than other oral hypoglycemic drugs [1].
Metformin now has a wide variety of indications. It has successfully been used in polycystic ovarian syndrome (PCOS), where insulin resistance is a key factor for the development of the metabolic disturbances. In this setting, it has a favorable effect not only on subfertility but also on cardiometabolic aberrations observed in this syndrome, such as hyperlipidemia and hypertension [2]. It is also used in the management of the metabolic syndrome [3] and diabetes prevention [4] in high-risk populations.
Metformin has recently received increased attention for its potential antitumorigenic effects that are thought to be independent of its hypoglycemic effects. This has been evaluated in multiple in vitro and in vivo studies and is now being tested in clinical trials as an adjuvant to classic chemotherapeutic regimens.
The notion that biguanides have anticarcinogenic properties is not new. Earlier studies involving phenformin, a biguanide that was only briefly used in humans secondary to its increased propensity to cause lactic acidosis, suggested that it led to better inhibition of tumor cell growth in vitro as well as in vivo, when added to conventional chemotherapeutic agents [5–7].
This review highlights basic aspects of the molecular biology of metformin as well as the AMPK/LKB1 pathway, which it is believed to be the major pathway of action of this medication. We also summarize new advances in basic science as well as intriguing results from recent clinical studies.
Metformin biology
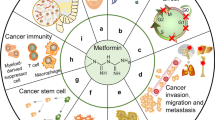
The anticarcinogenic effects of metformin have been attributed to several mechanisms that we will discuss further: (1) activation of LKB1/AMPK pathway, (2) induction of cell cycle arrest and/or apoptosis, (3) inhibition of protein synthesis, (4) reduction in circulating insulin levels, (5) inhibition of the unfolded protein response (UPR), (6) activation of the immune system, and (7) eradication of cancer stem cells (Fig. 1).

Possible mechanisms by which metformin may be able to inhibit cancer growth. By activating AMP-activated protein kinase (AMPK), it inhibits mammalian target of rapamycin (mTOR) downstream signaling, induces cell cycle arrest, and inhibits protein synthesis in cancer cells. It also inhibits the unfolded protein response (UPR), leading to apoptosis. Additionally, it decreases circulating insulin levels, inhibits angiogenesis, and exerts a toxic effect on cancer stem cells
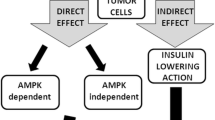
The potentially beneficial effects of metformin against cancer are believed to be mediated mainly by AMP-activated protein kinase (AMPK), a molecule that is a major player in the regulation of metabolism and growth, for both normal and cancer cells. However, more recent data have suggested that the metformin can also regulate cancer cell biology in an AMPK-independent manner.
The major link between AMPK, cancer, and metformin is believed to be the AMPK upstream kinase LKB1 (Fig. 1). Most researchers so far have adopted a simplified model in which metformin exerts its antitumorigenic effects by activating LKB1 and its downstream target AMPK, which, in turn, suppresses the activity of the mammalian target of rapamycin (mTOR), a signaling pathway with a central role in cancer cell growth and cancer pathogenesis.
LKB1 is a tumor suppressor protein [8] that is mutated in Peutz-Jeghers syndrome, an inherited disorder characterized by hamartomas and increased susceptibility to gastrointestinal malignancies. Interestingly, it is also somatically inactivated in 30–50% of sporadically occurring lung adenocarcinomas, 20% of squamous cell carcinomas, and 10% of large cell carcinomas of the lung as well as other tumors. The effect of metformin on the LKB1/AMPK pathway was first studied in glucose homeostasis in liver, where it was shown that loss of LKB1 function resulted in hyperglycemia with increased gluconeogenic and lipogenic gene expression. The authors suggested that the glucose lowering effects of metformin were mediated by activation of LKB1 in the liver, and this was proposed as the major mechanism through which metformin helped with glycemic control in diabetic patients [9]. Some authors have suggested that molecular studies of LKB1 expression and mTOR pathway activation could help predict the effectiveness of metformin in human cancers [10] although this pathway seems to be only one of many by which metformin exerts its effects in vivo. For instance, it was recently shown that metformin regulated mTOR, independently of AMPK, and through the Rag family of transmembrane GTPases [11]. In addition, many malignancies acquire mutations of LKB1 during growth, and therefore LKB1 expression may correlate poorly with effectiveness of metformin or other LKB1 activators. These observations generated the hypothesis that metformin, in addition to its role in glucose metabolism, may have a role in cancer biogenesis, and this in turn led to numerous in vitro and in vivo studies.
AMP-activated protein kinase and carcinogenesis
Otto Warburg as early as 1956 noted that cancer cells need to utilize glucose at a much higher rate compared to normal cells [12]. It has been hypothesized that, as a result, they are more sensitive to nutrient starvation than nonmalignant cells. Indeed, most of the time, and especially in rapidly growing tumors, cancer cells need to be able to survive in a nutrient-deprived environment. AMPK is a Ser/Thr protein kinase that is a major regulator of cell metabolism. AMPK is activated by a high AMP/ATP ratio that essentially signifies lack of or decrease in available energy resources for the cell. After its activation, it inhibits anabolic pathways and activates catabolic pathways. In this way, it allows the cell to survive during periods of metabolic stress [13]. Therefore, although in normal cells AMPK activation leads to energy conservation and ultimately increased survival, rapidly growing cancer cells are not able to sustain this AMPK-induced limit on utilization of available resources.
AMPK has the ability to regulate protein metabolism, cell polarity, growth, and apoptosis indirectly by tilting the metabolic balance toward energy conservation, but also directly by several other mechanisms.
Inhibition of protein synthesis by AMPK is achieved by two distinct mechanisms. AMPK activation results in inhibition of both elongation factor-2 [14] and mTOR pathway. The latter is more clinically relevant to cancer development since the mTOR signaling pathway is believed to be a central player in cell growth signaling [15]. It has been shown that AMPK phosphorylates tuberous sclerosis complex-2 (TSC-2) that in turns inhibits mTOR signaling [16]. AMPK is also able to directly phosphorylate co-signaling molecules that bind to mTOR and inhibit its action [17]. Thus, the mTOR pathway can be perceived as a major link between metformin, AMPK, and cancer biology.
