
Abstract
Data from preclinical studies propose nicotinamide adenine dinucleotide (NAD+) as a neuroprotective and bioenergetics stimulant agent to treat Alzheimer’s disease (AD); however, there seems to be inconsistency between behavioral and molecular outcomes. We performed this systematic review to provide a better understanding of the effects of NAD+ in rodent AD models and to summarize the literature.
Studies were identified by searching PubMed, EMBASE, Scopus, Google Scholar, and the reference lists of relevant review articles published through December 2020. The search strategy was restricted to articles about NAD+, its derivatives, and their association with cognitive function in AD rodent models. The initial search yielded 320 articles, of which 11 publications were included in our systematic review.
Based on the primary outcomes, it was revealed that NAD+ improves learning and memory. The secondary endpoints also showed neuroprotective effects of NAD+ on different AD models. The proposed neuroprotective mechanisms included, but were not limited to, the attenuation of the oxidative stress, inflammation, and apoptosis, while enhancing the mitochondrial function.
The current systematic review summarizes the preclinical studies on NAD+ precursors and provides evidence favoring the pro-cognitive effects of such components in rodent models of AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the current aging society, the number of patients suffering from Alzheimer’s disease (AD) is rapidly increasing, and without therapeutic breakthroughs is estimated to reach 80 million by 2040 (Li et al. 2016). As an irreversible chronic neurodegenerative disease, AD is characterized by progressive cognitive deficits, memory loss, and personality changes taking place with advancing age (Dong et al. 2019). Neuropathological hallmarks of AD are mainly associated with the accumulation of extracellular senile amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs), consisting of hyperphosphorylated tau proteins (Sadigh-Eteghad et al. 2015). Studies have shown that extensive distribution of Aβ oligomers and NFTs within neurons is associated with the generation of oxygen free radicals and the inhibition of synaptic transmission and long-term potentiation (LTP) (Lesné et al. 2006; Wilkinson and Landreth 2006). Synaptic dysfunction, and especially the degeneration of cholinergic neurons, also leads to cognitive impairment and deficits in memory and learning and in emotional resilience (Chen and Mobley 2019). Oxidative stress, apoptosis, inflammation, autophagy, and mitochondrial dysfunction have also been widely shown to be involved in the progress of the disease (Bostancıklıoğlu 2019; Butterfield and Halliwell 2019). Thus, it seems that novel multi-targeted treatments are needed to prevent or slow disease progression.
Nicotinamide adenine dinucleotide (NAD+) is a crucial cofactor involved in a wide spectrum of biological functions including oxidative phosphorylation, mitochondrial function and bioenergetics, cell proliferation, and calcium homeostasis (Wei et al. 2017). Also, NAD+ is essential for appropriate neuronal function and survival in the central nervous system (Fang et al. 2014). NAD+ enters mammalian mitochondria via two transporters, SLC25A51 and SLC25A52 (Luongo et al. 2020). It has been shown that during the process of pathological aging and neurodegenerative disease, mitochondrial dysfunction is associated with a reduction in NAD+, which in turn prevents cellular respiration, leading to reduced adenosine triphosphate (ATP) levels and neuronal death (Hosseini et al. 2019). Since neurons have a high demand for energy and are very sensitive to NAD+ deficiency, maintaining intracellular levels of NAD+ through its precursors such as nicotinamide riboside (NR), nicotinamide mononucleotide (NMN), and nicotinamide (NAM) is essential. In addition, these agents could be therapeutic candidates for metabolic dysfunction in age-related neurodegenerative diseases (Liu et al. 2008). Several studies have examined the possible therapeutic effects of NAD+ precursors in preclinical models of AD (Gong et al. 2013; Long et al. 2015). In the current systematic review, we summarize the effects of NAD+ precursors in preclinical studies of AD and further discuss the neuroprotective and pro-cognitive effects of NAD+ precursors.
Methods
Search Strategy
The literature was systematically searched from four independent databases, i.e. PubMed, EMBASE, Scopus, and Google Scholar, for studies published no later than December 2020. Specific Medical Subject Headings (MeSH) and keywords that were searched included the following: [(Alzheimer Disease) OR (AD)] AND [(nicotinamide) OR (nicotinamide-beta-riboside) OR (niacin) OR (Nicotinamide adenine dinucleotide) OR (NAD+) OR (Nicotinamide Mononucleotide)] AND [(rodent) OR (rat) OR (mice) OR (mouse) OR (Rattus)].
The search was limited to original English-language articles, with no date restrictions. The reference lists of all included studies were further searched to avoid missing any relevant publications.
