
Abstract
Introduction
Metformin, an oral hypoglycemic agent, was introduced in the clinical practice for the treatment of type 2 diabetes mellitus more than a half-century ago. Over the years, several studies demonstrated that diabetic patients treated with metformin have a lower incidence of cancer, raising the hypothesis that the spectrum of clinical applications of the drug could be expanded also to cancer therapy. Following these initial findings, a large number of studies were performed aimed at elucidating the effects of metformin on different types of tumor, at explaining its direct and indirect anti-cancer mechanisms and at identifying the molecular pathways targeted by the drug. Several clinical trials were also performed aimed at evaluating the potential anti-cancer effect of metformin among diabetic and non-diabetic patients affected by different types of cancer. While the results of several clinical studies are encouraging, a considerable number of other investigations do not support a role of metformin as an anti-cancer agent, and highlight variables possibly accounting for discrepancies.
Aim
We hereby review the results of in vitro and in vivo studies addressing the issue of the anti-cancer effects of metformin.
Conclusions
If in vitro data appear solid, the results provided by in vivo studies are somehow controversial. In this view, larger studies are needed to fully elucidate the role of metformin on cancer development and progression, as well as the specific clinical settings in which metformin could become an anti-cancer drug.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The history of Metformin (1,1-dimethylbiguanide) dates back to 1920s to the use of Galega officinalis (goat’s rue or French Lilac) as a botanic medicine, which was already known as a remedy for polyuria in medieval Europe [1]. In the 1950s, biguanides were developed as therapeutic agents [2] and their derivates, such as metformin, were introduced in the clinical practice, initially in Europe and Canada (since the 1970s), and then in the United States where the Food and Drug Administration approved it in 1994. Metformin has a glucose-lowering effect and reduces hyper-insulinemia [3–5] by improving insulin sensitivity in peripheral tissues. Nowadays, metformin in its different formulations is the most widely used drug in the biguanide class for the treatment of Type 2 Diabetes Mellitus (T2DM) because of its high efficacy and safety and also for its relatively low cost [6]. Other metabolic disorders in which metformin was proven of therapeutic efficacy include PCOS [7, 8] and NAFLD [9] two often associated clinical conditions. More recently, identified actions of metformin are represented by its ability to reduce some [10] but not other circulating markers of inflammation and a TSH-lowering effect [11, 12].
The link between diabetes and cancer, and the role of metformin
Epidemiologic data demonstrate that T2DM is associated with a higher risk for developing several types of cancer [2]. Moreover, T2DM is often associated with obesity, which, in turn, is an independent risk factor for the occurrence of cancer [13]. T2DM is characterized by insulin resistance and hyper-insulinemia, the latter was shown to favor the development of breast cancer in experimental animal models [14]. Patients with T2DM also display high serum levels of insulin-like growth factor (IGF)-1, a potent mitogen that can contribute to carcinogenesis [15].
Interest in metformin as a potential anti-cancer agent was raised by studies reporting that diabetic patients given metformin had a lower incidence of cancer, as compared with diabetic patients treated with other drugs. Nearly, 10 years ago Evans et al. [16] first reported the unexpected finding of a lower prevalence of cancer in diabetic patients treated with metformin. In agreement with this study, Bowker et al. reported that, compared with other anti-diabetic treatments, diabetic patients receiving metformin had a lower cancer-specific mortality [17]. Further studies showed that metformin can inhibit the growth of cancer cells and reduce the risk of developing several solid tumors [18], such as colorectal, liver, pancreatic, stomach, esophagus and breast cancer [19, 20].
The mechanistic effects of metformin
The discovery that metformin has anti-cancer effects raised attention on the potentially involved mechanisms. The first step was directed at understanding whether the anti-diabetic and anti-cancer effects would involve the same or different pathways.
Mechanistic effects of metformin in diabetes
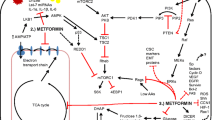
The primary mechanism of action of metformin in diabetes occurs through a reduced output of glucose from the liver. This effect is mediated by the AMP-activated serine–threonine protein kinase (AMPK), an intracellular sensor of energy and nutrient availability, and a regulator of energy homeostasis [21]. AMPK is a key therapeutic target in patients with diabetes, as it regulates lipid, cholesterol and glucose metabolism in specialized tissues such as liver, muscle, and adipose tissue [22]. Metformin targets the mitochondrion by inhibiting the respiratory chain complex I [23, 24] with subsequent decrease in the production of ATP and increase in the accumulation of ADP and AMP [21]. Both AMP and ADP regulate AMPK function by preventing its de-phosphorylation and inactivation [25]. Activated AMPK switches off ATP-consuming pathways and switches on pathways for ATP generation. The phosphorylation of the AMPK catalytic subunit is mediated by LKB1 (a protein called after liver kinase B1, the product of the corresponding tumor suppressor gene). Thus, metformin, by activating AMPK, induces a decrease in energy-consuming processes, such as gluconeogenesis, protein and fatty acid synthesis. In patients with T2DM, the final result of these metabolic changes is a reduction of blood glucose levels and of insulin resistance [26, 27].