AMPK activation assists in adaptation to metabolic stress not only by inhibiting cell growth and proliferation but also by inducing autophagy. Autophagy is a catabolic process involving the degradation of a cell’s own components through the lysosomal machinery, and it is a major survival mechanism by which a starving cell reallocates nutrients from unnecessary processes to more essential ones. The mechanisms by which AMPK is able to achieve this have not been elucidated yet and are a focus of intense research. For instance, AMPK phosphorylates the cyclin-dependent kinase inhibitor p27 and stabilizes it thereby permitting cells to survive growth factor withdrawal and metabolic stress through autophagy [18]. In addition, AMPK activation induces phosphorylation of p53, and this is required to initiate AMPK-dependent cell-cycle arrest [19]. Another possible mechanism by which AMPK may regulate cell growth involves reducing the cytoplasmic-to-nuclear ratio of the RNA-binding protein HuR, which in turn reduces mRNA stability of critical cell cycle regulators such as cyclins A and B1 [20].
Finally, AMPK is also essential for the transcriptional activity of the hypoxia-inducible factor 1 (HIF-1), a transcription factor that is critical for hypoxic induction of genes necessary for adaptation to a hypoxic environment, such as that found inside a solid tumor [21]. Of note, mTOR is a known upstream activator of HIF-1 [22, 23]; therefore, inhibition of mTOR can also indirectly and independently of AMPK inhibit HIF-1 signaling.
Effects on cell cycle and protein synthesis
Anisimov et al. was the first to show that metformin significantly decreased the incidence and size and increased mean latency to development of mammary adenocarcinomas in transgenic HER-2/neu-positive mice [24]. Thereafter, the key discovery that metformin action in glucose metabolism is mediated through the tumor suppressor gene LKB1, an activator of AMPK, sparked the interest, and with it further research, on a potential link between metformin and cancer [9]. It has been studied in vitro and in vivo in various common malignancies mostly breast and also prostate, colorectal, pancreatic, and others.
Breast cancer has been extensively studied as a potential therapeutic target for metformin. Metformin was able to inhibit breast cancer cell growth in vitro in an AMPK-dependent manner, and this growth inhibition was associated with decreased mTOR activation [25]. In two studies, metformin was able to inhibit translation initiation in breast cancer cell lines with variable ER receptor and erb-B2 oncogene status, by decreasing phosphorylation of S6 kinase, ribosomal protein S6, and eIF4E-binding protein 1, and this in turn was associated with mTOR inhibition. This led to cell cycle arrest and cell growth inhibition. The effects of metformin on translation were mediated by AMPK, as treatment of cells with the AMPK inhibitor compound C prevented the inhibition of translation [26, 27]. Metformin was able not only to inhibit cell growth but also to induce cell death in vitro and in vivo in triple-negative (ER/PR/Her-2 neu) breast cancer cells [28]. This profound effect of metformin in this specific subtype of cancer is interesting, especially if one considers that triple-negative breast cancer is more common in more obese and diabetic women [29]. Some authors have proposed that acquired resistance to newer tyrosine kinase inhibitors (TKIs) for HER2-positive breast cancer such as lapatinib, which is mediated through the activation of the mTOR pathway, may be delayed or attenuated with the use of metformin [30]. It has also been suggested that metformin could prevent or reduce cardiotoxicity of HER-2 inhibitors such as trastuzumab [31], since AMPK activation is essential for sparing of normal cardiac cells after trastuzumab treatment [32]. The authors suggested that newer generation inhibitors of Her-2, such as lapatinib, in addition to blocking Her-2, were also able to activate AMPK, where as trastuzumab was not, therefore depriving cardiac myocytes from an essential stress-related survival mechanism.
Furthermore, metformin suppressed HER2 (erbB-2) oncoprotein overexpression via inhibition of mTOR, an effect that was not entirely dependent on AMPK and that was enhanced after co-incubation with agents that block reactive oxygen species (ROS) production (e.g., N-acetylcysteine) [33]. The importance of the latter finding in this setting remains unclear. It is known for instance that metformin directly scavenges ROS and regulates the intracellular production of superoxide anion [34] and that ROS regulate insulin-induced expression of prometastatic molecules such as VEGF and HIF [35]. This could suggest a complex and possibly negative feedback loop between metformin/AMPK and ROS that leads to impaired response to oxidative stress in cancer cells.
AMPK activation is also responsible for the inhibition of aromatase. Metformin was able to inhibit aromatase in human breast adipose stromal cells in vitro resulting in the inhibition of estrogen production, and the potential for prevention of breast cancer development as well as use as an adjunct to treatment of hormonally responsive cells [36]. Finally, in a genome wide analysis of human breast cancer cells, metformin suppressed expression of genes coding for ribosomal proteins and numerous mitosis-related gene families including kinesins, tubulins, histones, and others underlying the global effect that this drug has on the regulation of genes related to cell growth [37].
In addition to breast cancer, a number of other malignancies have been studied in vitro as well as in vivo using animal models. In low-density cultures of a rat glioma cell line, where cells were still able to rapidly proliferate (log phase), metformin blocked cell cycle progression in G (0)/G (1) phase without inducing significant cell death. In confluent cultures on the other hand, where cells had reached the plateau phase of growth, metformin caused massive induction of caspase-dependent apoptosis. Interestingly, metformin-triggered apoptosis was completely prevented by agents that block mitochondrial permeability. The antiglioma effect of metformin was reduced by compound C, an inhibitor of AMPK, and was mimicked by the AMPK agonist AICAR [38]. Activation of AMPK by AICAR resulted in potent suppressive effects on renal cell carcinoma (RCC) growth, while combinations of AICAR and metformin with statins were potent inducers of apoptosis in such cells and demonstrated potent suppressive effects on RCC tumorigenicity in vitro [39]. Likewise, in pancreatic cancer cells, metformin was able to induce apoptosis, by activation of the caspase pathway, and to reduce epidermal growth factor receptor (EGFR) and phosphorylated mitogen-activated protein kinase (P-MAPK) levels [40]. Schneider et al. demonstrated that hamsters treated with metformin and high-fat diet did not develop pancreatic cancer after exposure to a pancreatic carcinogen and had significantly less premalignant lesions compared to controls [41]. Metformin reduced levels of cyclin D in prostate cancer cells in vitro and in vivo and blocked cell cycle in G(0)/G(1) phase. Interestingly, this effect was independent of AMKP, and it was only observed in malignant cells and not normal prostate cells [42]. Using colon cancer cells lines, Buzzai et al. showed that metformin was able to selectively inhibit p53-deficient cell growth and induce autophagy in vitro and in vivo in tumor xenografts, providing another potential mechanism for the action of this drug [43]. Results in mouse models of lung cancer are also encouraging. Metformin was able to reduce overall tumor burden by up to 53% when administered orally and up to 72% when administered intraperitoneally in a tobacco carcinogen-induced model of lung cancer [44]. Interestingly, the steady-state levels of metformin in plasma were comparable to the ones achievable in patients. In addition, the authors demonstrated that metformin did not activate AMPK in lung tissue but inhibited phosphorylation of insulin-like growth factor-I receptor/insulin receptor (IGF-1R/IR), Akt, extracellular signal-regulated kinase (ERK), and mTOR. They concluded that metformin indirectly inhibited mTOR in lung tissue by decreasing activation of these molecules and not by activating AMPK.