Inclusion and Exclusion Criteria
Original research articles published in English having an administration regimen of NAD+ precursors were included. We did include all dosages, administration routes, and administration time intervals of the drugs.
Non-rodent models, ex pathogenesis and development or in vitro (primary culture or cell line) experiments, non-relevant treatments, duplicate articles, reviews, theses, and books were excluded from the study.
Data Extraction and Quality Assessment
Based on the inclusion and exclusion criteria, two investigators independently screened the titles/abstracts and the full texts, where necessary, and disagreements were resolved by a third party, i.e. senior researcher. The articles were listed based on (i) authors and publication year; (ii) animal characteristics including species, sex, weight, and age; (iii) duration of treatment, dosage, and administration route; and (iv) the model used in the study (Table 1).
To examine the internal validity of the selected studies (e.g., selection, performance, detection, and attrition bias) and other study quality measures (e.g., reporting quality and power), a modified version of the Collaborative Approach to Meta Analysis and Review of Animal Data from Experimental Studies (CAMARADES) quality checklist was used. The items in the list comprised (i) publication in a peer-reviewed journal, (ii) randomization to treatment or control, (iii) allocation concealment, (iv) blinded assessment of an outcome, (v) statement of inclusion and exclusion of animals in the study, (vi) sample size calculation, (vii) statement of compliance with regulatory requirements, and (viii) statement of possible conflicts of interest.
Results
Study Selection
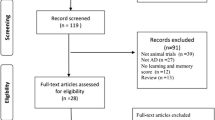
We evaluated the results of 11 studies investigating the effects of NAD+ precursors in the experimental AD models. A PRISMA [Preferred Reporting Items for Systematic Reviews and Meta-Analyses] flowchart and the study selection process are presented in Fig. 1. Overall, 247 articles were found after excluding the replicated studies. Of these, 205 articles were excluded based on the title and the abstract. Full texts of 42 articles were further read in order to assess their eligibility, excluding the non-relevant papers based on exclusion criteria. Finally, 11 qualified articles were included in the systematic review.

PRISMA flowchart and the study selection process of the systematic review
Quality of Included Studies
All investigated articles are published in peer-reviewed journals. A disclosure statement for conflict of interest was present in 91.67% of the studies. Thirty-three percent of the studies reported random allocation. Compliance with animal welfare regulations was reported in 83.34% of the studies. The sample size calculation was mentioned in none of the publications. The model and animal exclusion criteria and blind outcome assessment were reported in 16.67% and 8.33% of the papers, respectively (Fig. 2).

Evaluation of the included studies based on the modified CAMARADES quality checklist
AD Models in Included Studies
The 11 included studies involved five main types of AD models: (i) amyloid precursor protein (APP), (ii) presenilin 1 (PS1), (iii) tau transgenic models, (iv) streptozotocin (STZ), and (v) Aβ1–42-induced surgical models.
Primary Outcomes
Behavioral Tests
Among the included studies, eight investigated the effects of NAD+ precursors on behavioral outcomes (Table 2). Tasks for behavioral studies included the Morris water maze (MWM), elevated plus maze (EPM), Y-maze, novel object recognition (NOR), passive avoidance (PA), fear conditioning (FC), and open field (OF) tests. These tasks assess learning, memory, and depression- and anxiety-like behaviors in various experimental models of AD.
The results of a study by Liu et al. ( 2013) showed that NAM administration for 8 months significantly reduced goal latencies and path lengths, and markedly increased the time spent in the target quadrant in the MWM task. In the OF test, mice treated with NAM exhibited more vertical counts and stereotyped movements. However, no significant relation was established for the distance traveled and the ambulatory counts between NAM-treated 3xTg-AD and control groups (Liu et al. 2013). In another study, NR supplementation was revealed to improve recognition memory performance in Tg2576 mice, as assessed by the NOR test (Gong et al. 2013). According to Green et al. (2008), NAM treatment via an oral route in AD mice led to a noticeable improvement in spatial memory and prevented contextual fear memory deficits. Also, in the same study, no difference was found between NA-treated 3xTg-AD animals and the vehicle group in the NOR test. As reported by Hou et al., AD induction impairs spatial learning, recognition, and working memory in animals, while NR treatment reverses such deficits (Hou et al. 2018). NR was also found to increase cued and context freezing times and decrease anxiety-like behavior in AD mice. In a study by Vakilinezhad et al. (2018), NAM-loaded phosphatidylserine-solid lipid nanoparticles (PS-SLN) alleviated spatial learning and memory deficits in AD rats. In other studies, NMN treatment counteracted learning deficits and ameliorated MWM performance (Yao et al. 2017; Zhang et al. 2016). According to Xie et al., supplementation of NR improved contextual fear memory, but not cue fear memory, in APP/PSA1 mice (Xie et al. 2019). Also, there was no significant difference in anxiety behavior and short-term memory between the NR and the control group in APP/PSA1 mice.
