
Abstract
Mutations in the fms-like tyrosine kinase 3 (FLT3) gene (internal tandem duplication (ITD) and point mutation in the tyrosine kinase domain, FLT3/D835) as well as the nucleophosmin (NPM1) gene are the most common abnormalities in adult acute myeloid leukemia (AML). Their significance in pediatric AML is still unclear. In this study we evaluated the frequency of FLT3 and NPM1 mutations in childhood AML. We also examined clinical features and outcome of these patients. FLT3 and NPM1 mutations were analysed in 42 and 37 childhood AML patients, respectively, using polymerase chain reaction (PCR) and direct sequencing. FLT3 mutations were detected in 4/42 patients (9.5%). The frequencies of FLT3/ITD and FLT3/D835 were the same, 2/42 (4.7%). NMP1 mutations were found in 1/37 patients (2.7%). FLT3 gene mutations were correlated with induction failure. Here we report the results of the study of FLT3 and NPM1 gene mutations in childhood AML patients in Serbia. Low frequencies of these molecular markers point out that these abnormalities are rare in this cohort of patients. Comparative study of data on NPM1 mutations in childhood AML revealed that various NPM1 gene mutation types are associated with childhood AML. Our findings as well as previously reported data, contributes to a hypothesis of different biology and etiology of adult and childhood AML. More extensive studies of NPM1 and FLT3 mutations in childhood AML are needed to determine their biological and clinical importance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
FLT3 is a member of the third class receptor tyrosine kinase family. It plays a central role in hematopoietic growth regulation of early progenitor cell [1]. The presence of mutation in the FLT3 gene results in FLT3 ligand independent kinase activation leading to increased and uncontrolled cell proliferation. The most common mutations in FLT3 gene are FLT3/ITD of the region coding juxtamembrane (JM) domain and missense point mutations in tyrosine kinase domen (TKD) [2].
Internal tandem duplication of FLT3 gene (FLT3/ITD) is present in approximately 25% of adult acute myeloid leukemia (AML) and is an adverse prognostic factor [3]. The frequency among children is much lower [4, 5].
The most frequent FLT3 point mutation occurs in the second TKD, codon 835. Missense point mutations that substitute the aspartic acid in codon 835 have been reported in about 7% of adult AML patient and less than 4% pediatric AML patients. FLT3/TKD mutations were not found to be an adverse prognostic factor [2, 5–7].
Nucleophosmin (NPM1) is a nucleocytoplasmic shuttling protein with numerous functions such as preventing protein aggregation in the nucleolus and regulation of the assembly and transport of preribosomal particle through the nuclear membrane [8]. Mutations of the NPM1 gene have been reported in 35% of adult AML patients. Even higher frequency is found in AML with a normal karyotype (50–60%) [9]. More than 40 different mutations involving exon 12 of the NPM1 gene have been identified in AML [10]. All of them lead to frameshift and elongation of the protein, which is aberrantly retained in cytoplasm [9]. The presence of NPM1 mutations is associated with favorable outcome and increased event-free survival (EFS) and overall survival (OS), but only in cases when they are not in cooperation with FLT3/ITD mutations.
The incidence of NPM1 mutations found in the studies of pediatric AML is much lower [11]. Due to a small number of reports, the clinical impact of NPM1 gene mutations in these patients remained uncertain.
In this study, we evaluated the frequency of FLT3/ITD, FLT3/D835, and NPM1 gene mutations in Serbian children with AML. The biological and clinical features and the prognostic significance were also assessed. Additionally, data of previous studies on FLT3 and NPM1 gene mutation frequencies and types in childhood AML were compared.
Material and methods
Patients
Ninety-two patients with de novo AML diagnosed since January 1997 till June 2007, at the University Children’s Hospital and Mother and Child Healthcare Institute “Dr Vukan Cupic”, Belgrade, Serbia. Out of these 92 patients, presence for FLT3 and NMP1 gene mutations were analyzed in 42 (45%) and 37 (40%), respectively. All patients tested for NPM gene mutations were also tested for FLT3 mutation. The AML diagnosis was made according to FAB classification. Immunophenotyping was performed by flow cytometry and/or APAAP methods using monoclonal antibodies minimally against CD13, CD33, CD3, CD10, CD19, CD61, and glikophorin A. Cytogenetic G banding analyses was performed with standard method. The non APL children received induction and consolidation regimen derived from Berlin Frankfurt Munster (BFM) AML study group. Our policy treatment until 2006 was that children receive no BMT in first remission. Children with acute promyelocytic leukemia (APL) were treated according to Italian AIDA protocol, based on combined use of all transretinoic acid (ATRA) and idarubicin chemotherapy.