Mechanistic effects of metformin in cancer
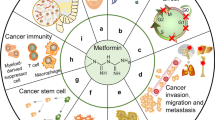
Since the first description of the anti-cancer effect of metformin, it became evident that more than one mechanism was involved. In particular, the observation that the anti-cancer effect of metformin also occurs in non-diabetic patients [28, 29] supported the hypothesis that, besides an indirect effect via its insulin lowering action, the drug influences other signaling pathways. [22, 30–33]. The direct and indirect mechanistic effects of metformin on tumor cells are summarized in Fig. 1.

Metformin anti-cancer effects. The direct and indirect action of Metformin on tumor cells by AMPK-dependent and AMPK-independent mechanisms leads to obtain the anti-cancer effects of the drug
The AMPK-dependent pathway
The inhibition of the LKB1-mediated-mTOR signaling pathway is a major candidate for the anti-cancer effect of the drug. The activation of the LKB1–AMPK pathway inhibits the mammalian target of rapamycin complex 1 (mTORC1), a kinase being activated in the majority of human cancers [34]. The sequence of events is as follows: AMPK phosphorylates the tuberous sclerosis complex protein 2 (TSC2), which results in the accumulation of Rheb-GDP (a protein with GTPase activity) and the subsequent inhibition of mTORC1 [35]. Although the LKB1, AMPK, and mTOR pathway is considered a potential pharmaceutical target in tumors, due to its crucial role in the regulation of cancer cell metabolism, growth, and proliferation [34], other mechanisms are also involved in the anti-cancer effect of metformin.
The AMPK-independent pathways
Several experimental data support the concept that AMPK-independent pathways are involved in the anti-cancer effect of metformin [36]. The drug inhibits hepatic gluconeogenesis by a mechanism, which is independent from the LKB1–AMPK pathway and involves a decrease in the hepatic energy state [37]. Metformin also inhibits the proliferation of human leukemia cells by inducing a cell cycle arrest in the G0/G1 or S-G2/M phase, and promotes the apoptosis of these neoplastic cells [38]. In prostate cancer cell lines, the drug exerts an anti-proliferation effect by an AMPK-independent pathway, which involves the induction of a p53 target gene (REDD1) [39]. In breast cancer cells, metformin interferes with purine/pyrimidine and glutathione synthesis, a pathway which is upstream and AMPK independent [40]. Taken together, these findings indicate that multiple molecular pathways account for the anti-proliferative effect of metformin on cancer cells.
The NF-kB pathway
The attention on the role of nuclear-factor-κB (NF-κB) in human cancer stems from the growing interest for tumor-related inflammation. NF-κB is involved in innate and adaptive immune responses, but it is also constitutively activated in human tumors [41, 42]. NF-κB activation results both in enhanced proliferation and invasiveness of cancer cells, and in their resistance to apoptosis induced by tumor surveillance mechanisms [41]. In vascular endothelial cells, metformin inhibits the activation of NF-κB induced by TNF-α [43]. Further to this observation, it was shown that in smooth muscle cells metformin suppresses the phosphorylation of three MAP kinases (p38, JNK, and Erk) and of Akt, all involved in the NF-κB pathway [44].
In vitro and in vivo experiments supporting the anti-cancer effect of metformin
A consistent number of in vitro studies evaluated the effect of metformin on cell cycle, cell proliferation, apoptosis and metastatic potential. Zakikhani et al. demonstrated that metformin exerts an anti-mitogenic effect in breast cancer cells [45] by activating AMPK and by inhibiting the MAPK pathway, a major mediator of the insulin-IGF signaling, which promotes cell growth. Carmignani et al. showed that the drug exerts an anti-cancer effect in glioblastoma cells by blocking the LKB1–AMPK–mTOR–S6K1 pathway [46]. Metformin exhibits a dose- and time-dependent anti-proliferation effect on intrahepatic cholangiocarcinoma cell lines, which appear to be mediated by different mechanisms including induction of apoptosis and cell cycle arrest. The in vitro anti-proliferative effect of metformin, although by different mechanisms, was consistently reported in several cancer cell lines derived from breast [47], colon [48], ovary, [49] pancreas [50], lung [51], and prostate [52] tumors.