Secondary Outcomes
Neuroprotective Mechanisms
Aβ- and Tau-Related Mechanisms
Among the 11 studies included in the review, eight investigated the effects of NAD+ precursors on Aβ plaque aggregation and/or tau hyperphosphorylation. Liu et al. (2013) illustrated that NAM treatment decreased the levels of p-tau and Aβ accumulation in the CA1 region of the hippocampus (HIP), subiculum, and the cerebral cortex in 3xTg mice. It was also reported that administration of NR for 3 months reduced Aβ production (Gong et al. 2013). Although Green and colleagues did not observe any effect of NAM treatment on Aβ pathology, they reported decreased Thr231-p-tau levels in 3xTg-AD mice (Green et al. 2008).
Similarly, evidence from the study by Hou et al. indicated that NR-treated AD mice demonstrated a decreased level of p-tau pathology, but no impact was observed on Aβ accumulation (Hou et al. 2018). Another study revealed that NAM prevented tau hyperphosphorylation in a STZ-induced rat model of AD (Vakilinezhad et al. 2018). Xie et al. demonstrated that an NR regimen inhibited Aβ accumulation while also blocking astrocyte migration towards zones of Aβ accumulation (Xie et al. 2019). A marked reduction in the levels of Aβ1-40, Aβ1-42 and APP expression was observed in the cortex and HIP of NMN-treated transgenic AD mice (Yao et al. 2017). Kim and Yang reported that NAM pretreatment decreased the expression of APP and PS1 in brain tissue (Kim and Yang 2017).
Under physiological conditions, microtubule-associated protein 2 (MAP2) is located predominantly in the cell body and dendrites, while tau is found mostly in axons. MAP2 is considered a main component of the neuronal cytoskeleton, with a key role in different stages of nervous system development, formation, and regeneration. Tau triggers tubulin polymerization and induces microtubule stabilization (Manczak et al. 2018). Tau is phosphorylated by two major kinases, glycogen synthase kinase 3 (GSK3) and cyclin-dependent kinase 5 (cdk5) (Hu et al. 2011). The accumulation of phosphorylated tau in the hippocampal area is responsible for the loss of dendritic protein MAP2, which in turn results in neuronal death, and consequently impaired learning and memory (Kandimalla et al. 2018). Based on the results of the study by Green et al., oral administration of NA cannot modulate the activity of GSK3β and cdk5, but can enhance learning and memory via the upregulation of structural and synaptic proteins (Green et al. 2008). In fact, decreased acetylated α-tubulin was observed in tangle-bearing neurons in the AD brain. Interestingly, NA intervention was shown to increase acetyl-α-tubulin levels significantly; however, it had no effect on the steady-state levels of total monomeric or dimeric α-tubulin. This study also found a significant increase in MAP2c in NA-treated AD mice compared to the vehicle group (Green et al. 2008).
Although MAP2 shares highly homologous carboxyl-terminal sequences with tau, these microtubules have different fates in the course of NFT formation in the AD brain. It has been demonstrated that tau forms filaments when aggregated, while MAP2 aggregates to granules. One study found that MAP2 was not implicated in the growth process of NFT in the AD brain (Xie et al. 2015).
Anti-inflammatory Effects
Among the included studies, five articles assessed the anti-inflammatory effect of NAD+ precursors in AD mice. Inflammation in neurodegenerative diseases is characterized by activated microglia, reactive astrocytes, and increased release of inflammatory cytokines (Kim et al. 2004; Waetzig et al. 2005). Glial fibrillary acidic protein (GFAP) and ionized calcium-binding adaptor molecule 1 (IBA1) are specific markers for activated astrocytes and microglia, respectively (Kaur et al. 2015). Hou et al. provided evidence that NR reduced GFAP and IBA1 in both the cortex and HIP regions and normalized the levels of pro-inflammatory and inflammatory cytokines including interleukin (IL)-1α, IL-1β, tumor necrosis factor (TNF)-α, MCP-1, MIP-1α, and RANTES in AD mice (Hou et al. 2018). Another study (Xie et al. 2019) showed that supplementation of NR decreased the density of GFAP-positive cells in the HIP of APP/PS1 mice, but had no effect on the density of IBA1-positive cells in the cortex and hippocampal dentate gyrus. Also, Yao et al. found that treatment with NMN inhibited c-Jun N-terminal kinase (JNK) activation, leading to suppression of glial cell activation. This further reduced the levels of proinflammatory cytokines such as IL-6, IL-1β, and TNF-α (Yao et al. 2017). NAM treatment was also shown to inhibit poly (ADP-ribose) polymerase-1 (PARP-1) overactivation (Turunc Bayrakdar et al. 2014) and reduce the mRNA and protein levels of nuclear factor kappa B (NF-κ B) expression (Kim and Yang 2017; Turunc Bayrakdar et al. 2014).
