
Abstract
Metachromatic leukodystrophy (MLD) is a neurodegenerative disorder characterized by progressive demyelination due to deficiency of the enzyme arylsulfatase A (ARSA) in leukocytes, and consequently leads to impaired degradation and accumulation of cerebroside-3-sulfate (sulfatide). This study aimed to sequence the ARSA gene in a total of 43 patients with metachromatic leukodystrophy descendant from 40 Egyptian families. In addition, four carrier parents from two families with children who had died from MLD came to the clinic for genetic analysis. Prenatal diagnosis was performed for four families with molecularly diagnosed MLD sibs. Different mutations were characterized in our cohort, including missense, nonsense, splice, and deletion. Overall, 21 different mutations in the ARSA gene were detected, with 12 novel mutations, i.e. p.Arg60Pro, p.Tyr65*, p.Val112Asp, p.Arg116*, p.Gly124Asp, p.Pro193Ser, p.Gln238*, p.Gln456*, p.Thr276Lys, and p.Gly311Arg, in addition to two new acceptor splice-site mutations 685-1G > A and c.954_956 delCTT. The amniotic fluid samples revealed two carrier fetuses with heterozygous monoallelic mutations, and two affected fetuses had the homozygous biallelic mutations. In conclusion, the current study sheds light on the underlying ARSA gene defect, with an expansion of the mutation spectrum. To our knowledge, this is the first molecular study of MLD among the Egyptian population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metachromatic leukodystrophy (MLD) (MIM #250100) is a severe neurodegenerative disorder inherited as an autosomal recessive trait, with estimated worldwide prevalence of 1.45 per 100,000 births (Giugliani 2012). MLD is caused by deficient activity of the enzyme arylsulfatase A (ARSA) and leads to sulfatide storage within the lysosomes (Cesani et al., 2016). ARSA catalyzes the initial step of the metabolic pathway, sphingolipid 3′-O-sulfogalactosylceramide, known as sulfatide, which accumulates in myelin-producing cells and causes progressive demyelination and neurodegeneration (Ozkan and Ozkara 2016, Dehghan Manshadi et al. 2017 and Issa et al. 2018).
Patients with MLD present with progressive physical and mental deterioration, clumsiness, frequent falls, seizures, and hypotonia. MLD patients are classified into three types, i.e. late infantile, juvenile, and adult forms, based on the age at onset (Biffi et al. 2008). The most severe type is the late infantile onset, which is associated with poor prognosis (death typically occurs within 5–6 years) and manifests as regression of motor skills, gait abnormalities, seizures, ataxia, hypotonia, extensor planters, and optic atrophy (Lynch et al. 2019). The juvenile form manifests between 4 and 15 years of age. It is further subdivided into early juvenile and late juvenile subtypes, depending on whether the onset is before or after the age of 6 years, while the adult form manifests after the age of 15 (Kehrer et al. 2014). Magnetic resonance imaging (MRI) of the brain and biochemical assay of ARSA enzymatic activity in leukocytes are used to confirm the diagnosis (Luzi et al. 2013). The first responsible gene defect is ARSA (MIM #607574; GenBank accession number, NG_009260), which includes eight exons and maps to chromosome 22q13.33, covering 3.2 kb of genomic DNA (Dehghan Manshadi et al. 2017). The gene is translated into 509 amino acid precursors (GenBank accession number NP_000478). Numerous different mutations have been identified in the ARSA gene in the Human Gene Mutation Database, including deletions, splice-site mutations, and mostly missense mutations (Lugowska et al. 2005). The second causative gene defect in MLD patients is the SapB protein derived from the 524 amino acid precursor proteins, encoded by the prosaposin gene (PSAP; MIM #176801). The PSAP gene is located on chromosome 10q22.1and consists of 15 exons (Cesani et al. 2016).
The ARSA pseudo-deficiency (PD) allele also has common polymorphisms that result in lower than average ARSA activity (Gieselmann et al. 1989). The ARSA-PD allele is characterized by two in-cis A > G transitions. The first c.1049A > G transition alters one of the N-glycosylation positions at 350 (p.N350S), resulting in partial mistargeting (approx. 55%) of the enzyme (Harvey et al. 1998). The second *96A > G transition occurs in the first functional polyadenylation signal, leading to a severe deficiency of the major 2.1 kb ARSA-mRNA species (Gieselmann et al. 1989), with a consequent reduction in the amount of ARSA protein produced. However, the presence of the modifier PD allele, in either the homozygous or compound heterozygous state, with the disease allele is thought to lower the enzyme activity and possibly increase disease severity (Cesani et al. 2016).
In this study, we investigated 43 patients descendant from 40 families of Egyptian origin by mutation analysis of the complete coding sequences of the ARSA gene. To our knowledge, this is the first molecular study of MLD among Egyptian patients.
Patients and Methods
The study included 43 Egyptian MLD patients. Their mean age at presentation was 2.8 years (range 1–7 years). Four parents with a history of deceased MLD children based only on enzyme activity came in for genetic counseling and carrier detection. Additionally, prenatal diagnosis was performed for four mothers with affected patients. The diagnosis was based on clinical features, brain MRI, and ARSA enzyme deficiency, followed by molecular testing. All samples were collected after obtaining the guardians’ informed consent using a form approved by the ethical committee of the National Research Centre, Egypt.
ARSA Enzymatic Assay
ARSA activity was estimated as μmol/gpt/h protein in leukocytes, using p-nitrocatechol sulfate (Wenger et al. 1991).
Molecular Analysis
Genomic DNA was extracted from leukocytes of patients and healthy control subjects, using the standard method, and all eight ARSA gene exons and exon–intron boundaries were amplified with primer pairs generated using Primer3 (https://frodo.wi.mit.edu/). Primer sequences and conditions for amplification for each fragment are available upon request. In addition, DNA was extracted from the amniotic fluid (AF) samples of four pregnant mothers with MLD sibs using the QIAamp DNA Blood Mini Kit (QIAGEN Inc., Valencia, CA, USA), according to the manufacturer’s protocol. The PCR products were purified using the ExoSAP PCR Cleanup Kit (Fermentas, Germany), sequenced in both directions using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA), and analyzed on the ABI Prism 3500 Genetic Analyzer (Applied Biosystems). Sequencing results were then analyzed on the NCBI website (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and compared with the cDNA sequence of the ARSA gene (NM_000487.5). The novel gene variations were confirmed after ruling out polymorphism by the analysis of the exome sequencing data of 50 Egyptian healthy individuals.
In Silico Sequence Variant Validation
To predict the putative effect of the novel missense variants, we used different prediction algorithms including SIFT [Sorting Intolerant From Tolerant] (Kumar et al. 2009), PolyPhen-2 [Polymorphism Phenotyping v2] (Adzhubei et al. 2010) (https://genetics.bwh.harvard.edu/pph2), MutationTaster (https://www.mutationtaster.org/), and I-Mutant2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html). In addition, the dbSNP (https://www.ncbi.nlm.nih.gov/snp/), 1000 Genomes (https://browser.1000genomes.org/Homosapiens/Info/Index), Exome Aggregation Consortium (ExAC) (https://gnomad.broadinstitute.org/gene/ENSG00000100299?dataset=gnomad_r2_1), and gnomAD databases were accessed for confirmation of the novel and previously reported variations.
Results
This study included 43 patients from 40 Egyptian families; 27 were female and 16 were male (1.7:1). Thirty-two patients (76.2%) were descendants of consanguineous marriages. Thirty-three patients (76.7%) were classified as late infantile type, and ten (23.3%) were of the juvenile type. All patients presented with regression of motor and mental development. Nine patients (20.9%) showed minor dysmorphic features in the form of depressed nasal bridge, upwards slanting of the eyelids, nystagmus, and low-set ears. Hypertonia was present in 38 patients (90.5%), strabismus in four patients (9.3%), gait disturbance in 10 (23.8%), ataxia in 2 (4.7%), spastic paraplegia in 2 (4.7%), EEG abnormality in 23 (54.8%), microcephaly in 10 (23.3%), cortical atrophy in 7 (16.3%), and white matter demyelination in all patients (Figs. 1 and 2). The biochemical analysis of the arylsulfatase A enzyme activity in leukocytes (expressed as μmol/gpt/h) was deficient in all 43 patients and ranged between 0 and 5 μmol/gpt/h.