Metformin treatment also suppressed mitochondrial electron transport. Metformin-treated cells were able to compensate for this suppression of oxidative phosphorylation by increasing their rate of glycolysis in a p53-dependent manner. The fact that biguanides inhibit the respiratory chain complex I was known from earlier studies on hepatocytes [45]. This attribute of metformin may contribute to its selective toxicity against cancer cells, which have increased needs for energy production. In agreement with these results, addition of metformin to 2-deoxyglucose (2DG) inhibited mitochondrial respiration and glycolysis in prostate cancer cells leading to severe depletion in ATP. The combination of the two drugs was much more harmful for cancer cells than the treatment with metformin or 2DG alone, leading to 96% inhibition of cell viability in prostate cancer cells. Reduction in cell viability was mediated through p53- and AMPK-dependent apoptosis, inhibition of 2DG-induced autophagy and decrease in beclin 1 expression, a molecule involved in cell cycle regulation [46].
Effects on hyperglycemia
An important mechanism of action for metformin in cancer could be the decrease in circulating insulin levels. Multiple studies have shown that type II diabetes and obesity are associated with an increased risk of various types of cancer, especially breast, colon, pancreatic, and endometrial [47–51].
It is widely known that cancer cells express insulin as well as insulin-like growth factor (IGF) receptors (IGF-R) and that, besides its metabolic effect, IGF-R promotes proliferation and metastasis [52]. Cancer cells in particular have a constitutively high glucose uptake, independently of IGF-R activation [53]. In fact, this is the principle behind FDG-positron emission tomography, which detects tissue with high rates of glucose uptake. It is believed therefore that IGF-R activation leads to increased survival and proliferation regardless of glucose uptake and that hyperglycemia, indirectly but stimulating insulin release, contributes to IGF-R activation and cell growth. Insulin also plays a role in reducing insulin-like growth factor binding protein (IGFBP)-I, IGFBPII, and IGFBP-III. The IGFBP-I binds IGF and inhibits its action, leading to an increase in free or bioactive IGF-I levels [54].
Hyperinsulinemia may promote tumor growth by various indirect mechanisms too, such as proliferation of epithelial tissue, increasing bioavailability of steroid sex hormones and serum levels of insulin-like growth factors (IGF), as well as disrupting the homeostasis of adipokines, which are cytokines selectively secreted by adipose tissue and thought to be implicated in cancer pathogenesis [55]. In addition, IGF activation promotes vascular smooth muscle cell proliferation and migration, promoting angiogenesis which could contribute to tumor growth [56].
Metformin was recently shown to directly inhibit insulin-induced malignant as well as benign cell growth in an AMPK/mTOR-dependent manner [57, 58]. However, it remains unknown, to what extend this may contribute to the observed effects of metformin on cancer cells. However, if the observed antitumorigenic properties of biguanides can be primarily attributed to their antihyperglycemic/antihyperinsulinemic properties, then the development of targeted therapies aiming to suppress the IGF-R axis should be undertaken.
Other mechanisms of action
Another mechanism that has been proposed for the potential antitumorigenic effect of metformin is the inhibition of the unfolded protein response (UPR). The UPR is a survival mechanism of the cell. When cells are exposed to various stressors, including energy deprivation, similar to what happens in the cells of solid malignancies, the protein folding capacity of the endoplasmic reticulum (ER) is increased, and the overall folding demand of the cell is decreased [59]. If this fails to happen, the ER is “overwhelmed” and apoptotic pathways are activated leading to cell death. It was recently shown, using gene expression profiling techniques, that metformin was able to inhibit activators of the UPR and lead to massive cell death in glucose-deprived cultures of human colon, fibrosarcoma, renal, and stomach cancer [60].
Pearce et al. provided evidence that tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF-6) regulates CD8 T-cell development after infection by modulating fatty acid metabolism [61]. Metformin was able to correct defects in fatty acid metabolism in TRAF-6-deficient mice promoting T-cell generation and protective immunity after infection and was able to considerably improve the efficacy of an experimental cancer vaccine. Although these authors were not able to elucidate the exact mechanism by which metformin altered T cell biology, in this setting, these results point out that metformin may have a role as an adjuvant in cancer immunotherapy. Dilman et al. demonstrated that administration of phenformin and clofibrate to breast cancer patients who underwent radical mastectomy delayed hypersensitivity reaction to DNCB, tuberculin, and candidin, increased T lymphocyte count, and improved the reaction of lymphocyte blast transformation suggesting an immunomodulating action of biguanides [7].
Hirsch et al., using a xenograph mouse model for breast cancer, showed that low doses of metformin inhibit cellular transformation and selectively kill cancer stem cells. Cancer stem cells are a group of cells within a tumor that resist chemotherapeutic drugs and can regenerate the various cell types in the tumor, thereby providing a highly resistant source for cancer cells and leading to relapse of the disease [62]. The authors demonstrated that metformin in combination with chemotherapy did not only reduce tumor mass but reduced relapse rates much more effectively than chemotherapy alone. In support of these results, Vazquez et al. showed that metformin acted synergistically with the anti-Her-2 monoclonal antibody trastuzumab to eliminate stem/progenitor cell populations in HER2-gene-amplified breast carcinoma cells [63]. Using a trastuzumab resistance model, addition of metformin, led to decreased cell growth, which was attributed to down-regulation of the percentage of trastuzumab-refractory cells displaying the CD44(pos)/CD24(neg/low) stem cell/progenitor immunophenotype. The same group of authors, in an effort to further elucidate the precise mechanism of cancer stem cell targeting by metformin, demonstrated that metformin was able to suppress epithelial to mesenchymal transition (EMT) status of these cells [64]. EMT is a key developmental program that is often activated during cancer invasion and metastasis and is essential for the acquisition of stem cell and cellular motility properties in normal as well as in malignant cells [65]. They showed that metformin, by suppressing a number of transcription factors as well as TGFb, efficiently eliminated the breast cancer stem cell phenotype in cell populations bearing either mesenchymal CD44posCD24neg/low-enriched) or epithelial (CD44posCD24pos-enriched) cells with tumor-initiating potential. The same group showed that metformin exposure not only inhibited TGFb-promoted loss of the epithelial marker E-cadherin in MCF-7 breast cancer cells, but also prevented further TGF-induced cell scattering and accumulation of the mesenchymal marker vimentin in Madin-Darby canine kidney cells [66].
