
Abstract
Mutations in the fms-like tyrosine kinase 3 (FLT3) gene, such as internal tandem duplication (FLT3/ITD) in the juxtamembrane domain and point mutations in the tyrosine kinase domain, are the most common abnormalities in acute myeloid leukemia (AML). FLT3/ITD and FLT3/D835 mutations were analyzed in 113 Serbian adult AML patients using polymerase chain reaction. Twenty patients were found to be FLT3/ITD positive (17.7%). The mutations occurred most frequently in M5 and M0 subtypes of AML. They were mainly associated with the normal karyotype. All patients harboring FLT3/ITD had a higher number of white blood cells than patients without it (p = 0.027). FLT3/ITD mutations were associated with lower complete remission (CR) rate (χ 2 = 5.706; p = 0.017) and shorter overall survival (OS; Log rank = 8.76; p = 0.0031). As for disease-free survival, the difference between FLT3/ITD-positive and FLT3/ITD-negative patients was not statistically significant (Log rank = 0.78; p = 0.3764). In multivariate analysis, the presence of FLT3/ITD mutations was the most significant prognostic factor for both OS and CR rate (p = 0.0287; relative risk = 1.73; 95% CI = 1.06–2.82). However, in the group of patients with the intermediate-risk karyotype, the mere presence of FLT3/ITD was not associated with inferior clinical outcome. FLT3/D835 point mutation was found in four patients (3.5%) only. Follow-up of the FLT3/ITD-positive patients revealed stability of this mutation during the course of the disease. However, changes in the pattern of FLT3/D835 mutations in initial and relapsed AML were observed. Our results indicate an association of FLT3/ITD with the adverse outcome in AML patients treated with standard induction chemotherapy. Because FLT3/ITD mutation is a target for specific therapeutic inhibition, its early detection could be helpful in clinical practice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
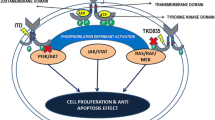
The fms-like tyrosine kinase 3 (FLT3) is a member of the class III receptor tyrosine kinase, with a structure that resembles KIT, FMS, and the platelet-derived growth factor receptor [21, 28, 39]. It is predominantly expressed on hematopoietic progenitor cells, but it is also found in a variety of hematological malignancies including acute myeloid leukemia (AML), B-precursor cell acute lymphoblastic leukemia (ALL), a fraction of T cell ALL, myelodysplastic syndrome in leukemic transformation, and chronic myelogenous leukemia in blast crisis [8, 29]. FLT3 plays an important role in stem cell proliferation, differentiation, and survival. In normal hematopoesis, FLT3 ligand binding to the FLT3 receptor causes dimerization of the receptor, autophosphorylation, activation of tyrosine kinase, and induction of multiple intracellular signaling pathways, which are involved in cell proliferation and leukemogenesis [10, 15].
The presence of a mutation in the FLT3 gene results in FLT3 ligand-independent kinase activation, leading to increased and uncontrolled cell proliferation [15, 23].
The most common mutation in the FLT3 gene is an internal tandem duplication (FLT3/ITD) of the region coding for the juxtamembrane (JM) domain of the FLT3 receptor [14]. It can be found in approximately 20–30% of adult AML patients [20]. The frequency among children is much lower, approximately 5–22% [43]. The presence of a FLT3/ITD mutation is clearly associated with poor prognosis in both groups of the patients.
The second most common type of FLT3 mutations are mis-sense point mutations in the tyrosine kinase domain (TKD). These mutations also promote constitutive phosphorylation of the receptor [2, 36, 42]. The most frequent are point mutations and deletions of codons 835/836 in second TKD, but there are reports on the presence of a 6-bp insertion between codons 840 and 841 (FLT3-840GS) [36] and two novel point mutations N841I and N841Y [16]. Mis-sense mutations were also identified in the JM domain [37] and in the first TKD of FLT3 [25, 31].