Pre-clinical in vivo experiments confirmed the anti-cancer effect of metformin in mouse models of tumor xenografts [53] and chemically induced cancers. Cancer stem cells (CSC) were also identified as a target of the anti-cancer effect of metformin. Bao et al. showed that metformin attenuates the CSC phenotypes, functions and mediators [54]. The drug reduces the expansions of CSC clones by inducing apoptosis and by inhibiting CSC mediators and markers. By these mechanisms, metformin attenuates the growth of tumors in animal models.
Other lines of research showed that metformin regulates the epithelial–mesenchymal transition (EMT) status, an essential differentiation program in early embryonic development that is modified in cancer to mediate acquisition of malignant properties including stemness [55–57]. Metformin decreases the expression of key drivers of EMT including the transcription factors ZEB1, TWIST1 and SNAI2 (Slug), and the pleiotropic cytokines TGFβs [58, 59] in several cell types. The inhibition of these components of EMT by metformin causes an inhibition of cell invasiveness without affecting cell migration [60]. Metformin was also shown to reduce the expression of miR-34a and its direct EMT targets Notch, Slug, and Snail [61].
A recent line of research investigates the inhibition of NFκB activity by metformin. The available studies addressing this issue may be summarized as follows: (1) In vitro experiments by Tan et al. demonstrated that the invasiveness of endometrial cancer cells was significantly attenuated by sera from women with polycystic ovary syndrome in treatment with metformin [62], (2) Metformin inhibits the NFκB inflammatory signaling in mouse pancreatic tumors, as evidenced by the blocking effect on NFκB phosphorylation and by the decreased mRNA expression of the downstream genes MCP-1, TGF-β1, TNF-α, and IL-1β [63], (3) Metformin inhibits the TNF-α-induced secretion of CXCL8, a chemokine with well-established pro-tumorigenic actions [64, 65].
In vivo studies showing an anti-cancer effect of metformin
As reviewed in the previous section, over the past two decades experimental data raised interest on the potential benefits of metformin treatment in human malignancies. If in vitro data appear solid and encouraging, the results provided by in vivo studies are somehow controversial. Table 1 summarizes the studies performed both in diabetic and in non-diabetic patients with various types of cancer, evaluating the possible anti-cancer effect of metformin.
Studies in diabetic patients with cancer
As previously discussed, in 2005 Evans et al. first reported that the overall incidence of cancer was lower in diabetic patients given metformin compared with patients treated with other anti-diabetic drugs [16]. They also found that a longer duration of metformin treatment and a larger number of prescriptions correlated with a lower incidence of cancer. The anti-cancer effect of metformin, in terms of reduced cancer incidence and increased survival, was subsequently reported in diabetic patients affected by other types of cancer [18, 66–69]. The beneficial effect of metformin was found to be dose dependent and persisted when the biguanide was administered in combination with insulin. The latter observation suggested that the anti-cancer effect of metformin was, at least in part, insulin independent [17, 70–72]. A recent meta-analysis evaluating 13,008 subjects with cancer and concurrent T2DM showed that metformin administration, compared to treatment with other glucose-lowering drugs, has beneficial effects in terms of overall survival and cancer-specific survival [73].
A few studies focused their attention on tumors of the gastric-intestinal tract and of the thyroid gland. In a population survey performed in North America, Sehdev et al. showed that metformin reduces the risk of colorectal cancer in patients with diabetes [74]. Furthermore, it was recently reported that an increased cumulative duration of metformin treatment decreases recurrence rate, all-cause mortality, and cancer-specific mortality in diabetic patients affected by gastric cancer [75]. In a retrospective study, Klubo-Gwiezdzinska et al. found that the size of thyroid tumors was significantly smaller in patients with diabetes treated with metformin as compared with those not receiving the drug [76]. In a large series of diabetic Taiwanese patients, treatment with metformin was shown to reduce the risk of thyroid cancer [77]. The protective effect of metformin was maintained after disregarding age and sex of patients, and did not change after excluding users of insulin, sulfonylurea, or insulin plus sulfonylurea. A previous diagnosis of other types of cancer that might introduce a searching bias did not affect the results [77].
Studies in non-diabetic patients with cancer
Several clinical trials were performed in non-diabetic patients with breast cancer. The first pilot randomized trial demonstrated a reduction of Ki67 and of other cancer-specific biomarkers in women with breast cancer treated with metformin [28]. Further support to the anti-cancer effect of metformin derived from the observation that non-diabetic women with breast cancer treated with a higher dose of metformin (1.5 g/day) experienced a more profound decrease of biochemical prognostic markers (insulin level, HOMA-IR index, testosterone level, and free androgen index) compared with those taking a lower dose of the drug (1 g/day) [29]. In a recent trial, non-diabetic women with breast cancer (T1–3, N0–3, M0) were randomized to receive metformin vs. placebo after surgical treatment [78]. The results showed that a 6-month metformin treatment significantly improved several metabolic factors associated with poor breast cancer outcomes (weight, insulin, glucose, leptin, and C-reactive protein). This observation is in agreement with previous data indicating that metformin treatment in newly diagnosed, untreated, non-diabetic breast cancer patients results in clinical and cellular changes consistent with beneficial anti-cancer effects. In particular, the treatment with metformin before surgery resulted in a decrease of weight, BMI, HOMA index and breast cancer cell proliferation index (Ki67) [79].