Brain MRI. T2-weighted MRI of the brain in MLD patient (15) typically reveals white matter hyperintensities (arrow)

Brain MRI. T2-weighted MRI of the brain in MLD patient 31 revealed white matter hyperintensities (arrow)
Molecular Results
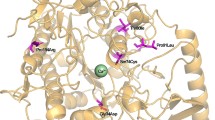
Sequencing of all coding regions of the ARSA gene for the 43 patients and their available parents revealed a total of 21 causative mutations with 12 novel variants (Table 1). The majority of the ARSA-MLD allele types varied between single base substitution, nonsense mutation, small deletion, and splice-site mutation. The six new missense variants were NM_000487.5 p.Arg60Pro, p.Val112Asp, p.Gly124Asp, p.Pro193Ser, p.Thr276Lys, and p.Gly311Arg, four new stop codon mutations (p.Try65*, Arg116*, p.Gln238*, p.Gln456*), one new acceptor-splice acceptor site mutation 685-1 G > A that would be expected to disrupt splicing, and one in-frame deletion c.954_956delCTT. Most of the new characterized missense mutations showed a change in the characteristic chemical nature of the resulting amino acid, including a change from hydrophilic polar to hydrophobic non-polar amino acid of (Arg60Pro), from hydrophobic non-polar to hydrophilic polar of each p.Val112Asp, p.Gly124Asp, p.Pro193Ser, and p.Gly311Arg, and from polar uncharged to polar positive p.Thr276Lys (Figs. 3 and 4). Interestingly, most of the characterized mutations were found in the homozygous state (36/43, 83.7%), with the exception of seven (7/43, 16.3%) patients who had a compound heterozygous causative variant (Table 1). In addition, four parents with children who had died from MLD and were requesting genetic counseling, were found to be carriers (Table 2). Prenatal diagnosis was also performed in four families and revealed that two affected fetuses had the homozygous biallelic mutation and two were carrier fetuses with heterozygous monoallelic mutations (Table 3). All novel mutations were predicted for the functional impact of an amino acid based on the alignment of highly similar orthologous and/or paralogous protein sequences and predicted to be “disease-causing” by in silico analysis (Table 4). Also, the novel potential mutations were defined by exclusion from the Human Gene Mutation Database (https://www.hgmd.cf.ac.uk) and the previously reported mutations on PubMed (https://www.ncbi.nlm.nih.gov/PubMed/).