Mis-sense point mutations that substitute the aspartic acid in codon 835 have been reported in about 7% of AML patients [2, 42]. Although these point mutations, similar to the FLT3/ITD mutations, cause ligand-independent activation of the FLT3 receptor, their impact on the clinical outcome is not clear. In some studies, FLT3/D835 mutations are associated with shorter disease-free survival (DFS) and relative reduction in overall survival (OS) [2, 42]. On the other hand, in one large study by Thiede et al. [38] on 979 AML patients, no significant differences in DFS or OS could be established between patients harboring FLT3/D835 and patients without the mutation. There is a possibility that the less clear impact of the FLT3/D835 mutation on prognosis is due to a low number of patients with this mutation available for analysis or due to its different influence on cell signaling compared to FLT3/ITD [12].
We have examined biological and hematological characteristics of 113 adult AML patients treated in the Institute of Hematology, Clinical Center of Serbia. We have also analyzed the impact of FLT3 mutations on clinical outcome in these patients, who were followed up for 52 months, with some patients following up for 6 years.
Materials and methods
Patients and treatment
Bone marrow or peripheral blood was obtained at diagnosis from 113 patients (66 female and 47 male) after signed informed consent. Patients with acute promyelocytic leukemia received treatment according to the Pethema/LPA99 protocol (induction therapy: idarubicin 12 mg/m2 on days 2, 4, 6, and 8 and all-transretinoic acid [ATRA] 45 mg/m2 on day 1 until complete remission [CR]), consolidation with idarubicin, novantrone, and ATRA for four cycles, and maintenance chemotherapy with 6-mercaptopurin, methotrexate, and ATRA [30]. All other AML patients received induction and consolidation chemotherapy with standard doses of daunorubicin and cytarabine according to the protocol 3 + 7 (Ara-C 200 mg/m2 for 7 days in continuous infusion and 3 days doxorubicin 50 mg/m2 (cycle I) followed by a second cycle of protocol ADE in which etoposide 200 mg/m2 was added for 5 days. After the ADE protocol, the patients received two courses of consolidation therapy of MACE (amsacrine, cytarabine, etoposide) and MIDAC (mitozantrone, cytarabine) according to the MRC10 protocol [40]. Patients who achieved CR were off from the treatment except for four patients: one who underwent an allogeneic stem cell transplant from an unrelated donor and three patients that were transplanted from a matched sibling donor.
Cytogenetic analyses
Cytogenetic G-banding analysis was performed with standard methods. The definition and descriptions of karyotypes followed the International System for Human Cytogenetic Nomenclature [22]. As cytogenetic finding is thought to be one of the most important prognostic factor for AML patients, they were stratified in three groups: the favorable-risk group consisting of patients with karyotypes of t(8;21), t(15;17), or inv(16), an adverse-risk group defined by -5, del (5q), -7, abnormal 3q, or the presence of a complex karyotype with more than three aberrations, and an intermediate-risk group, which included patients with a normal karyotype and patients with a trisomy of chromosome 8, 21, or 22, alterations in the 11q23 region, del (7q), and del (9q) [11].
Flow cytometry
Immunophenotypic analysis was carried out with flow cytometry FACScalibur (Becton Dickinson Immunocytometry Systems, San Jose, CA) using monoclonal antibodies against human common leukocyte antigens CD45, HLADR, CD34, CD33, CD13, CD117 (c-kit), CD15, CD11b, CD11c, CD41, CD61, and glicophorin A. Analysis was done using the Cell Quest software package (Becton Dickinson). Using flow cytometry and cytological findings, we classified patients in eight subgroups according to French–American–British (FAB) classification.
Detection of FLT3 mutations by PCR
Genomic deoxyribonucleic acid (DNA) was isolated from mononuclear cell preparations using GFX genomic Blood DNA Purification Kit (Amersham Biosciences, GE Healthcare, UK) according to the manufacturer’s recommendations.
As the location of FLT3/ITDs is restricted to exons 14 and 15, the polymerase chain reaction (PCR) amplification was carried out as previously described [17], and its products were resolved on a 4% agarose gel stained with ethidium bromide. Each sample displaying an additional PCR product (longer than 325 bp) was considered as the one containing the internal tandem duplication. Longer PCR products were directly sequenced by the same primers used for amplification.