Two recent studies specifically evaluated the anti-tumor effect of metformin in non-diabetic women with breast cancer starting from the diagnostic biopsy until surgery. In the first study, metformin treatment significantly reduced the insulin receptor expression in tumor cell. The phosphorylation status of protein kinase B (PKB)/Akt (S473), the extracellular signal-regulated kinase 1/2 (ERK1/2, T202/Y204), AMPK (T172) and acetyl coenzyme [80] were also reduced. In the same clinical setting, Cazzaniga et al. compared, after 4 weeks of preoperative metformin, the apoptosis levels in core biopsies and surgical specimens of non-diabetic women with breast cancer. They found a heterogeneous effect of metformin on the modulation of apoptosis, which was dependent upon the degree of individual insulin resistance [81]. This finding would suggest for the first time that metformin exerts a dual effect of on breast cancer growth according to the insulin resistance status [81]. Moving from this observation, the same group of investigators evaluated the effect of metformin according to several biomarkers of insulin resistance (HOMA index, BMI, C-peptide, IGF-I, IGFBP-1, IGFBP-3, free IGF-I, C-reactive protein, adiponectin) and to the tumor subtype. They found that the effect of metformin on the tumor proliferation index Ki67 varies according to both host and tumor characteristics [82].
In vivo studies showing no anti-cancer effect of metformin
Overall, the above reported data suggested that metformin could be the drug of choice in the treatment of patients with cancer and concurrent T2DM, with promising results also for the treatment of oncologic non-diabetic patients. However, a beneficial effect of metformin on tumor progression was not consistently reported in all studies.
In 2014, Oppong et al. analyzed a cohort of patients with T2DM receiving systemic chemotherapy for invasive breast cancer. After a follow-up of 87 months, there were no differences in terms of recurrence-free survival, overall survival and contralateral breast cancer in metformin users vs. nonusers [83]. Ko et al. evaluated the potential benefit of metformin treatment in women who were new users of the drug compared with a control group of new users of sulfonylureas. They found no association with a decreased risk of developing endometrial cancer in women treated with metformin [84]. Negative results, indicating no beneficial effect on cancer, were also reported in recent studies performed in patients with endometrial, lung, prostate, and breast cancer [85–88].
Factors possibly accounting for the discrepant in vivo results
In 2012, Suissa et al. first reported that time-related biases (immortal time bias, time-window bias, and time-lag bias), which potentially lead to an overestimation of the protective effects of metformin, were present in several observational studies reporting a decreased incidence or of cancer mortality in patients taking metformin [89]. This observation may explain the discrepant results obtained in different studies. Time-related biases were taken into account in four recent studies. The results of these more controlled investigations are summarized as follows: (1) the reduction in cancer incidence and cancer mortality observed in diabetic patients taking metformin is of modest relevance and does not affect all populations equally [90], (2) in a cohort of patients with a new diagnosis of diabetes, there was no evidence that metformin use is associated with reduced cancer risk in comparison to sulfonylurea use [91], (3) in a retrospective observational study considering a total of 22,556 diabetic patients, the authors failed to found a reduced cancer risk in metformin users [92], (4) among 1057 patients with T2DM before the diagnosis of breast cancer, the cancer-specific mortality was inversely related to the duration of metformin use, supporting an inverse association between long-term metformin treatment and breast cancer-specific mortality [93]. Thus, it would appear that the discrepancy among different studies is not solved even when time-related biases are taken into account.
The study by Decensi et al. might shed light on the controversy. These authors reported a significant correlation between decreased cancer risk and metformin treatment for hepatocellular and pancreatic cancers, whereas no significant relationship was found for colon, breast or prostate cancers [94]. This finding suggests that metformin elicits cancer site-specific effects. In other words, the effect of metformin could be tumor specific (which would be in line with the results of some in vitro studies) and/or the effect of metformin could be more or less relevant in relation with a given stadium of the malignancy. This appears at the moment the most likely explanation for the discrepant results obtained by in vivo studies evaluating the anti-tumor effect of metformin.
Conclusions
During the past decade, we witnessed a progressive transition of the pharmacological effects of metformin from a well-established anti-diabetic drug to a potential anti-cancer drug. Current knowledge supports the concept that metformin is able to modify the clinical course of human cancer.