Some authors have proposed vitamin B12 deficiency, a well-documented effect of this drug, as a potential mechanism of action in cancer, since inactivation of B12 in vitro and lack of B12 in vivo has been associated with increased malignant cell death and improved response to chemotherapy, respectively [67, 68].
Finally, there are some studies that suggest that metformin, along with its antitumorigenic effect in vitro and in vivo may also promote tumor survival in vivo indirectly, specifically by enhancing angiogenesis. Metformin was able to increase secretion of vascular endothelial growth factor (VEGF) in an AMPK-dependent manner in estrogen receptor (ER)-negative cells in vitro. In addition, tumors in ER-negative mice treated with metformin had increased vascular density compared to controls [69]. These results are reasonable from a physiologic standpoint. One would expect an activator of AMPK, a “starvation-induced” molecule, to promote changes in the microenvironment of tissues that along with energy conservation (such as cell cycle arrest and decrease in protein synthesis) would also facilitate transfer of nutrients. In addition, these results suggest that metformin may have other, yet unrecognized and, at times, opposing actions. However, other studies have contradicted these results. For instance, metformin increased circulating levels of thrombospondin (TSP), a well-established antiangiogenic factor, in the serum of women with polycystic ovarian syndrome (PCOS) [70]. This effect was depended upon the NF-kappa B and Erk1/2/Erk5 signaling pathways. Metformin use was associated with decreased vascularization and levels of TGF-beta in a murine model of inflammatory angiogenesis after implantation of polyester–polyurethane sponges in Swiss mice [71].
A point of criticism in most preclinical and in vitro studies is that the dose of metformin used was much higher than the standard dose in clinical practice, at times as much as 50-fold higher. However, it has been shown in vivo that metformin accumulates in tissues of diabetic mice in concentrations several fold higher than those in blood [72], suggesting that it may be possible to reach therapeutic levels when used for cancer treatment. Therefore, it is uncertain whether metformin will reproduce some of these experimental benefits when utilized in human clinical trials.
In summary, although typically the antitumorigenic effect of metformin has been attributed to its ability to activate the LKB1/AMPK/mTOR pathway as well as inhibition of insulin-induced growth mechanisms, in reality the mechanism of action of this drug is much more complex potentially involving multiple protein kinases and other molecules with key roles in cell growth and survival.
Metformin and clinical studies
After the discovery that metformin exerts its effects through the LKB1/AMPK/mTOR pathway, several clinical studies followed that attempted to test the hypothesis that metformin use is potentially associated with an improvement in cancer incidence and survival (Table 1).
Evans et al. first found an association between metformin use and decreased cancer incidence [73]. The unadjusted odds ratio in that study was 0.79 (CI 0.67–0.93), and a dose-related effect was suggested, although it did not reach statistical significance. In another large retrospective study, cancer mortality rate per 1,000 person-years of follow-up was 4.9% for sulfonylureas, 3.5% for metformin and 5.8% for insulin users [74]. Retrospective analysis of data from the ZODIAC trial, whose primary outcomes were diabetes-related complications, revealed that cancer-related mortality was lower in patients taking metformin (HR for cancer mortality was 0.43 (95% CI 0.23–0.80) [75]. Although out of 1,353 patients only about 122 had a malignancy related death, this study had a relatively long mean follow up period of 9.6 years. Exposure to metformin for more than 36 months was associated with a significant reduction in the risk of cancer (adjusted OR with 95% CI: 0.28 (0.13–0.57), P < 0.001), in a small (n = 198), single-center retrospective study [76]. In another large retrospective study by Libby et al., cancer was diagnosed among 7.3% of 4,085 metformin users compared with 11.6% of 4,085 controls, with median times to cancer of 3.5 and 2.6 years, respectively (P < 0.001, HR 0.46 (95% CI 0.40–0.53). Of note, in this study, data on social status and hemoglobin A1C were available and were adjusted for in the analysis part of the study. Currie et al., in one of the largest observational studies so far, identified more than 60,000 diabetic patients and showed that patients on insulin or insulin secretagogues were more likely to develop solid tumors than those on metformin with a HR of 1.36 (95% CI 1.19–1.54) for sulfonylurea monotherapy, and 1.42 (95% CI 1.27–1.60) for insulin-based regimens and that metformin use was associated with decreased incidence of colon or pancreas cancer, but did not affect breast or prostate cancer [77]. Secondary analysis of data from two large randomized trials in diabetic patients (ADOPT and RECORD) revealed no difference in cancer incidence between patients treated with metformin and patients treated with rosiglitazone, without refuting the possibility of a difference compared with sulfonylureas [78]. A recent large, cross-sectional prospective study by Mantzoros et al. that included more than 7,000 patients found a higher prevalence of malignancies (66/1308, 5.1%) in subjects with type 2 diabetes mellitus compared to nondiabetic subjects (185/6211, 3.0%) (odds ratio, 1.64; 95% confidence interval, 1.12–2.41) before and after adjustment for several confounding factors [79]. Patients on metformin had a nonsignificant trend toward lower prevalence of malignancies, comparable with that among nondiabetic patients, whereas those on any other oral combination treatment had a twofold higher risk for malignancies even after adjusting for possible confounders. Interestingly, inclusion of metformin in these regimens decreased the prevalence of malignancies, and this was the first study to suggest an independent effect of metformin in decreasing cancer incidence, although in some of the subgroup analyses the difference was not statistically significant. In the prospective analyses, diabetic patients in general and diabetic patients treated with insulin (either as monotherapy or in combination with other treatments) had a twofold and fourfold, respectively, higher mortality rate than nondiabetic patients, even after adjustment for potential confounders (incidence of cancer deaths in patients with type 2 diabetes mellitus [2.6%] versus the incidence of cancer deaths in patients without type 2 diabetes mellitus [1.2%]). Yang et al. as well as Monami et al. also found an association between metformin and decreased cancer incidence (Table 1). Of note, the first group suggested that the beneficial effects of metformin were particularly evident in diabetic patients with low HDL-C, whose main lipoprotein, APOA-I, also activates AMPK [80, 81].