Analysis of the FLT3/D835 mutation was carried out as follows: exon 20 of the FLT3 gene was amplified by genomic DNA PCR as previously reported [42]. The PCR products digested with EcoRV (Biolabs, England) were resolved on the 8% polyacrylamide gel.
Statistical analysis
Data are presented as medians with range or as absolute numbers with percentages. OS was calculated from the first day of therapy to death. CR duration was calculated from the date of CR to relapse. DFS for patients who had achieved CR was measured from the date of CR to relapse or death. OS and DFS curves were computed according to the Kaplan–Meier method using the SPSS computer software and the rates of survival were compared by log-rank test. Relative risks (RR) and 95% confidence intervals (CI) were estimated with the multivariate Cox’s regression model.
Results
Prevalence and association of FLT3 mutations with FAB subtypes and biological characteristics
Of 113 AML patients studied, 20 had the FLT3/ITD mutation (17.7%). The FLT3/D835 mutation was detected only in 4 of 113 (3.5%) patients (Table 1). One patient with FAB subtype M5 showed both FLT3/ITD and FLT3/D835 mutations. The patients ranged in age from 16 to 80 years with a median age of 50 years. There was no statistically significant difference in age and gender between the patients harboring mutations in the FLT3 gene and patients with no FLT3 mutation.
Patients were classified according to the FAB criteria [4]: 12 M0 (11%), 15 M1 (13%), 23 M2 (20%), 13 M3 (12%), 1 M3 var (1%), 23 M4 (20%), 17 M5a (15%), 2 M5b (2%), 2 M6 (2%), and 2 M7 (2%), and three patients had biphenotypic leukemia (2%; Table 1). The majority of FLT3 mutations were detected in patients with M0 6 of 12 (50%), M5 8 of 19 (40.3%), M3 3 of 14 (21.4%), M2 4 of 23 (18.0%), and M4 3 of 23 (12.5%; Table 1).
Of the 113 patients, cytogenetic analysis was successful in 99 patients. They were divided in three risk groups according to cytogenetics (favorable, intermediate, and adverse).
Sixty patients had abnormalities associated with intermediate risk, 25 were stratified in the favorable-risk group (14 patients t(15;17) and 11 inv (16)), and 14 were with adverse-risk karyotypes. The rate of FLT3 mutations was higher among patients with the intermediate-risk karyotype (13 of 21; 62%) compared to patients with the adverse-risk karyotype (3 of 21; 14%). In the intermediate-risk group, 12 patients had the normal karyotype, and one had trisomy 8.
White blood cell (WBC) counts were significantly higher in patients harboring FLT3/ITD (mean 67.3 × 109/l) than in patients without it (mean 28 × 109/l; p = 0.027). There was not a significant difference in the mean number of WBCs between patients with the FLT3/D835 mutation and patients with no FLT3 mutations.
There was a statistically significant increase in percentage of bone marrow blasts depending on the presence of the FLT3/ITD mutation in subtypes M3, M4, and M5 (p = 0.018; Table 2).
Impact of FLT3 mutations on clinical outcome in patients with AML
CR was obtained in 8 of 24 (33.3%) patients with FLT3 mutations. The rate of CR in those patients was significantly lower when compared with the patients who had no FLT3 mutations (CR 54 of 89, 60.7%). This difference was statistically significant (χ 2 = 5.706; p = 0.017).
Median duration of remission in the whole group of patients was 12 months (mean 17.5), vs 10 months (mean 13.5) in FLT3/ITD-positive patients. There was no statistically significant difference in DFS between FLT3/ITD-negative and FLT3/ITD-positive patients (mean = 13.75; median = 10.00; Log rank = 0.78; p = 0.3764; Fig. 1).

Kaplan–Meier curves of the effects of FLT3/ITDs on DFS of all AML patients included in the study. Solid line, FLT3/ITD-positive patients; dashed line, FLT3/ITD-negative patients
Median survival was 4 months (95% CI 2.09–5.91) for patients with FLT3/ITD mutation comparing to 12 months (95% CI 8.79–15.21) for the patients with no FLT3/ITD mutation.