Detection of the FLT3/ITD mutations
To detect FLT3 mutations, genomic DNA was isolated from a bone marrow using QIAamp DNA Blood Mini Kit (Qiagen, Gremany). The PCR amplification was carried out as previously described [12] and its products were resolved on 4% agarose gel stained with ethidium bromide. Each sample displaying an additional PCR product (longer than 325 bp) was considered as containing the internal tandem duplication. Additional bands were extracted from the gel using QIAquick Gel Extraction Kit (Qiagen, Germany) and were directly sequenced.
Detection of FLT3/TKD mutations
Analysis of the FLT3/D835 mutation was carried out as follows: exon 20 of the FLT3 gene was amplified by genomic DNA PCR as previously reported [2]. The PCR products digested with EcoRV (Biolabs, England) were resolved on the 8% polyacrilamide gel.
Detection of NPM1 gene mutations
For the screening of NPM mutations, we amplified genomic DNA corresponding to exon 12 of the NPM gene using PCR method as previously described [9]. Amplified PCR products were purified using QIAquick PCR Purification Kit (Qiagen, Germany), and directly sequenced using internal reverse primer NPM1_1112R [9].
Statistics
Fisher exact test, the chi squares test, the Kaplan–Meier estimator and long rank test for comparison of EFS were used for statistical analyses.
Results
The presenting characteristics of patients are detailed in Table 1.
FLT3 gene mutations were detected in 4/42 patients (9.5%). The frequencies of FLT3/ITD and FLT3/D835 were the same, 2/42 (4.7%). One patient had a mutation in NPM1 gene, but at the same time, carried FLT3/D835 mutation.
FLT3/ITD mutation was found in one boy with microgranular variant of APL and he is in continuous long term remission. Another patient harboring FLT3/ITD mutation never achieved complete remission. Sequence analyses of FLT3/ITD positive patients showed that both ITDs were in-frame involving only juxtamembrane domain of FLT3 protein, namely exon 14. The open reading frame of the transcripts in one patient was preserved by faithful in-frame duplication (24 bp long) and in the other by the insertion of the nucleotides at the ITD junction (the size of duplication was 48 bp and the insertion was 4 bp long).
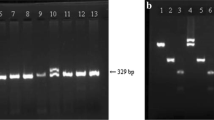
FLT3/D835 mutation was found in one patient with FAB M4 morphology, t(11;17)(q23,q21) and resistant disease who received BMT but died after 19 months. No APL clinical features were present. The other patient, relapsed after 1 year and reversed its mutational status from FLT3/D835 to FLT3/ITD. The ITD duplication found in the relapsed sample was 107 bp long and it is presented in Fig. 1. This patient was also a carrier of NPM1 mutation, which did not change its status during the course of the disease.

Detection of FLT3-ITD mutation. a Line 1 PCR product of the FLT3-ITD in a relapsed sample (duplication of 107 bp); line 2 DNA ladder (Low Range MassRuler, Fermentas); line 3 PCR product of the wt-FLT3 (325 bp). b Partial sequence chromatogram of FLT3-ITD mutation (duplication of 107 bp)
In our cohort of childhood AML patients, NPM1 mutation was found in 1/37 analyzed patients (2.7%). The mutation was of the Q type [13]. Similar to all NPM1 mutations in AML, Q mutation is a heterozygous one, consisting of 4 bp insertion between position nt 964 and nt 965 (Genebank accession NM_002520). Opposite to the previous reports of the Q mutation (4 bp insertion was AGGA), in our case the sequence of 4 bp insertion was CGGA, but the amino acid sequence remained the same (286-DLWQRMEEVSLRK instead of wt sequence 286-DLWQWRKSL).
The mean WBC count at presentation in FLT3 negative patients was 53.6 × 109/l (range 1.4–275 × 109/l). The mean WBC count at presentation in FLT3/ITD positive patients was 114.0 × 109/l (67.2–250 × 109/l), which is not statistically higher comparing to FLT3 negative patients (P = 0.114).
Induction failure was observed in four out of 30 patients in whom remission status was achieved after induction therapy, comparing to two out of four patients with FLT3 mutations (P = 0.047) (Table 1). After median follow up of 63 months in FLT3 negative patients the EFS is 45%, CI 42.23–82.62. Overall survival for this cohort group is 44% CI 40.89–78.63 and for APL group of patients OS is 39% CI 6.74–70.93.