Other authors have examined whether metformin use is associated with better outcomes after chemotherapy in various types of cancer. Diabetic patients who had taken metformin had a significantly lower risk of pancreatic cancer compared with those who had not taken metformin (odds ratio, 0.38; 95% CI, 0.22–0.69; P = 0 0.001) [82]. More recently, Bodmer et al. found a decreased incidence of breast cancer in patients taking metformin for more than 5 years, but not for a shorter period of time [83]. Jiralespong et al. identified approximately 155 diabetic and 2,374 nondiabetic patients with breast cancer and showed that diabetic patients receiving metformin and neoadjuvant taxane chemotherapy have a higher pathologic complete remission (pCR) rate than do diabetics not receiving metformin [84].The rate of pCR among groups was also significantly different: 24% in the metformin group, 8.0% in the nonmetformin group, and 16% in the nondiabetic group.
More recently, Hosono et al. [85], in a randomized pilot study involving a small (n = 26) number of nondiabetic patients demonstrated that low-dose metformin (250 mg/day) suppressed formation of aberrant crypt foci (ACF), a surrogate marker for colon cancer, after only 1 month of administration. Immunostaining for markers of cell proliferation and apoptosis revealed that inhibition of cell proliferation and not increase in apoptosis was responsible for ACF suppression.
Finally, Berstein et al. analyzed the receptor status of breast carcinomas in 90 postmenopausal women affected with diabetes mellitus type 2 who had been cured, for not less than 1 year prior to surgery, with different modes of antidiabetic therapy, including a dietary treatment only, sulfonylurea preparations, insulin therapy, and metformin as a monotherapy or in combination with sulfonylurea derivatives [86]. This group found no differences in estrogen receptors occurrence in tumor tissue in different treatment groups. However, the frequency of progesterone receptor-positive mammary carcinomas in women who were treated with metformin, irrespective of whether it was combined with sulfonylurea preparations, was significantly higher than in the sulfonylurea only group (P = 0.043) and in the combined group of patients treated with either sulfonylurea or insulin (P = 0.041). The significance of these results remains unclear, given the lack of in vivo data that could suggest a mechanism through which metformin of sulfonylureas may be able to alter the receptor status of breast cancer cells.
Limitations of clinical studies and the role of insulin and oral hypoglycemics
In most of these retrospective studies, a number of limitations were present. Their retrospective design makes them unsuitable for drawing firm conclusions on the matter because of the complex nature of the management of diabetes. Individuals receiving oral hypoglycemic agents can be new to pharmacological treatment, or they may have switched from other therapies or take various combinations of medications with or without insulin. This further complicates data analysis since it is challenging to draw conclusions about the individual effects of metformin. Furthermore, tumorigenesis is a slow process, and for any protective or harmful effect to be seen, sustained exposure to certain doses of these medications is required. The short periods of follow-up in some studies argue against a causal relationship.
In addition, in several of these studies, metformin was compared with insulin, insulin secretagogues, and thiazolidinediones (TZDs); therefore, it is difficult to conclude whether the beneficial effect of metformin in various settings could be attributed to an inherent antitumorigenic effect of the drug or to the hypothesized tumor-promoting effects of insulin or other oral antidiabetic medications. The latter is an important point, since there are several epidemiological data to suggest a possible connection between insulin, especially insulin glargine, insulin secretagogues, and cancer [74, 76, 77, 87, 88].
A biologic basis for this effect has been discussed earlier in this review. Data regarding TZDs are conflicting with studies showing no association [89], a protective [90, 91], or a harmful [92] effect on cancer incidence.
Another criticism is that most of these studies that included insulin, oral hypoglycemics, and metformin were underpowered to detect small to moderate effect sizes, and that was especially true for site-specific malignancies. Therefore, including cancer endpoints (incidence, mortality, etc.) in general may miss site-specific associations, but in the same time, conducting subgroup analyses for specific sites, frequently compromised power and validity of these associations. In addition, important details about confounding factors such as diabetes control, obesity, duration, or dose of medications, diagnosis of cancer, and others were not available in several cases or were derived from indirect sources such as self-reported use, questionnaires, and prescribing records. Some of these data are very difficult to collect because of the retrospective nature of these studies. For instance, diabetes control and BMI are dynamic and may change over time. This becomes particularly important in cancers such as colon, breast, pancreatic, and others that have a strong association with diabetes and obesity. Some of these studies had significant methodological faults such as differences in baseline characteristics of patients [87, 88], leading to allocation bias.
Reverse causation also cannot be excluded. For instance, in cases of pancreatic cancer, worsening glycemic control is a common phenomenon that precedes the diagnosis of the actual tumor. These patients may have required intensification of diabetic therapy several months before the diagnosis of their malignancy.
A recent consensus statement from the American Diabetes Association and the American Cancer Society, who, after comprehensively reviewing the epidemiologic evidence connecting diabetes and diabetes treatments with cancer incidence or prognosis, concluded that “Although still limited, early evidence suggests that metformin is associated with a lower risk of cancer and that exogenous insulin is associated with an increased cancer risk. Further research is needed to clarify these issues and evaluate if insulin glargine is more strongly associated with cancer risk compared with other insulins.” [93].