The OS curve (Fig. 2) suggested a significantly shorter OS in AML patients carrying the FLT3/ITD mutation comparing to those without it. The difference was statistically significant (Log rank = 8.76; p = 0.0031).

Kaplan–Meier curves of the effects of FLT3/ITDs on OS of all AML patients included in the study. Solid line, FLT3/ITD-positive patients; dashed line, FLT3/ITD-negative patients
In multivariate analysis, the presence of the mutation was the most significant prognostic factor for both OS and CR rate (p = 0.0287; RR = 1.73; 95% CI = 1.06–2.82).
As the intermediate-risk disease represents the largest group of patients lacking additional prognostic markers, we have analyzed these patients separately. No statistical significance in DFS was observed between FLT3/ITD-negative and FLT3/ITD-positive patients (Log rank = 0.29; p = 0.5904; Fig. 3). In this group of patients, OS was shortened in the patients with the FLT3/ITD mutation compared to the ones with no FLT3/ITD (5 vs 12 months), but this difference did not reach statistical significance (p = 0.0723 by the log-rank test; Fig. 4). Multivariate analysis showed that the FLT3/ITD state is not an independent prognostic factor for DFS or OS in the intermediate risk group of AML patients (p = 0.6176 and p = 0.0878, respectively).

Kaplan–Meier curves of the effects of FLT3/ITDs on DFS of AML patients with the intermediate-risk karyotype. Solid line, FLT3/ITD-positive patients; dashed line, FLT3/ITD-negative patients

Kaplan–Meier curves of the effects of FLT3/ITDs on OS of AML patients with the intermediate-risk karyotype. Solid line, FLT3/ITD-positive patients; dashed line, FLT3/ITD-negative patients
Because of the limited number of patients with the FLT3/D835 mutation (only four cases), reliable conclusions on survival data could not be drawn.
One patient with AML, subtype M5, with hyperleukocytosis (185 × 109/l) had both FLT3/ITD and FLT3/D835 mutations at the same time. That patient did not achieve CR and died 4 months after diagnosis.
Patients harboring FLT3 mutations were followed up in this study. Most of those who achieved CR (8 of 24) maintained the same FLT3 status at presentation and relapse (six of eight). All of the FLT3/ITD-positive patients preserved the same pattern of the mutation during the course of the disease. However, one patient with the FLT3/D835 mutation and inv(16) lost the FLT3/D835 mutation at relapse (after 7 months). This patient survived for 14 months with paliative treatment [7]. Another patient with a M5a subtype of the disease and normal cytogenetics was positive for the FLT3/D835 mutation at the diagnosis and lost this type of mutation during relapse (after 8 months) but acquired two different FLT3/ITD (duplications of 63 bp and 46 bp) mutations. The patient survived for 26 months.
Discussion
Mutations in the FLT3 gene are one of the most frequent somatic alterations in AML patients [18, 42]. A number of reports have shown that some of these mutations are an independent adverse prognostic factor in AML. This has led to the general belief that FLT3 mutations are an important molecular target for the development of specific therapeutics against the constitutively activated FLT3 receptor. Different treatment protocols reported in various studies of prognostic significance of FLT3 mutations in patients with AML were useful in designing treatment protocols used in several ongoing or planned trails. Therefore, early detection of FLT3 mutations became a standard diagnostic procedure for AML patients in several clinical centers.
We have investigated the prevalence of FLT3/ITD and FLT3/D835 mutations in a cohort of 113 AML patients treated in the Institute of Hematology, Clinical Center of Serbia, and their possible prognostic significance. The incidence of the FLT3/ITD mutation in our cohort of AML patients (17.7%) is marginally lower than the ones found in UK (27%) [18], Dutch (22%) [27], and German studies (23.5%) [32]. The incidence rate of the FLT3/ITD mutation in this study corresponds with the report of Yamamoto et al. (18.9%) [42]. The rate of the FLT3/D835 mutation (3.5%) was lower than in other series [2, 42]. It is possible that it is due to either a slightly different distribution of FAB subtypes of AML patients in this study compared to the others, or a limited number of patients.