The anti-tumor activity of metformin appears to be elicited by several indirect (through lowering insulin levels) and direct mechanisms acting on tumor cells. The latter encompass the more extensively characterized actions of metformin on both AMPK-dependent (mTOR) and AMPK-independent mechanisms. Alternative actions include an effect of metformin on tumor stem cells and the Nf-Kb pathway.
Following initial enthusiasm derived from experimental and early clinical studies, it is now clear that the field of clinical applications of metformin in cancer therapy is more limited than previously believed. Indeed, while in vitro studies consistently point toward anti-cancer effects of metformin, discrepant results are reported by in vivo data. Several studies indicate that the therapeutic benefit of metformin treatment in cancer patients is not universally obtained, but rather they may be more or less evident in specific clinical settings. The latter include both disease-related (i.e., type of tumor, clinical stage) and patient-related (i.e., insulin resistance, age, sex) factors. The retrospective design, the limited number of patients enrolled, the length of the follow-up period and the presence of time-related biases of most currently available studies should be taken into account for interpreting the discrepancy. The most recent advance in this field is represented by clinical trials in which metformin was prescribed to non-diabetic patients with cancer.
Future large-series longitudinal studies in both diabetic and non-diabetic patients with cancer will be required for fully elucidating the role of metformin on cancer development and progression, as well as the specific clinical settings in which metformin could become a prescribed anticancer drug.
References
Bailey CJ, Day C (2004) Metformin: its botanical background. Pract Diab Int 21:115–117
Malek M, Aghili R, Emami Z, Khamseh ME (2013) Risk of cancer in diabetes: the effect of metformin. ISRN Endocrinol 2013:636927
Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, Shulman GI (1998) Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med 338:867–872
Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, Moller DE, Thorell A, Goodyear LJ (2002) Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51:2074–2081
Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE (1995) Metabolic effects of metformin in noninsulin-dependent diabetes mellitus. N Engl J Med 333:550–554
Pala L, Rotella CM (2014) The “slower” the better. J Endocrinol Invest 37:497–498
Glintborg D, Mumm H, Altinok ML, Richelsen B, Bruun JM, Andersen M (2014) Adiponectin, interleukin-6, monocyte chemoattractant protein-1, and regional fat mass during 12-month randomized treatment with metformin and/or oral contraceptives in polycystic ovary syndrome. J Endocrinol Invest 37:757–764
Ghazeeri G, Abbas HA, Skaff B, Harajly S, Awwad J (2015) Inadequacy of initiating rosuvastatin then metformin on biochemical profile of polycystic ovarian syndrome patients. J Endocrinol Invest [Epub ahead of print]
Vassilatou E (2014) Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J Gastroenterol 20:8351–8363
Esteghamati A, Rezvani S, Khajeh E, Ebadi M, Nakhjavani M, Noshad S (2014) Comparative effects of metformin and pioglitazone on YKL-40 in type 2 diabetes: a randomized clinical trial. J Endocrinol Invest (epub ahead of print)
Cappelli C, Rotondi M, Pirola I, Agosti B, Formenti A, Zarra E, Valentini U, Leporati P, Chiovato L, Castellano M (2012) Thyreotropin levels in diabetic patients on metformin treatment. Eur J Endocrinol 167:261–265
Rotondi M, Cappelli C, Magri F, Botta R, Dionisio R, Iacobello C, De Cata P, Nappi RE, Castellano M, Chiovato L (2011) Thyroidal effect of metformin treatment in patients with polycystic ovary syndrome. Clin Endocrinol (Oxf) 75:378–381
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ (2003) Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348:1625–1638
Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, Carboni JM, Gottardis MM, Pennisi PA, Molinolo AA, Kurshan N, Mejia W, Santopietro S, Yakar S, Wood TL, LeRoith D (2010) Insulin mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res 70:741–751
Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R (2009) Diabetes and cancer. Endocr Relat Cancer 16:1103–1123
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330:1304–1305
Bowker SL, Majumdar SR, Veugelers P, Johnson JA (2006) Increased cancerrelated mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 29:254–258
Bo S, Benso A, Durazzo M, Ghigo E (2012) Does use of metformin protect against cancer in type 2 diabetes mellitus? J Endocrinol Invest 35:231–235
Mei ZB, Zhang ZJ, Liu CY, Liu Y, Cui A, Liang ZL, Wang GH, Cui L (2014) Survival benefits of metformin for colorectal cancer patients with diabetes: a systematic review and meta-analysis. PLoS One 9:e91818
Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A (2013) Metformin therapy and risk of cancer in patients with type 2 diabetes. PLoS One 8:e71583
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Shackelford DB, Shaw RJ (2009) The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 9:563–575
El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X (2000) Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275:223–228
Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its antidiabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348:607–614
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262
Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–1646
Towler MC, Hardie DG (2007) AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100:328–341
Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L, Jellema G, Deharo S, Hardie DG, Pusztai L, Moulder-Thompson S, Dewar JA, Thompson AM (2011) Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res Treat 128:783–794
Campagnoli C, Pasanisi P, Abbà C, Ambroggio S, Biglia N, Brucato T, Colombero R, Danese S, Donadio M, Venturelli E, Zito G, Berrino F (2012) Effect of different doses of metformin on serum testosterone and insulin in non-diabetic women with breast cancer: a randomized study. Clin Breast Cancer 12:175–182
Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N (2007) Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 67:10804–10812
Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, Marette A, Kozma SC, Thomas G (2010) Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase dependent manner. Cell Metab 11:390–401
Saeedi R, Parsons HL, Wambolt RB, Paulson K, Sharma V, Dyck JR, Brownsey RW, Allard MF (2008) Metabolic actions of metformin in the heart can occur by AMPK independent mechanisms. Am J Physiol Heart Circ Physiol 294:2497–2506
Treins C, Murdaca J, Van Obberghen E, Giorgetti-Peraldi S (2006) AMPK activation inhibits the expression of HIF-1alpha induced by insulin and IGF-1. Biochem Biophys Res Commun 342:1197–1202
Chiang GG, Abraham RT (2007) Targeting the mTOR signaling network in cancer. Trends Mol Med 13:433–442
Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590
Miller RA, Birnbaum MJ (2010) An energetic tale of AMPK-independent effects of metformin. J Clin Invest 120:2267–2270
Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 120:2355–2369
Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, Sénégas I, Peyraud R, Peyriga L, Théodoro F, Dumon E, Martineau Y, Danet-Desnoyers G, Bono F, Rocher C, Levade T, Manenti S, Junot C, Portais JC, Alet N, Récher C, Selak MA, Carroll M, Sarry JE (2013) Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 27:2129–2138
Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F (2011) Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res 71:4366–4372
Corominas-Faja B, Quirantes-Piné R, Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Martin-Castillo B, Micol V, Joven J, Segura-Carretero A, Menendez JA (2012) Metabolomic fingerprint reveals that metformin impairs onecarbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging (Albany NY) 4:480–498
Pacifico F, Leonardi A (2006) NF-kappaB in solid tumors. Biochem Pharmacol 72:1142–1152
Gerondakis S, Fulford TS, Messina NL, Grumont RJ (2014) NF-κB control of T cell development. Nat Immunol 15:15–25
Hattori Y, Suzuki K, Hattori S, Kasai K (2006) Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 47:1183–1188
Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, Schönbeck U, Libby P (2006) Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol 26:611–617
Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M (2006) Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 66:10269–10273
Carmignani M, Volpe AR, Aldea M, Soritau O, Irimie A, Florian IS, Tomuleasa C, Baritchii A, Petrushev B, Crisan G, Valle G (2014) Glioblastoma stem cells: a new target for metformin and arsenic trioxide. J Biol Regul Homeost Agents 28:1–15
Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, Barbosa AM, Dekker RF, Fortes ZB, Khaper N (2014) Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One 9:e98207
Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB (2007) Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 67:6745–6752
Erices R, Bravo ML, Gonzalez P, Oliva B, Racordon D, Garrido M, Ibañez C, Kato S, Brañes J, Pizarro J, Barriga MI, Barra A, Bravo E, Alonso C, Bustamente E, Cuello MA, Owen GI (2013) Metformin, at concentrations corresponding to the treatment of diabetes, potentiates the cytotoxic effects of carboplatin in cultures of ovarian cancer cells. Reprod Sci 20:1433–1446
Wang LW, Li ZS, Zou DW, Jin ZD, Gao J, Xu GM (2008) Metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol 14:7192–7198