Clinical trials of metformin in cancer therapy
There are currently several clinical trials under way that aim to test the efficacy of metformin as an adjuvant to conventional chemotherapy as well as in combination with new, targeted agents in various settings such as breast, prostate cancer, and other solid malignancies (Table 2). The largest one so far (NCT01101438) is a phase III randomized multicenter trial that aims to recruit 3582 participants and test whether metformin is superior to placebo in patients with early stage, nonmetastatic (T1-3,N0-3,M0) breast cancer. Patients will receive metformin twice daily (dose is not mentioned) for 5 years after their diagnosis. Patients with prior use of metformin, insulin, or other oral hypoglycemics will be excluded. Primary outcome will be invasive disease-free survival (DFS), and secondary outcomes will include overall survival (OS), DFS, and relevant medical endpoints (e.g., new diabetes, new cardiovascular hospitalizations). Two phase II trials involving pancreatic cancer are also under way. So far gemcitabine in combination with other agents has shown a modest but statistically significant benefit on overall survival in patients with advanced disease [94]. The first of these trials (NCT01167738) will enroll 82 patients and will investigate whether a 6-month daily dose of metformin in addition to combination chemotherapy (cisplatin, epirubicin, capecitabine, and gemcitabine) leads to prolonged 6-month progression-free survival (PFS) compared to combination chemotherapy alone in patients with metastatic pancreatic cancer. Patients with prior metformin use will be excluded. The second one (NCT01210911) will enroll 120 patients and will investigate whether a 1,000-mg twice daily dose of metformin for 6 months in combination with erlotinib and gemcitabine is more effective than the combination of these two agents in prolonging PFS. Prior users of metformin and patients with uncontrolled diabetes (fasting serum glucose higher than twice the upper limit of normal) will be excluded. However, in both of these trials, prior users of other antidiabetic medications or insulin will not be excluded. Careful randomization will be required if one is to make any conclusions about the potential effects of metformin in this case, since limitations similar to the ones mentioned above will again apply. Moreover, glycemic control during metformin treatment should be carefully monitored and accounted for in the analysis part of the study. Finally, a phase II trial involving castration-resistant prostate cancer patients (NCT01215032) will recruit 106 patients and will test whether metformin in addition to androgen derivation therapy (ADT) will lead to improved PSA response (decline of 50% or more, confirmed by a second value 4 or more weeks apart). Metformin will be given twice daily for 12 months. In this study, diabetic patients or prior users of metformin will be excluded, and glycemic control will be included as a secondary outcome.
The results of these trials will be of great interest for various reasons. First, in some of these studies, metformin will be tested for the first time in nondiabetic patients. Diabetes and higher serum insulin levels are a well-recognized risk factor for numerous malignancies including pancreatic [95], prostate [96], as well as breast cancer [97]. If insulin is in fact directly involved in the pathogenesis of these cancers, it is reasonable to expect a greater effect size if metformin was tested in diabetic patients and a smaller one in nondiabetics. Significant differences in nondiabetic patients will be very encouraging and will prompt further research in diabetic patients at least for the subset of malignancies where diabetes is a risk factor. In addition, not involving diabetic patients will put the emphasis on the inherent, direct antitumorigenic effects of metformin and not on its indirect ones.
Suggestions for further research
Metformin could be considered for primary prevention trials, for which it would be an excellent candidate, since it is an inexpensive and relatively well-tolerated medication, as well as an adjuvant to current treatment options. However, at this point, several unanswered questions remain. If more evidence supporting an anticancer effect that is independent of the antihypoglycemic effect emerges from basic research and current clinical trials, then further research should be undertaken to explore the major mechanisms responsible for this. Some authors have proposed to compare the efficacy of metformin with that of specific IGF-1 receptor inhibitors [98]. If metformin were to be effective despite the concurrent use of these molecules, this could suggest a mechanism of action independent of its insulin lowering effects. If this were to be the case, a reasonable next step would be the development of more specific drugs that would exploit this mechanism in a more effective way. Subsequently, the optimal dosing regimen and route of administration would need to be determined since current dosing achieves adequate levels of this medication in the liver but is unclear to what extended metformin would penetrate variable malignant tissues.
It would be helpful to determine patient or tumor characteristics that may predict efficacy of biguanides. For instance, activation status of the LKB1/AMPK/mTOR pathway, baseline insulin levels, body mass index, etc. could be relevant. Malignancies of the breast and colon that have a strong association with obesity and hyperinsulinemia are attractive settings to study metformin. Prevention of postcastration hyperinsulinemia which may be responsible for driving growth of prostate cancer cells may be inhibited by metformin, making this malignancy another potential target for further research.
In addition, one must consider the potential implications of tampering with glucose and insulin metabolism in adults with no preexisting diabetes or glucose intolerance. One cannot assume that regimens optimized for diabetes control will be effective or safe for potential oncology indications.
In light of the results of recent studies, metformin could prove to be an important addition in our arsenal against cancer. However, data from randomized trials are lacking, and results from clinical and basic studies so far, although promising, have been at times either controversial or with several limitations.
References
United Kingdom Diabetes study group, United Kingdom Prospective Diabetes Study (UKPDS). 13: Relative efficacy of randomly allocated diet, sulphonylurea, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ, 1995. 310(6972):83–8.
Diamanti-Kandarakis E, Economou F., Palimeri S, Christakou C, Metformin in polycystic ovary syndrome. Ann N Y Acad Sci. 2010;1205:192–8.
Bianchi C, Penno G, Romero F, Del Prato S, Miccoli R. Treating the metabolic syndrome. Expert Rev Cardiovasc Ther. 2007;5(3):491–506.
Knowler WC, Barett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Eng J Med. 2002;346(6):393–403.
Shikhman AR, Lebedev K, Dil’man VM. Inhibiting effect of phenformin (phenethyl biguanide) on the growth of Ehrlich carcinoma. Vopr Onkol. 1981;27(2):67–9.
Cohen MH, Strauss B. Enhancement of the antitumor effect of 1, 3-bis(2-chloroethyl)-l-nitrosourea (BCNU) by phenylethylbiguanide (phenformin). Oncology. 1976;33(5):257–9.
Dilman VM, Berstein L, Ostroumova MN, Fedorov SN, Poroshina TE, Tsyrlina EV, Buslaeva VP, Semiglazov VF, Seleznev IK, YuF Bobrov, Vasilyeva IA, Kondratjev VB, Nemirovsky VS, Nikiforov YF. Metabolic immunodepression and metabolic immunotherapy: an attempt of improvement in immunologic response in breast cancer patients by correction of metabolic disturbances. Oncology. 1982;39(1):9–13.
Huang X, Wullschleger S, Shapiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412(2):e3–5.
Shaw RJ, Lamia K, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–6.
Memmott RM, Dennis P. LKB1 and mammalian target of rapamycin as predictive factors for the anticancer efficacy of metformin. J Clin Oncol. 2009;27(34):e226.
Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, Marette A, Kozma SC, Thomas G. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11(5):390–401.
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
Steinberg GR, Kemp B. AMPK in health and disease. Physiol Rev. 2009;89(3):1025–78.
Browne GJ, Finn S, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem. 2004;279(13):12220–31.
Schmelzle T, Hall M. TOR, a central controller of cell growth. Cell. 2000;103(2):253–62.
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90.
Gwinn DM, Shackelford D, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Moll Cell. 2008;30(2):214–26.
Liang J, Shao S, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9(2):218–24.
Jones RG, Plas D, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Moll Cell. 2005;18(3):283–93.