Schnittger et al. [32] and Kiyoi et al. [17] have found that the incidence of FLT3/ITD mutations in patients with t(15;17) are 28.6 and 20.3%, respectively. It seems that there is an increased incidence of FLT3 mutations in patients with promyelocytic leukemia–retinoic acid receptor rearrangement.
In our study, the highest rate of FLT3/ITD mutations was detected in FAB M5 (seven patients) and M0 patients (six patients). This confirms the results of some other studies [13, 38]. FLT3 is persistently expressed during the process of monocyte differentiation, and the FLT3-L is required for complete differentiation of monocytes from CD34+ cells [9, 26]. According to these facts, we can presume that constitutive activation of FLT3 might be associated with monocyte differentiation.
We have detected both FLT3/ITD and FLT3/D835 mutations in favorable and intermediate-risk cytogenetic categories. FLT3/ITD was also detected in an adverse-risk category. Cytogenetic analyses of 21 patients with FLT3 mutations showed that 12 of them had the normal karyotype (57%). These results are similar to most published series [1, 20, 32]. It is well known that the expression of the constitutively activated FLT3 receptor in transgenic mice displayed a myeloproliferative disease phenotype only [3]. These findings suggest that at least one additional mutation besides the one in the FLT3 gene is indispensable for leukemic transformation. Therefore, it is essential to further investigate molecular alterations in patients with AML and the normal karyotype.
Our study demonstrates that the presence of FLT3/ITD mutation at diagnosis predicts the poor outcome of induction chemotherapy and attainment of CR. Moreover, it was the most significant factor predicting for OS in our patients (p = 0.0287). Decreased OS has been reported in numerous studies of AML patients harboring the FLT3/ITD mutation, including the meta-analysis of Yanada et al. [41]. However, in several studies, the presence of the FLT3/ITD mutation did not effect OS. According to the authors explanations, it could be due to a higher percentage of elderly patients or patients with therapy-related AML, myelodysplastic syndrome, or AML with preceding myelodysplasia included in the study, short follow-up, or the application of more intensive induction chemotherapy [5, 20, 35].
Several studies have shown that OS in the group of AML patients with an intermediate-risk karyotype is not different between those with and without FLT3/ITD mutation [32, 38]. Additionally, in the study of Schnittger et al. [32], the multivariate analysis showed that the FLT3/ITD state is not an independent prognostic factor for DFS or OS in the intermediate risk group of AML patients. The results of our study are similar to those observed by Schnittger et al. [32].
We have followed up our group of FLT3-positive patients to investigate whether the mutation can be used as a marker of minimal residual disease. Most of them (75%) maintained the same FLT3 status at presentation and relapse, including all of the FLT3/ITD-positive patients. We have found that only two patients with the FLT3/D835 mutation lost the mutation at relapse of the disease. At the same time, one of them acquired the FLT3/ITD mutation at relapse. Heterogeneous patterns of FLT3/D835 mutations at presentation and relapse have also been reported in AML patients [19, 33, 34]
Opposite to our findings, Kottaridis et al. found that 5 of 20 (25%) of the patients who had been FLT3/ITD positive at the presentation of the disease had lost their mutation at relapse. The same findings were published by Nakano et al. [24]. This loss of the FLT3/ITD mutation was not confirmed in the large study of Schnittger et al. [32], who reported that all 25 patients who could be analyzed at diagnosis and at relapse carried the FLT3 mutation at both time points. Our study is in concordance with these results.
We, however, observed that 50% of the FLT3/D835-positive patients lost the mutation in relapse. This finding is in concordance with the report of Shih et al. [34]. Moreover, one patient did not only loose the FLT3/D835 mutation at relapse but gained two different FLT3/ITD mutations. In a recent study by Cloos et al. [6], the authors suggested that the acquisition of FLT3/ITD in relapsed AML can be explained by oligoclonality in the samples from an initial diagnosis. A lot of subclones are undetectable by the PCR assay. We can hypothesize that in the case of this patient, there were at least three subclones present (FLT3/D835-positive and two minor FLT3/ITD subclones under the detection limit). During initial therapy, the D835-positive clone was eradicated only.