Mitochondrial Function and Bioenergetics
Mitochondrial dysfunction is a prominent feature of the AD brain, and leads to morphological and functional abnormalities and limits electron transport chain and ATP production (Bonda et al. 2010). A decreased level of ATP reduces the neuron’s ability to maintain ionic gradients, which further hinders the production and propagation of action potentials, and thus, neurotransmission (Butterfield and Halliwell 2019). Mitochondrial morphology is modulated by a dynamic balance between fission and fusion. Mitochondrial fusion is regulated by protein optic atrophy 1 (Opa1) and mitofusin proteins (Mfn1 and Mfn2), and its fission is regulated by dynamin-related protein (Drp1) and fission protein 1 (FIS1) (Zhu et al. 2013). It was demonstrated that the balance between mitochondrial fission and fusion is impaired in AD (Wang et al. 2009). Long et al. found that the rate of mitochondrial fission was higher in transgenic AD models than that in control animals. Mitochondrial dynamics were shown to be improved in NMN-treated AD mice (Long et al. 2015).
Four studies also investigated the effects of NAD+ precursor supplementation on the levels of NAD+, and all reported increased levels of NAD+ in AD models (Gong et al. 2013; Hou et al. 2018; Liu et al. 2013; Zhang et al. 2016).
The study by Liu et al. (2013) revealed that NAM treatment normalized mitochondrial dynamics and improved autophagy-lysosome procession via enhanced lysosome/autolysosome acidification to reduce autophagosome accumulation. NAM treatment was also found to activate neuroplasticity-related kinases (p-Akt, MAPK/ERK42, and cyclic AMP response element-binding protein [CREB]) and elevate sirtuin 1 (SIRT1) protein levels in the brains of AD mice. Gong et al. (2013) reported that NR improved synaptic plasticity and facilitated proliferator-activated receptor-γ coactivator 1α (PGC-1α) and β-secretase 1 degradation. Furthermore, it promoted the expression of energy metabolism genes citrate synthase, cytochrome c subunit Vic, aconitase, pyruvate dehydrogenase kinase 3, phosphoglycerate kinase, and glucose phosphate isomerase 1. Kim and Yang showed that the administration of NAM in AD mice increased the expression of SIRT1 (Kim and Yang 2017).
Oxidative Stress and Synaptic Factors
Oxidative stress is an imbalance between the production of reactive oxygen species (ROS)/reactive nitrogen species (RNS) and the counteracting antioxidant mechanisms. It is a major contributor to the pathogenesis and development of AD (Ton et al. 2020). Turunc Bayrakdar et al. (2014) reported that NA markedly reduced oxidative stress elements including protein carbonyls, ROS, and malondialdehyde (MDA), and increased antioxidant enzyme activity in the rat brain. This study also found that mRNA and protein levels of apoptosis markers were decreased in the HIP and cortex of AD rats treated with NA (Turunc Bayrakdar et al. 2014).
NAD+ processor administration may affect the synaptic factors in the brain of AD animals. It was shown that the expression of MAP2c was increased after NA administration in AD animals (Green et al. 2008). Moreover, Yao et al. reported that NMN ameliorated synaptic loss and increased the levels of postsynaptic density protein-95 (PSD-95) and synaptophysin (SYP) expression in the cortex and HIP of AD mice (Yao et al. 2017). In another study, Wang et al. (Zhang et al. 2016) revealed that treatment of AD rats with NMN increased LTP, restored the level of ATP, and decreased ROS and superoxide levels in the mitochondria (Fig. 3).