Discussion
Internal tandem duplication of FLT3 gene (FLT3/ITD) is present in approximately 25% of adult AML [3]. The frequency among children is much lower, approximately 5–16.5% (Table 2). However, higher incidence of FLT3/ITD in APL patients was found in several studies [14, 15]. Our study showed relatively low frequency of FLT3/ITD mutations in childhood AML (4.7%). The detected frequency of FLT3/D835 of 4.7% is comparable to other studies (Table 2).
Most studies of children with AML showed that FLT3/ITD mutations are associated with older age, higher WBC, FAB subgroups M1, M2 and M3, normal kariotype, induction failure, and poor outcome [4]. Clinical characteristics of one of our patients with FLT3/ITD are similar to these observations.
In most studies, FLT3/ITD is found to be the strongest independent predictor of outcome, because even patients with t(15;17), a favorable prognostic factor, had a very poor prognosis [4]. But in one case reported by Iwai et al. [16] patient positive for t(15;17) and FLT3/ITD had a very good outcome. Our patient positive for t(15;17) and FLT3/ITD (24 bp duplication) also had good outcome of the disease.
Frequency of FLT3/D835 is relatively low in adult AML patients, as well as in pediatric population and prognostic significance of this mutation has not been established. There are some reports that FLT3/TKD mutation was not an adverse prognostic factor [5, 7]. One of our patients with FLT3/D835 had an adverse outcome, never achieving complete remission. Interestingly, another child with FLT3/D835 and positive for NPM1 mutation relapsed. In the relapse of the disease, FLT3 mutation status changed to FLT3/ITD positive but the same NMP1 mutation was detected at the presentation and the relapse of the disease. Our findings contribute to the hypothesis that FLT3 mutation is not a stable molecular marker [6].
In our study, NPM1 mutation was found in only one of the 37 patient tested (2.7%). Mutations in the NPM1 gene are the single most frequent abnormality in adult AML [10]. NPM1 mutations are distinctly less common in childhood AML ranging from 0 to 6.5% (Table 3). Particularly low incidence of these mutations in Asian population are reported by Chou et al. (2%) and Shimada et al. (0%) (Table 3). These data may suggest the presence of racial variation in susceptibility to these mutations. Our finding is similar to the Asian studies, but the number of analyzed patients is rather small to draw any definite conclusion.
In adults, as well as in childhood patients, the presence of the NPM1 mutations are associated with normal karyotype, FLT3/ITD positive status, older age, high remission induction rate and improved survival, particularly in patients lacking FLT3/ITD [14]. One of the patients included in this study was FLT3/D835-NPM1 double positive. Whitman et al. [17] have recently reported a significant number of patients with similar finding.
The NPM1 mutation we found was of Q type. Type A mutation (tandem duplication of TCTG) was found in approximately 80% of adult cases [14]. On the other hand, Brown et al. [18] found that only 40% of children were type A mutants, and 35% of cases represent novel mutations never reported in adults. The similar findings are reported by Cazzaniga et al. and Thiede et al. [11, 19]. Our finding of unusual type Q mutation is in concordance with these findings.
Comparative study of data on NPM1 mutations in childhood AML (Table 3) revealed that only 46/617 analyzed patients were positive (7.4%). Also, we observed that the frequency of the type A mutation was only 33.3%, and the frequency of all other types was 66.7%.
Evident discrepancy in frequency and type of NPM1 mutation between adult and childhood AML, contributes to a hypothesis of different biology and etiology of the disease in these two groups of patients.
Small number of patients with the FLT3 and NPM1 mutations were analyzed in this study but we believe that data presented in this study could contribute to a growing knowledge of frequency and clinical significance of FLT3 and NMP1 mutations in childhood AML. This study also points out that molecular markers should be studied in a number of different groups of patients in order to make definite conclusion of its medical relevance.
References
Drexler HG. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cell. Leukemia. 1996;10:588–9.
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematological malignancies. Blood. 2001;97:2434–9.
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, et al. The presence of FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–9.
Zwaan CM, Meshinchi S, Radich JP, Veerman AJ, Huismans DR, et al. FLT3 internal tandem mutations in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood. 2003;102:2387–94.
Liang DC, Shih LY, Hung IJ, Yang CP, Chen SH, et al. FLT3-TKD mutation in childhood acute myeloid leukemia. Leukemia. 2003;17:883–6.