Our results suggest that the FLT3/ITD but not the FLT3/D835 mutation can be used as a valid marker of minimal residual disease. However, this should be treated with cautiousness because of the limited number of analyzed cases.
We suggest that an early detection of FLT3 mutations should become a standard diagnostic procedure for AML patients because they are a molecular target for specific therapeutics. In a number of ongoing clinical trials, conventional chemotherapy is combined with FLT3 inhibitors in an attempt to realize a synergistic effect against leukemia cells harboring the FLT3 mutation. The success of these trails will finally determine the importance of FLT3 mutations for clinical practice.
References
Abu-Duhier FM, Goodeve AC, Wilson GA et al (2000) FLT3 internal tandem duplications in adult acute myeloid leukemia define a high-risk group. Br J Haematol 111:190–195
Abu-Duhier FM, Goodeve AC, Wilson MA et al (2001) Identification of novel FLT3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol 113:983–988
Baldwin BR, Tse KF, Small D (2001) Transgenic mice expressing a constitutively activated FLT3 receptor display a myeloproliferative disease phenotype. Blood 98:801
Bennet JM, Catovsky D, Daniel MT et al (1985) Proposed revised criteria for the classification of acute myeloid leukemia: a report of the French–American–British Cooperative Group. Ann Intern Med 103:620–629
Boissel N, Cayuela JM, Preudhomme C et al (2002) Prognostic significance of FLT3 internal tandem repeat in patients with de novo acute myeloid leukemia treated with reinforced courses of chemotherapy. Leukemia 16:1699–1704
Cloos J, Goemans BH, Hess CJ et al (2006) Stabillity and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia 20:1217–1220
Colovic M, Jurisic V, Pavlovic S et al (2006) FLT3/D835 mutation and inversion of chromosome 16 in leukemic transformation of myelofibrosis. Eur J Intern Med 17:434–435
Drexler HG (1996) Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 10:1584–1591
Gabbianelli M, Pelosi E, Montesoro E et al (1995) Multilevel effects of flt3 ligand on human hematopoiesis: expansion of putative stem cells and proliferation of granulomonocytic progenitors/monocytic precursors. Blood 86:1661–1670
Gilliland DG, Griffin JD (2002) The roles of FLT3 in hematopoiesis and leukemia. Blood 100:1532–1542
Grimwade D, Walker K, Oliver F et al (1998) The importance of diagnostic cytogenetics on outcome in AML: analysis of 1.612 patients entered into MRC AML trial. The Medical Reserch Council Adult and Children’s Leukemia Working Parties. Blood 92:2322–2333
Grundler R, Miething C, Thiede C et al (2005) FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantational model. Blood 150:4792–4800
Haferlach T, Schoch C, Schnittger S et al (2002) Distinct genetic patterns can be identified in acute monoblastic and acute monocytic leukaemia (FAB AML M5a and M5b): a study of 124 patients. Br J Haematol 118:426–431
Hayakawa F, Towatari M, Kiyoi H et al (2000) Tandem duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 19:624–631
Heldin CH (1995) Dimerization of cell surface receptors in signal transduction. Cell 80:213–223
Jiang J, Paez JG, Lee JC et al (2004) Identification and characterization of a novel activating mutation of the FLT3 tyrosine kinase in AML. Blood 104:1855–1858
Kiyoi H, Naoe T, Yokota S et al (1997) Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocitic leukemia. Leukemia 11:1447–1452
Kottaridis PD, Gale RE, Frew ME et al (2001) The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 98:1752–1759
Kottaridis PD, Gale RE, Langabeer SE et al (2002) Studies of FLT3 mutations in paired presentation and relapse samples from patient with acute myloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood 100:2393–2398
Kottaridis PD, Gale RE, Linch DC (2003) Prognostic imlications of the presence of FLT3 mutations in patients with acute myeloid leukemia. Leuk Lymphoma 44:905–913
Matthews W, Jordan CT, Wiegand GW et al (1991) A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell 65:1143–1152
Mitelman F (ed) (1995) An international system for human cytogenetic nomenclature. Karger, Basel, Switzerland