Schematic diagram showing the molecular mechanisms involved in the pathogenesis of Alzheimer’s disease (AD) and the role played by NAD+ as a neuroprotective regimen. Amyloid-beta (Aβ) triggers a cascade of events including mitochondrial dysfunction, oxidative stress, p-tau, and activation of microglia and JNK. Oxidative stress disrupts neuronal mitochondrial function, increases p-tau and Aβ production, apoptosis, and neuroinflammation. Hyperphosphorylated tau protein is the main constituent of the NFTs. These events lead to neural dysfunction, followed by neural death, and finally, memory impairment. NAD+ exerts neuroprotective effects through the modulation of multiple points in the complex AD cascade. NAD+: nicotinamide adenine dinucleotide, NFTs: neurofibrillary tangles
Discussion
This systematic review supports the efficacy of NAD+ precursors for improving both behavioral and neuroprotective outcomes in animal models of AD. NAD+ is an energy substrate and cofactor for several enzymes, including sirtuins, ADP-ribosyl cyclase (CD38), PARP-1, and NAD+-dependent dehydrogenases (Davila et al. 2018; Ying 2008). NAM, NAD+, and nicotinic acid are incorporated into cells using a transporter. NMN is converted to NR by CD73 5′-ectonucleotidase in the extracellular space. Cell entrance of NR can be performed through nucleoside transporters (Mericskay 2016). Recently, a new NMN-specific transporter, i.e. Slc12a8, was identified in mammals (Grozio et al. 2019).
Because intracellular NAD+ is an essential mediator of cell survival, (Hosseini et al. 2019), its restoration can play an important role in reducing degeneration. A growing body of evidence has shown that NAD+ plays an important role in brain metabolism, and has a crucial influence on neurotransmission, synaptic plasticity, learning, and memory (Wang et al. 2015). As demonstrated by the present review, treatment with NAD+ precursors restored NAD+ levels in the brain of rodent models of AD (Gong et al. 2013; Hou et al. 2018; Liu et al. 2013; Zhang et al. 2016). Also, the administration of precursors increased rodents’ spatial memory (Green et al. 2008; Hou et al. 2018; Liu et al. 2013; Vakilinezhad et al. 2018; Yao et al. 2017; Zhang et al. 2016), contextual learning, and freezing behavior (Hou et al. 2018; Xie et al. 2019). These treatments were also shown to alleviate AD-related dementia by inhibiting Aβ production and reducing tau phosphorylation (Gong et al. 2013; Liu et al. 2013; Xie et al. 2019).
As mentioned above, neuroinflammation is known as a major contributor to AD pathogenesis (Kim et al. 2004; Waetzig et al. 2005). Aβ fragments are able to induce activation of JNK, leading to phosphorylation of both tau and APP (Minogue et al. 2003; Vogel et al. 2009). Clinical studies have reported increased expression of phosphorylated JNK in the brains of AD patients (Killick et al. 2014; Zhu et al. 2001). It has been demonstrated that JNK promotes neuroinflammatory processes in AD by activating microglial cells (Mehan et al. 2011). The activated form of JNK is involved in beta‐site APP‐cleaving enzyme (BACE1) expression (Guglielmotto et al. 2011; Rahman et al. 2012), which plays a role in the generation of Aβ. Also, the abnormal activation of microglia leads to the generation of proinflammatory mediators, neuronal apoptosis, inhibition of neurogenesis, and decreased hippocampal volume, all of which are associated with AD (Wang and Colonna 2019). A recent study showed that the downregulation of JNK3 genetically led to a decrease in Aβ levels and ameliorated cognitive impairment (Yoon et al. 2012). In addition, the hyperactivation of PARP-1, a nuclear enzyme, and the accumulation of poly (ADP-ribose) (PAR) have been proven to exist in the brains of AD patients (Martire et al. 2015). The pathological activation of PARP-1 promotes cell death via mechanisms involving NAD+ depletion. Therefore, PARP-1 knockdown or its pharmacological inhibition can notably increase cell survival partially by NAD+ modulation (Mandir et al. 2000). Collectively, the results of the included studies show that NAD+ mediators can prevent Aβ-induced neuroinflammation by inhibiting JNK, PARP-1, and glial cell activation (Hou et al. 2018; Rahman et al. 2012; Turunc Bayrakdar et al. 2014; Yao et al. 2017), suggesting anti-inflammatory effects of NAD+. PSD-95 and SYP proteins play critical roles in synapse maturation and synaptic plasticity (Popugaeva et al. 2017). JNK activation triggers Aβ-induced synaptic loss (Costello and Herron 2004; Sclip et al. 2014); in contrast, administration of precursors such as NMN elevates the expression of synaptic proteins via inhibition of JNK (Yao et al. 2017).