Colovic N, Tosic N, Aveic S, Djuric M, Milic N, et al. Importance of early detection and follow-up of FLT3 mutations in patients with acute myeloid leukemia. Ann Hematol. 2007;86:741–7.
Kang HJ, Hong SH, Kim IH, Park BK, Han KS, et al. Prognostic significance of FLT3 mutations in pediatric non-promyelocytic acute myeloid leukemia. Leuk Res. 2005;29:617–23.
Cordell JL, Pulford KA, Bigerna B, Roncador G, Banham A, et al. Detection of normal and chimeric nucleophosmin in human cells. Blood. 1999;93:632–42.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, et al. Cytolasmatic nucleophosmin in acute myelogenous leukemia with normal karyotype. N Engl J Med. 2005;352:254–66.
Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc + AML): biologic and clinical features. Blood. 2007;109:874–85.
Cazzaniga G, Dell`Oro MG, Mecucci C, Giarin E, Masetti R, et al. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood. 2005;106:1419–22.
Kiyoi H, Naoe T, Yokota S, Nakao M, Minami S, et al. Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia. 1997;11:1447–52.
Falini B, Marteli MP, Bolli N, Bonasso R, Ghia E, et al. Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood. 2006;108:1999–2005.
Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106:3733–9.
Arrigoni P, Berreta C, Silvestri D, Rossi V, Rizzari C, et al. FLT3 internal tandem duplication in childhood acute myeloid leukemia: association with hyperleukocytosis in acute promyelocytic leukemia. Br J Haematol. 2003;120:89–92.
Iwai T, Yokota S, Nakao M, Okamoto T, Taniwaki M, et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. Leukemia. 1999;13:38–43.
Whitman SP, Ruppert AS, Radmacher MD, Mrozek K, Paschka P, et al. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favourable prognostic significance. Blood. 2008;111:1552–9.
Brown P, McIntyre E, Rau R, Meshinchi S, Lacayo N, et al. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood. 2007;110:979–85.
Thiede C, Creutzig E, Reinhardt D, Ehninger G, Creutzig U. Different types of NPM1 mutations in children and adults: evidence for an effect of patient age on the prevalence of the TCTG-tandem duplication in NPM1-exon 12. Leukemia. 2007;21:366–7.
Kondo M, Horibe K, Takahashi Y, Matsumoto K, Fukuda M, et al. Prognostic value of internal tandem duplication of the FLT3 gene in childhood acute myelogenous leukemia. Med Pediatr Oncol. 1999;33:525–9.
Meshinchi S, Woods WG, Stirewalt DL, Sweetser DA, Buckley JD, et al. Prevalence and prognostic significance of FLT3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001;97:89–94.
Liang DC, Shih LY, Hung IJ, Yang CP, Chen SH, et al. Clinical relevance of internal tandem duplication of the FLT3 gene in childhood acute myeloid leukemia. Cancer. 2002;94:3292–8.
Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006;108:3654–61.
Xu F, Taki T, Yang HW, Hanada R, Hongo T, et al. Tandem duplication of the FLT3 gene is found in acute lymphoblastic leukemia as well as acute myeloid leukemia but not in myelodysplastic syndrome or juvenile chronic myelogenous leukemia in children. Br J Haematol. 1999;105:155–62.
Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107:4011–20.
Mullighan CG, Kennedy A, Zhou X, Radtke I, Philips LA, et al. Pediatric acute myeloid leukemia with NPM1 mutations is characterised by a gene expression profile with dysregulated HOX gen expression distinct from MLL-rearranged leukemias. Leukemia. 2007;21:2000–9.
Chou WC, Tantg JL, Lin LI, Yao M, Tsay W, et al. Nucleophosmin mutations in de novo acute myeloid leukemia the age-dependent incidences and stability during disease evolution. Cancer Res. 2006;66:3310–6.
Shimada A, Taki T, Kubota C, Tawa A, Horibe K, et al. No nucleophosmin mutations in pediatric acute myeloid leukemia with normal karyotype: a study of the Japanese Childhood AML Cooperative Study Group. Leukemia. 2007;21:1307.
Acknowledgment
This study is supported by grant 143 051 from the Ministry of Science of Serbia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Krstovski, N., Tosic, N., Janic, D. et al. Incidence of FLT3 and nucleophosmin gene mutations in childhood acute myeloid leukemia: Serbian experience and the review of the literature. Med Oncol 27, 640–645 (2010). https://doi.org/10.1007/s12032-009-9261-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12032-009-9261-5