Ashinuma H, Takiguchi Y, Kitazono S, Kitazono-Saitoh M, Kitamura A, Chiba T, Tada Y, Kurosu K, Sakaida E, Sekine I, Tanabe N, Iwama A, Yokosuka O, Tatsumi K (2012) Antiproliferative action of metformin in human lung cancer cell lines. Oncol Rep 28:8–14
Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F (2008) The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 27:3576–3586
Fujihara S, Kato K, Morishita A, Iwama H, Nishioka T, Chiyo T, Nishiyama N, Miyoshi H, Kobayashi M, Kobara H, Mori H, Okano K, Suzuki Y, Masaki T (2015) Antidiabetic drug metformin inhibits esophageal adenocarcinoma cell proliferation in vitro and in vivo. Int J Oncol 46:2172–2180
Bao B, Wang Z, Ali S, Ahmad A, Azmi AS, Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S, Sarkar FH (2012) Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) 5:355–364
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715
Moreno-Bueno G, Peinado H, Molina P, Olmeda D, Cubillo E, Santos V, Palacios J, Portillo F, Cano A (2009) The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat Protoc 4:1591–1613
Sarrió D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J (2008) Epithelialmesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res 68:989–997
Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA (2010) Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 9:3807–3814
Wang Y, Yao B, Wang Y, Zhang M, Fu S, Gao H, Peng R, Zhang L, Tang J (2014) Increased FoxM1 expression is a target for metformin in the suppression of EMT in prostate cancer. Int J Mol Med 33:1514–1522
Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P, Bertolotto C, Tartare-Deckert S, Ballotti R, Rocchi S (2013) Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther 12:1605–1615
Cifarelli V, Lashinger LM, Devlin KL, Dunlap SM, Huang J, Kaaks R, Pollak MN, Hursting SD (2015) Metformin and rapamycin reduce pancreatic cancer growth in obese prediabetic mice by distinct microRNA-regulated mechanisms. Diabetes 64:1632–1642
Tan BK, Adya R, Chen J, Lehnert H, Sant Cassia LJ, Randeva HS (2011) Metformin treatment exerts antiinvasive and antimetastatic effects in human endometrial carcinoma cells. J Clin Endocrinol Metab 96:808–816
Tan XL, Bhattacharyya KK, Dutta SK, Bamlet WR, Rabe KG, Wang E, Smyrk TC, Oberg AL, Petersen GM, Mukhopadhyay D (2015) Metformin suppresses pancreatic tumor growth with inhibition of NFκB/STAT3 inflammatory signaling. Pancreas 44:636–647
Rotondi M, Coperchini F, Pignatti P, Magri F, Chiovato L (2015) Metformin reverts the secretion of CXCL8 induced by TNF-α in primary cultures of human thyroid cells: an additional indirect anti-tumor effect of the drug. J Clin Endocrinol Metab 100:E427–E432
Rotondi M, Coperchini F, Chiovato L (2013) CXCL8 in thyroid disease: from basic notions to potential applications in clinical practice. Cytokine Growth Factor Rev 24:539–546
Bodmer M, Meier C, Krähenbühl S, Jick SS, Meier CR (2010) Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care 33:1304–1308
Bosco JL, Antonsen S, Sørensen HT, Pedersen L, Lash TL (2011) Metformin and incident breast cancer among diabetic women: a population-based case–control study in Denmark. Cancer Epidemiol Biomark Prev 20:101–111
Lee JH, Kim TI, Jeon SM, Hong SP, Cheon JH, Kim WH (2012) The effects of metformin on the survival of colorectal cancer patients with diabetes mellitus. Int J Cancer 131:752–759
Tan BX, Yao WX, Ge J, Peng XC, Du XB, Zhang R, Yao B, Xie K, Li LH, Dong H, Gao F, Zhao F, Hou JM, Su JM, Liu JY (2011) Prognostic influence of metformin as first-line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer 117:5103–5111
Baur DM, Klotsche J, Hamnvik OP, Sievers C, Pieper L, Wittchen HU, Stalla GK, Schmid RM, Kales SN, Mantzoros CS (2011) Type 2 diabetes mellitus and medications for type 2 diabetes mellitus are associated with risk for and mortality from cancer in a German primary care cohort. Metabolism 60:1363–1371
Monami M, Colombi C, Balzi D, Dicembrini I, Giannini S, Melani C, Vitale V, Romano D, Barchielli A, Marchionni N, Rotella CM, Mannucci E (2011) Metformin and cancer occurrence in insulintreated type 2 diabetic patients. Diabetes Care 34:129–131
Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A (2014) New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res 159:355–376
Yin M, Zhou J, Gorak EJ, Quddus F (2013) Metformin is associated with survival benefit in cancer patients with concurrent type 2 diabetes: a systematic review and meta-analysis. Oncologist 18:1248–1255
Sehdev A, Shih YC, Vekhter B, Bissonnette MB, Olopade OI, Polite BN (2015) Metformin for primary colorectal cancer prevention in patients with diabetes: a case–control study in a US population. Cancer 121:1071–1078