Wang W, Fan J, Yang X, Fürer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M. AMP-activated kinase regulates cytoplasmic HuR. Moll Cell Biol. 2002;22(10):3425–36.
Lee M, Hwang J, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003;278(41):39653–61.
Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Moll Cell Biol. 2002;22(20):7004–14.
Majumder PK, Febbo P, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10(6):594–601.
Anisimov VN, Berstein L, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40(8):685–93.
Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–73.
Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–12.
Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, Thor AD. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8(6):909–15.
Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8(13):2031–40.
Yang XR, So M, Rimm DL, Lissowska J, Brinton LA, Peplonska B, Hewitt SM, Anderson WF, Szeszenia-Dabrowska N, Bardin-Mikolajczak A, Zatonski W, Cartun R, Mandich D, Rymkiewicz G, Ligaj M, Lukaszek S, Kordek R, García-Closas M. Differences in risk factors for breast cancer molecular subtypes in a population-based study. Cancer Epidemiol Biomarkers Prev. 2007;16(3):439–43.
Vázquez-Martín A, Oliveras-Ferraros C, del Barco S, Martín-Castillo B, Menéndez JA. mTOR inhibitors and the anti-diabetic biguanide metformin: new insights into the molecular management of breast cancer resistance to the HER2 tyrosine kinase inhibitor lapatinib (Tykerb). Clin Transl Oncol. 2009;11(7):455–9.
Vazquez-Martin A, Oliveras-Ferraros C, del Barco S, Martin-Castillo B, Menendez JA. The antidiabetic drug metformin: a pharmaceutical AMPK activator to overcome breast cancer resistance to HER2 inhibitors while decreasing risk of cardiomyopathy. Ann Oncol. 2009;20(3):592–5.
Shell SA LL, Trusk PB, Pry KJ, Wappel RL, Bacus SS. Activation of AMPK is necessary for killing cancer cells and sparing cardiac cells. Cell Cycle. 2008;7(12):1769–75.
Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8(1):88–96.
Bonnefont-Rousselot D, Raji B, Walrand S, Gardès-Albert M, Jore D, Legrand A, Peynet J, Vasson MP. An intracellular modulation of free radical production could contribute to the beneficial effects of metformin towards oxidative stress. Metabolism. 2003;52(5):586–9.
Zhou Q, Liu L, Fu B, Hu X, Shi X, Fang J, Jiang BH. Reactive oxygen species regulate insulin-induced VEGF and HIF-1alpha expression through the activation of p70S6K1 in human prostate cancer cells. Carcinogenesis. 2007;28(1):28–37.
Brown KA, Hunger N, Docanto M, Simpson ER. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res Treat. 2010;Epub ahead of print.
Oliveras-Ferraros C, Vazquez-Martin A, Menendez JA. Genome-wide inhibitory impact of the AMPK activator metformin on [kinesins, tubulins, histones, auroras and polo-like kinases] M-phase cell cycle genes in human breast cancer cells. Cell Cycle. 2009;8(10):1633–6.
Isakovic A, Harhaji L, Stevanovic D, Markovic Z, Sumarac-Dumanovic M, Starcevic V, Micic D, Trajkovic V. Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci. 2007;64(10):1290–302.
Woodard J, Joshi S, Viollet B, Hay N, Platanias LC. AMPK as a therapeutic target in renal cell carcinoma. Cancer Biol Ther. 2010;10(11):[Epub ahead of print].
Wang LW, Liu Z, Zou DW, Jin ZD, Gao J, Xu GM. Metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol. 2008;14(47):7192–8.
Schneider MB, Matsuzaki H, Haorah J, Ulrich A, Standop J, Ding XZ, Adrian TE, Pour PM. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120(5):1263–70.
Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–86.
Buzzai M, Jones R, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745–52.
Memmott RM, Mercado J, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev Res. 2010;3(9):1066–76.
El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275(1):223–8.
Sahra IB, Tanti J, Bost F. The combination of metformin and 2-deoxyglucose inhibits autophagy and induces AMPK dependent apoptosis in prostate cancer cells. Autophagy. 2010;6(5).
Wolf I, Sadetzki S, Catane R, Karasik A, Kaufman B. Diabetes mellitus and breast cancer. Lancet Oncol. 2005;6(2):130–1.
Huxley R, Ansary-Moghaddam A, Berrington de González A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer. 2005;92(11):2076–83.
Larsson SC, Orsini N, Wolk A. Diabetes mellitus and risk of colorectal cancer: a meta-analysis. J Natl Cancer Inst. 2005;97(22):1679–87.
Friberg E, Orsini N, Mantzoros CS, Wolk A. Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia. 2007;50(7):1365–74.
Becker S, Dossus L, Kaaks R. Obesity related hyperinsulinaemia and hyperglycaemia and cancer development. Arch Physiol Biochem. 2009;115(2):86–9.
LeRoith D, Baserga R, Helman L, Roberts CT Jr. Insulin-like growth factors and cancer. Ann Intern Med. 1995;122(1):54–9.
Zhang H, Pelzer A, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res. 2007;67(1):391–7.
Ooi GT, Tseng L, Tran MQ, Rechler MM. Insulin rapidly decreases insulin-like growth factor-binding protein-1 gene transcription in streptozotocin-diabetic rats. Mol Endocrinol. 1992;6(12):2219–28.
Renehan AG, Frystyk J, Flyvbjerg A. Obesity and cancer risk: the role of the insulin-IGF axis. Trends Endocrinol Metab. 2006;17(8):328–36.
Clemmons DR, Maile L, Ling Y, Yarber J, Busby WH. Role of the integrin alphaVbeta3 in mediating increased smooth muscle cell responsiveness to IGF-I in response to hyperglycemic stress. Growth Horm IGF Res. 2007;17(4):265–70.
Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69(16):6539–45.
Tosca L, Ramé C, Chabrolle C, Tesseraud S, Dupont J. Metformin decreases IGF1-induced cell proliferation and protein synthesis through AMP-activated protein kinase in cultured bovine granulosa cells. Reproduction. 2010;139(2):409–18.
Schröder M, Kaufman R. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89.
Saito S, Furuno A, Sakurai J, Sakamoto A, Park HR, Shin-Ya K, Tsuruo T, Tomida A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009;69(10):4225–34.
Pearce EL, Walsh M, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–7.
Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69(19):7507–11.
Vazquez-Martin A, Olivera-Ferraros C, Barco SD, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2010;Epub ahead of print.