Increasing evidence suggests that oxidative stress triggers the generation of Aβ, p-tau, and mitochondrial dysfunction. Ultimately, the pathological cascade of events results in neuronal damage and memory impairment (Cassidy et al. 2020). Alterations in membrane phospholipid composition increase the levels of MDA, which is a known lipid peroxidation marker. This has been frequently reported in the brains of experimental AD models and cerebrospinal fluid of AD patients (Sultana and Butterfield 2010; Sultana et al. 2006). The administration of NAD+ has been shown to decrease cell death induced by oxidative stress (Alano et al. 2004). Also, activation of NAD+ has been associated with a reduction in amyloid toxicity in the brains of the AD animal models (Kim et al. 2007). Furthermore, studies on AD animals have reported a reduction in ROS levels following NA or NMN treatment (Turunc Bayrakdar et al. 2014; Zhang et al. 2016). In the study by Turunc Bayrakdar et al., NA increased antioxidant enzyme activity and decreased MDA in the prefrontal cortex and HIP (Turunc Bayrakdar et al. 2014).
Aβ oligomers disrupt the brain bioenergetics system via interference with mitochondrial function (Onyango et al. 2016). Mitochondrial dynamics (fission and fusion) are essential for the maintenance of mitochondrial integrity, energy metabolism, inhibition of ROS production, and cellular apoptosis (Wang et al. 2017). Mitochondrial fission disassociates functional forms from damaged mitochondria via mitophagy (Wang et al. 2009). In AD, Aβ overproduction has been linked to extreme mitochondrial fragmentation (fission), which leads to mitochondrial dysfunction as evidenced by the loss of ATP synthesis and increased ROS that eventually results in cell death (Flannery and Trushina 2019; Zhu et al. 2013). Long and colleagues showed that mitochondrial dynamics were impaired in neurons affected by AD, while NMN therapy improved the balance of fission and fusion in the affected neurons (Long et al. 2015). Several studies demonstrated that PGC-1a, the primary regulator of energy metabolism, facilitates the degradation of the BACE1 protein (Gong et al. 2010; Kwak et al. 2011). It was also shown that NR treatment upregulates the expression of PGC-1a and promotes BACE1 degradation in the AD brain (Gong et al. 2013). The evidence further suggests that the silencing of PGC-1a can eradicate the effects of NR on BACE1 degradation (Gong et al. 2013). CREB and SIRT1 exhibit regulatory effects on the genes involved in neuronal survival and function in an Aβ-induced oxidative stress condition (Pugazhenthi et al. 2011; Su et al. 2014). Treatment with NAM also increases CREB and SIRT1 expression and activates Akt kinase, which is another possible mechanism for its pro-cognitive effects (Liu et al. 2013).
Conclusion
The data from the included studies showed that NAD+ precursors appeared to be an effective anti-AD strategy for preventing neuropathological and behavioral symptoms induced by AD in preclinical trials. Such favorable effects are possibly modulated by reducing central Aβ and tau levels and improving brain bioenergetics, inflammation, and oxidative stress regulation.
References
Alano CC, Ying W, Swanson RA (2004) Poly (ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem 279:18895-18902
Bonda DJ, Wang X, Perry G, Smith MA, Zhu X (2010) Mitochondrial dynamics in Alzheimer’s disease Drugs & aging 27:181–192
Bostancıklıoğlu M (2019) An update on the interactions between Alzheimer's disease, autophagy and inflammation. Gene 705:157-166
Butterfield DA, Halliwell B (2019) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20:148-160
Cassidy L, Fernandez F, Johnson JB, Naiker M, Owoola AG, Broszczak DA (2020) Oxidative stress in alzheimer’s disease: A review on emergent natural polyphenolic therapeutics. Complement Ther Med 49:102294
Chen X-Q, Mobley WC (2019) Exploring the pathogenesis of Alzheimer disease in basal forebrain cholinergic neurons: Converging insights from alternative hypotheses. Front Neurosci 13:446
Costello DA, Herron CE (2004) The role of c-Jun N-terminal kinase in the Aβ-mediated impairment of LTP and regulation of synaptic transmission in the hippocampus. Neuropharmacology 46:655–662
Davila A et al (2018) Nicotinamide adenine dinucleotide is transported into mammalian mitochondria Elife 7:e33246
Dong M et al (2019) Alzheimer's Disease (AD) Detect & Prevent-presymptomatic AD detection and prevention
Fang EF et al (2014) Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction. Cell 157:882-896