Lee CK, Jung M, Jung I, Heo SJ, Jeong YH, An JY, Kim HI, Cheong JH, Hyung WJ, Noh SH, Kim HS, Rha SY, Chung HC (2015) Cumulative metformin use and its impact on survival in gastric cancer patients after gastrectomy. Ann Surg (epub ahead of print)
Klubo-Gwiezdzinska J, Costello J Jr, Patel A, Bauer A, Jensen K, Mete M, Burman KD, Wartofsky L, Vasko V (2013) Treatment with metformin is associated with higher remission rate in diabetic patients with thyroid cancer. J Clin Endocrinol Metab 98:3269–3279
Tseng CH (2014) Metformin reduces thyroid cancer risk in Taiwanese patients with type 2 diabetes. PLoS One 9:e109852
Goodwin PJ, Parulekar WR, Gelmon KA, Shepherd LE, Ligibel JA, Hershman DL, Rastogi P, Mayer IA, Hobday TJ, Lemieux J, Thompson AM, Pritchard KI, Whelan TJ, Mukherjee SD, Chalchal HI, Oja CD, Tonkin KS, Bernstein V, Chen BE, Stambolic V (2015) Effect of metformin vs placebo on and metabolic factors in NCIC CTG MA.32. J Natl Cancer Inst 4:107(3)
Niraula S, Dowling RJ, Ennis M, Chang MC, Done SJ, Hood N, Escallon J, Leong WL, McCready DR, Reedijk M, Stambolic V, Goodwin PJ (2012) Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat 135:821–830
Dowling RJ, Niraula S, Chang MC, Done SJ, Ennis M, McCready DR, Leong WL, Escallon JM, Reedijk M, Goodwin PJ, Stambolic V (2015) Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res 3:17–32
Cazzaniga M, DeCensi A, Pruneri G, Puntoni M, Bottiglieri L, Varricchio C, Guerrieri-Gonzaga A, Gentilini OD, Pagani G, Dell’Orto P, Lazzeroni M, Serrano D, Viale G, Bonanni B (2013) The effect of metformin on apoptosis in a breast cancer presurgical trial. Br J Cancer 109:2792–2797
DeCensi A, Puntoni M, Gandini S, Guerrieri-Gonzaga A, Johansson HA, Cazzaniga M, Pruneri G, Serrano D, Schwab M, Hofmann U, Mora S, Aristarco V, Macis D, Bassi F, Luini A, Lazzeroni M, Bonanni B, Pollak MN (2014) Differential effects of metformin on breast cancer proliferation according to markers of insulin resistance and tumor subtype in a randomized presurgical trial. Breast Cancer Res Treat 148:81–90
Oppong BA, Pharmer LA, Oskar S, Eaton A, Stempel M, Patil S, King TA (2014) The effect of metformin on breast cancer outcomes in patients with type 2 diabetes. Cancer Med 3:1025–1034
Ko EM, Stürmer T, Hong JL, Castillo WC, Bae-Jump V, Funk MJ (2015) Metformin and the risk of endometrial cancer: a population-based cohort study. Gynecol Oncol 136:341–347
Luo J, Beresford S, Chen C, Chlebowski R, Garcia L, Kuller L, Regier M, Wactawski-Wende J, Margolis KL (2014) Association between diabetes, diabetes treatment and risk of developing endometrial cancer. Br J Cancer 111:1432–1439
Sakoda LC, Ferrara A, Achacoso NS, Peng T, Ehrlich SF, Quesenberry CP Jr, Habel LA (2015) Metformin use and lung cancer risk in patients with diabetes. Cancer Prev Res (Phila) 8:174–179
Merrick GS, Bennett A, Couture T, Butler WM, Galbreath RW, Adamovich E (2015) Metformin does not predict for prostate cancer diagnosis, grade, or volume of disease after transperineal template-guided mapping biopsy. Am J Clin Oncol (epub ahead of print)
Soffer D, Shi J, Chung J, Schottinger JE, Wallner LP, Chlebowski RT, Lentz SE, Haque R (2015) Metformin and breast and gynecological cancer risk among women with diabetes. BMJ Open Diabetes Res Care 3:e000049
Suissa S, Azoulay L (2014) Metformin and cancer: mounting evidence against an association. Diabetes Care 37:1786–1788
Gandini S, Puntoni M, Heckman-Stoddard BM, Dunn BK, Ford L, DeCensi A, Szabo E (2014) Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Prev Res (Phila) 7:867–885
Tsilidis KK, Capothanassi D, Allen NE, Rizos EC, Lopez DS, van Veldhoven K, Sacerdote C, Ashby D, Vineis P, Tzoulaki I, Ioannidis JP (2014) Metformin does not affect cancer risk: a cohort study in the U.K. Clinical practice research datalink analyzed like an intention-to-treat trial. Diabetes Care 37:2522–2532
Kowall B, Rathmann W, Kostev K (2015) Are sulfonylurea and insulin therapies associated with a larger risk of cancer than metformin therapy? A retrospective database analysis. Diabetes Care 38:59–65
Vissers PA, Cardwell CR, van de Poll-Franse LV, Young IS, Pouwer F, Murray LJ (2015) The association between glucose-lowering drug use and mortality among breast cancer patients with type 2 diabetes. Breast Cancer Res Treat 150:427–437
Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S (2010) Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 3:1451–1461
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study formal consent is not required.
Rights and permissions
About this article

Cite this article
Coperchini, F., Leporati, P., Rotondi, M. et al. Expanding the therapeutic spectrum of metformin: from diabetes to cancer. J Endocrinol Invest 38, 1047–1055 (2015). https://doi.org/10.1007/s40618-015-0370-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-015-0370-z