Vazquez-Martin A, Olivera-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010;9(18):3807–14.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15.
Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFβ-induced epithelial-to-mesenchymal transition (EMT): From cancer stem cells to aging-associated fibrosis. Cell Cycle. 2010;9(22):4461–8.
Garcia A, Tisman G, Metformin. B(12), and enhanced breast cancer response to chemotherapy. J Clin Oncol. 2010;28(2):e19.
Goldhirsch A, Gelber R, Tattersall MN, Rudenstam CM, Cavalli F. Methotrexate/nitrous-oxide toxic interaction in perioperative chemotherapy for early breast cancer. Lancet. 1987;2(8551):151.
Phoenix KN, Vumbaca F, Claffey KP. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERalpha negative MDA-MB-435 breast cancer model. Breast Cancer Res Treat. 2009;113(1):101–11.
Tan BK, Adya R, Chen J, Farhatullah S, Heutling D, Mitchell D, Lehnert H, Randeva HS. Metformin decreases angiogenesis via NF-kappaB and Erk1/2/Erk5 pathways by increasing the antiangiogenic thrombospondin-1. Cardiovasc Res. 2009;83(3):566–74.
Xavier DO, Amaral L, Gomes MA, Rocha MA, Campos PR, Cota BD, Tafuri LS, Paiva AM, Silva JH, Andrade SP, Belo AV. Metformin inhibits inflammatory angiogenesis in a murine sponge model. Biomed Pharmacother. 2010;64(3):220–5.
Wilcock C, Bailey C. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica. 1994;24(1):49–57.
Evans JM, Donnelly L, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–5.
Bowker SL, Majumdar S, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29(2):254–8.
Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010;33(2):322–6.
Monami M, Lamanna C, Balzi D, Marchionni N, Mannucci E. Sulphonylureas and cancer: a case-control study. Acta Diabetol. 2009;46(4):279–84.
Currie CJ, Poole C, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52(9):1766–77.
Home PD, Kahn S, Jones NP, Noronha D, Beck-Nielsen H, Viberti G. Experience of malignancies with oral glucose-lowering drugs in the randomised controlled ADOPT (A Diabetes Outcome Progression Trial) and RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycaemia in Diabetes) clinical trials. Diabetologia. 2010;Epub ahead of print.
Baur DM, Klotsche J., Hamnvik OP, Sievers C, Pieper L, Wittchen HU, Stalla GK, Schmid RM, Kales SN, Mantzoros CS. Type 2 diabetes mellitus and medications for type 2 diabetes mellitus are associated with risk for and mortality from cancer in a German primary care cohort. Metabolism. 2010;Epub ahead of print.
Yang X, So WY, Ma RC, Kong AP, Lee HM, Yu LW, Chow CC, Ozaki R, Ko GT, Chan JC. Low HDL cholesterol, metformin use and cancer risk in Type 2 diabetes—the Hong Kong Diabetes Registry. Diabetes Care. 2010;Epub ahead of print.
Monami M, Colombi C, Balzi D, Dicembrini I, Giannini S, Melani C, Vitale V, Romano D, Barchielli A, Marchionni N, Rotella CM, Mannucci E. Metformin and Cancer occurence in insulin-treated type 2 diabetic patients. Diabetes care. 2010;Epub ahead of print.
Li D, Yeung S, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137(2):482–8.
Bodmer M, Meier C, Krähenbühl S, Jick SS, Meier CR. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care. 2010;33(6):1304–8.
Jiralerspong S, Palla S, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27(20):3297–302.
Hosono K, Endo H, Takahashi H, Sugiyama M, Sakai E, Uchiyama T, Suzuki K, Iida H, Sakamoto Y, Yoneda K, Koide T, Tokoro C, Abe Y, Inamori M, Nakagama H, Nakajima A. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev Res. 2010;3(9):1077–83.
Berstein LM, Boyarkina M, Tsyrlina EV, Turkevich EA, Semiglazov VF. More favorable progesterone receptor phenotype of breast cancer in diabetics treated with metformin. Med Oncol. 2010;Epub ahead of print.
Hemkens LG, Grouven U, Bender R, Günster C, Gutschmidt S, Selke GW, Sawicki PT. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia. 2009;52(9):1732–44.
Jonasson JM, Ljung R, Talbäck M, Haglund B, Gudbjörnsdòttir S, Steineck G. Insulin glargine use and short-term incidence of malignancies-a population-based follow-up study in Sweden. Diabetologia. 2009;52(9):1745–54.
Koro C, Barrett S, Qizilbash N. Cancer risks in thiazolidinedione users compared to other anti-diabetic agents. Pharmacoepidemiol Drug Saf. 2007;16(5):485–92.
Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, Kim L, Kim PJ, Owens RJ, Lang NP. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncol. 2007;25(12):1476–81.
Monami M, Lamanna C, Marchionni N, Mannucci E. Rosiglitazone and risk of cancer: a meta-analysis of randomized clinical trials. Diabetes Care. 2008;31(7):1455–60.
Ramos-Nino ME, MacLean C, Littenberg B. Association between cancer prevalence and use of thiazolidinediones: results from the Vermont Diabetes Information System. BMC Med. 2007;5(17).
Giovannucci E, Harlan D, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report. CA Cancer J Clin. 2010;60(4):207–21.
Heinemann V, Boeck S, Hinke A, Labianca R, Louvet C. Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer. 2008;8(82).
Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA. 1995;273(20):1605–9.
Albanes D, Weinstein S, Wright ME, Männistö S, Limburg PJ, Snyder K, Virtamo J. Serum insulin, glucose, indices of insulin resistance, and risk of prostate cancer. J Natl Cancer Inst. 2009;101(18):1272–9.
Gunter MJ, Hoover D, Yu H, Wassertheil-Smoller S, Rohan TE, Manson JE, Li J, Ho GY, Xue X, Anderson GL, Kaplan RC, Harris TG, Howard BV, Wylie-Rosett J, Burk RD, Strickler HD. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2009;101(1):48–60.
Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prev Res (Phila). 2010;3(9):1060–5.
Mackenzie MJ, Ernst S, Johnson C, Winquist E. A phase I study of temsirolimus and metformin in advanced solid tumours. Invest New Drugs. 2010;Epub ahead of print.
Conflict of interest
All authors state that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kourelis, T.V., Siegel, R.D. Metformin and cancer: new applications for an old drug. Med Oncol 29, 1314–1327 (2012). https://doi.org/10.1007/s12032-011-9846-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12032-011-9846-7