Flannery PJ, Trushina E (2019) Mitochondrial dynamics and transport in Alzheimer’s disease. Mol Cell Neurosci 98:109–120
Gong B, Chen F, Pan Y, Arrieta‐Cruz I, Yoshida Y, Haroutunian V, Pasinetti GM (2010) SCFFbx2‐E3‐ligase‐mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell 9:1018-1031
Gong B et al (2013) Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer's mouse models. Neurobiol Aging 34:1581-1588
Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, LaFerla FM (2008) Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci 28:11500-11510
Grozio A et al (2019) Slc12a8 is a nicotinamide mononucleotide transporter. Nat Metab 1:47-57
Guglielmotto M, Monteleone D, Giliberto L, Fornaro M, Borghi R, Tamagno E, Tabaton M (2011) Amyloid-β 42 activates the expression of BACE1 through the JNK pathway. J Alzheimers Dis 27:871-883
Hosseini L, Farokhi-Sisakht F, Badalzadeh R, Khabbaz A, Mahmoudi J, Sadigh-Eteghad S (2019) Nicotinamide mononucleotide and melatonin alleviate aging-induced cognitive impairment via modulation of mitochondrial function and apoptosis in the prefrontal cortex and hippocampus. Neuroscience 423:29–37
Hou Y et al (2018) NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci 115:E1876-E1885
Hu JP et al (2011) Valproate reduces tau phosphorylation via cyclin-dependent kinase 5 and glycogen synthase kinase 3 signaling pathways. Brain Res Bull 85:194-200
Kandimalla R, Manczak M, Yin X, Wang R, Reddy PH (2018) Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Human Mol Gen 1;27(1):30-40
Kaur H, Patro I, Tikoo K, Sandhir R (2015) Curcumin attenuates inflammatory response and cognitive deficits in experimental model of chronic epilepsy. Neurochem Int 89:40–50
Killick R et al (2014) Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt–PCP–JNK pathway. Mol Psychiatry 19:88-98
Kim D et al (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J 26:3169-3179
Kim EJ, Yang SJ (2017) Nicotinamide reduces amyloid precursor protein and presenilin 1 in brain tissues of amyloid beta-tail vein injected mice. Clin Nutr Res 6:130-135
Kim SH, Smith CJ, Van Eldik LJ (2004) Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1β production. Neurobiol Aging 25:431-439
Kwak YD, Wang R, Li JJ, Zhang YW, Xu H, Liao FF (2011) Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener 6:17
Lesné S et al (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440:352–357
Li N, Liu Y, Li W, Zhou L, Li Q, Wang X, He P (2016) A UPLC/MS-based metabolomics investigation of the protective effect of ginsenosides Rg1 and Rg2 in mice with Alzheimer's disease. J Ginseng Res 40:9-17
Liu D et al (2013) Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging 34:1564–1580
Liu D, Pitta M, Mattson MP (2008) Preventing NAD+ depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann N Y Acad Sci 1147:275
Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS, Schuh RA (2015) Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol 15:19
Luongo TS et al (2020) SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature 588:174-179
Manczak M, Kandimalla R, Yin X, Reddy PH (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet 27:1332-1342
Mandir AS et al (2000) NMDA but not non-NMDA excitotoxicity is mediated by Poly (ADP-ribose) polymerase. J Neurosci 20:8005-8011
Martire S, Mosca L, d’Erme M (2015) PARP-1 involvement in neurodegeneration: a focus on Alzheimer’s and Parkinson’s diseases. Mech Ageing Dev 146:53-64
Mehan S, Meena H, Sharma D, Sankhla R (2011) JNK: a stress-activated protein kinase therapeutic strategies and involvement in Alzheimer’s and various neurodegenerative abnormalities. J Mol Neurosci 43:376-390
Mericskay M (2016) Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch Cardiovasc Dis 109:207–215
Minogue AM, Schmid AW, Fogarty MP, Moore AC, Campbell VA, Herron CE, Lynch MA (2003) Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-β on long term potentiation and cell death in hippocampus a role for Interleuken-1β? J Biol Chem 278:27971–27980
Onyango IG, Dennis J, Khan SM (2016) Mitochondrial dysfunction in Alzheimer’s disease and the rationale for bioenergetics based therapies. Aging Dis 7:201
Popugaeva E, Pchitskaya E, Bezprozvanny I (2017) Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease–A therapeutic opportunity? Biochem Biophys Res Commun 483:998–1004
Pugazhenthi S, Wang M, Pham S, Sze C-I, Eckman CB (2011) Downregulation of CREB expression in Alzheimer’s brain and in Aβ-treated rat hippocampal neurons Molecular neurodegeneration 6:60