Nakao M, Yokota S, Iwai T et al (1996) Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 10:1911–1918
Nakano Y, Kiyoi H, Miyawaki S et al (1999) Molecular evolution of acute myeloid leukemia in relapse: unstable N-ras and FLT3 genes compared with p53 gene. Br J Haematol 104:659–664
Piccaluga PP, Biancchini M, Martinelli G (2003) Novel FLT3 point mutation in acute myeloid leukaemia. Lancet Oncol 4:604
Rappold I, Ziegler BL, Kohler I et al (1997) Functional and phenotypic characterisation of cord blood and bone marrow subsets expressing FLT3 (CD 135) receptor tyrosine kinase. Blood 90:111–125
Rombouts WJ, Blokland I, Lowenberg B, Ploemacher RE (2000) Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the Flt3 gene. Leukemia 14:675–683
Rosnet O, Marchetto S, de Lapeyriere O, Birnbaum D (1991) Murine Flt3, a gene encoding a novel tyrosine kinase receptor of the PDGFR/CSF1R family. Oncogene 6:1641–1650
Rosnet O, Buhring HJ, Marchetto S et al (1996) Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 10:238–248
Sanz MA, Martin G, Vellenga E et al (2005) Risk-adapted treatment of acute promyelocytic leukemia: updated results of the Spanish PETHEMA LPA99 Trial using ATRA and anthracycline monochemotherapy. J Clin Oncol 23:6515
Schittenhelm MM, Yee KWH, Tyner JW et al (2006) FLT3 K663Q is a novel AML-associated oncogenic kinase: determination of biochemical properties and sensitivity to Sunitinib (SU11248). Leukemia 20:2008–2014
Schnittger S, Schoch C, Dugas M et al (2002) Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia (AML): correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study, and usefulness as a marker for detection of minimal residual disease. Blood 100:59–66
Scholl S, Loncarevic IF, Krause C et al (2005) Analyses of minimal residual disease based on Flt3 mutations in allogeneic peripheral blood stem cell transplantation. Leuk Res 29:849–853
Shih LY, Huang CF, Wu JH et al (2004) Heterogenous patterns of FLT3 Asp(835) mutations in relapsed de novo acute myeloid leukemia: a comparative analysis of 120 paired diagnostic and relapse bone marrow samples. Clin Cancer Res 10:1326–1332
Small D (2006) FLT3 mutations: biology and treatment. Hematology 2006:178–184
Spiekermann K, Bagrintseva K, Schoch C et al (2002) A new and recurrent activating lenght mutation in exon 20 of the FLT3 gene in acute myeloid leukemia. Blood 100:3423–3425
Stirewalt DL, Mechinchi S, Kussick SJ et al (2004) Novel FLT3 point mutations within exon 14 found in patients with acute myeloid leukaemia. Br J Haematol 124:481–484
Thiede C, Steudel C, Mohr B et al (2002) Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99:4326–4335
Ulrich A, Schlessinger J (1990) Signal transduction by receptors with tyrosine kinase activity. Cell 61:203–212
Wheatley K, Burnett AK, Goldstone AH et al (1999) A simple robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukemia derived from MRC AML 10 trial. United Kingdom Medical Resourch Council’s Working Parties. Br J Haematol 107:69–79
Yanada M, Matsuo K, Suzuki T et al (2005) Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia 19:1345–1349
Yamamoto Y, Kiyoi H, Nakano Y et al (2001) Activating mutation of D835 within the activation loop of FLT3 in human hematological malignancies. Blood 97:2434–2439
Zwaan CM, Meshinchi S, Radich JP et al (2003) FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to celllular drug resistance. Blood 102:2387–2394
Acknowledgments
This study is supported by grant 145 061 from the Ministry of Science and Environmental Protection of Serbia. The experiments performed in this study comply with the current laws of Serbia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ms. Colovic and Ms. Tosic contributed equally to this work.
Rights and permissions
About this article
Cite this article
Colovic, N., Tosic, N., Aveic, S. et al. Importance of early detection and follow-up of FLT3 mutations in patients with acute myeloid leukemia. Ann Hematol 86, 741–747 (2007). https://doi.org/10.1007/s00277-007-0325-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-007-0325-3