Rahman M, Zhang Z, Mody AA, Su D-M, Das HK (2012) Intraperitoneal injection of JNK-specific inhibitor SP600125 inhibits the expression of presenilin-1 and Notch signaling in mouse brain without induction of apoptosis. Brain Res 1448:117-128
Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J (2015) Amyloid-beta: a crucial factor in Alzheimer's disease. Med Princ Pract 24:1-10
Sclip A et al (2014) c-Jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death Dis 5:e1019-e1019
Su S et al (2014) Sesamin ameliorates doxorubicin-induced cardiotoxicity: involvement of Sirt1 and Mn-SOD pathway. Toxicol Lett 224:257-263
Sultana R, Butterfield DA (2010) Role of oxidative stress in the progression of Alzheimer’s disease. J Alzheimers Dis 19:341–353
Sultana R, Perluigi M, Butterfield DA (2006) Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal 8:2021–2037
Ton AMM et al (2020) Oxidative stress and dementia in alzheimer’s patients: Effects of synbiotic supplementation. Oxidative Med Cell Longev 2020
Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A (2014) Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Aβ (1–42)-induced rat model of Alzheimer's disease. Free Radic Res 48:146-158
Vakilinezhad MA, Amini A, Javar HA, Zarandi BFBaB, Montaseri H, Dinarvand R (2018) Nicotinamide loaded functionalized solid lipid nanoparticles improves cognition in Alzheimer’s disease animal model by reducing Tau hyperphosphorylation DARU. J Pharm Sci 26:165–177
Vogel J, Anand V, Ludwig B, Nawoschik S, Dunlop J, Braithwaite S (2009) The JNK pathway amplifies and drives subcellular changes in tau phosphorylation. Neuropharmacology 57:539–550
Waetzig V et al (2005) c‐Jun N‐terminal kinases (JNKs) mediate pro‐inflammatory actions of microglia. Glia 50:235-246
Wang S, Colonna M (2019) Microglia in Alzheimer’s disease: a target for immunotherapy. J Leukoc Biol 106:219–227
Wang W et al (2017) Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum Mol Genet 26:4118–4131
Wang X, Hu X, Yang Y, Takata T, Sakurai T (2015) Systemic pyruvate administration markedly reduces neuronal death and cognitive impairment in a rat model of Alzheimer’s disease. Exp Neurol 271:145–154
Wang X, Hu X, Yang Y, Takata T, Sakurai T (2016) Nicotinamide mononucleotide protects against β-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res 15;1643:1-9
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090–9103
Wei CC et al (2017) NAD replenishment with nicotinamide mononucleotide protects blood–brain barrier integrity and attenuates delayed tissue plasminogen activator‐induced haemorrhagic transformation after cerebral ischaemia. Br J Pharmacol 174:3823-3836
Wilkinson BL, Landreth GE (2006) The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J Neuroinflammation 3:30
Xie C, Soeda Y, Shinzaki Y, In Y, Tomoo K, Ihara Y, Miyasaka T (2015) Identification of key amino acids responsible for the distinct aggregation properties of microtubule‐associated protein 2 and tau. J Neurochem 135:19-26
Xie X, Gao Y, Zeng M, Wang Y, Wei T-F, Lu Y-B, Zhang W-P (2019) Nicotinamide ribose ameliorates cognitive impairment of aged and Alzheimer’s disease model mice. Metab Brain Dis 34:353–366
Yao Z, Yang W, Gao Z, Jia P (2017) Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci Lett 647:133–140
Ying W (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10:179-206
Yoon SO et al (2012) JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron 75:824-837
Zhang TT, Li W, Meng G, Wang P, Liao W (2016) Strategies for transporting nanoparticles across the blood–brain barrier. Biomater Sci 4:219-229
Zhu X, Perry G, Smith MA, Wang X (2013) Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis 33:S253–S262
Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA (2001) Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem 76:435–441
Author information
Authors and Affiliations
Contributions
LH, JM, and SS-E designed the project. FP and HS-P performed the literature review and extracted data. LH and S S-E wrote the manuscript. S S-E supervised the work. JM and S S-E edited the final version.
Corresponding author
Ethics declarations
Consent for Publication
The authors give the publisher permission to publish the work.
Competing Interests
The authors declare they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hosseini, L., Mahmoudi, J., Pashazadeh, F. et al. Protective Effects of Nicotinamide Adenine Dinucleotide and Related Precursors in Alzheimer’s Disease: A Systematic Review of Preclinical Studies. J Mol Neurosci 71, 1425–1435 (2021). https://doi.org/10.1007/s12031-021-01842-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-021-01842-6