The distribution of the alleles is reported in relation to the ARSA gene. The ARSA gene maps to chromosome 22q13 covering 3.2 kb of genomic DNA. The map of the ARSA gene depicts the positions of eight exons (blue boxes numbered 1–8) and between numbered introns (pale blue lines). All exonic variants are given above the gene schema, all intronic below. The red arrows point to the novel mutations detected in this study, and the black arrows point to the reported known variants

Portion of the sequencing electrophoregrams displaying the ARSA gene variants identified in our patients. The arrow indicates the site of the variant



Discussion
Metachromatic leukodystrophy is a lysosomal storage disease, with more than 264 mutations reported in different populations (https://www.LOVD.nl/ARSA). The diagnosis of MLD depends on the presence of the clinical phenotypic characteristics, the deficiency of the ARSA activity in leukocytes, and the molecular testing of the ARSA gene (Liaw et al. 2015). In the current study, we report on the clinical, biochemical, and molecular findings in 43 patients descendant from 40 Egyptian families.
The late infantile variant accounted for 76.7% of the studied group, while 23.3% were of the juvenile type. The late infantile subtype is the most common and presents with severe clinical features, including gait disturbances, frequent falls, and regression of motor and mental functions (Cesani et al. 2016; Wang et al. 2019). Five patients showed mild dysmorphic features in the form of depressed nasal bridge, upward slanting of eyelids, nystagmus, and low-set ears. Nerve conduction studies showed delayed motor and/or sensory conduction velocity, consistent with demyelinating peripheral neuropathy, in all the investigated patients. Cameron et al. (2004) emphasized that slowing of the nerve conduction velocity is the hallmark of MLD.
In this study, the biochemical analysis of the arylsulfatase A enzyme activity in leukocytes was deficient compared to controls. This reduction occurs in certain conditions including classical MLD, the presence of PD allele, compound heterozygosity for the pathogenic variant, and PD alleles of the ARSA gene without white matter disease (Wang et al. 2016). The presence of the PD alleles with other pathogenic variants in the ARSA gene in the same patient leads to greater deficiency of enzyme activity and possibly increased disease severity (Regis et al. 2004). Based on MRI, all patients showed white matter demyelination, and only four had cortical atrophy consistent with previous reports (Grossi et al. 2008 and Wang et al. 2019).
The sequencing of MLD patients in the present study identified 12 novel causative variants in the ARSA gene. MLD is known as a heterogeneous disease, and ARSA gene variants are not distributed homogeneously, but tend to cluster around exons 2 and 4. It is worth mentioning that p.Asn284Ser was encountered in eight patients belonging to eight unrelated families, showing the highest density of all identified mutations (8/43, 18.6%), which may be evidence of a founder effect in MLD patients with a common ancestor in an Egyptian population. This mutation, previously reported in an Italian patient, prevents the correct positioning of the sulfate group of the substrate and consequently its hydrolysis, leading to low residual ARSA activity compared to controls (Qu et al. 1999).
Similarly, the novel c.712C > T mutation detected in six patients (6/43, 13.95%) suggests that it might be a founder mutation of the ARSA gene in Egyptian families with MLD, and results in severe reduction in enzyme activity due to premature termination at codon Gln238*. In addition, the p.Trp195Cys mutation found in four patients (4/43, 9.3%) was previously reported in a MLD patient from Iran, with 2.8% reduced enzyme activity (Dehghan Manshadi et al. 2017). The new p.Thr276Lys mutation encountered in four patients has not been reported in the current public databases dbSNP (https://www.ncbi.nlm.nih.gov/SNP/) and gnomAD (https://gnomad.broadinstitute.org/gene/ENSG00000100299?dataset = gnomad_r2_1), nor has it been described in the 1000 Genomes Project data sets (https://www.1000genomes.org), but it was reported in the same codon Thr276, with the substitution of lysine to methionine, in six patients of Lebanese descent (Harvey et al. 1993). The infrequently encountered mutations among our study group such as c.459 + 1G > A, p.Pro426Leu, and p.Iso179Ser were found to be common in the European population, and p.Gly99Asp, p.Gly245Arg, and p.Thr409Ile were found in Japanese populations (Lugowska et al. 2005 and Zhi-Hong et al. 2016). This may be due to differences in ethnic origin.
Most of the characterized mutations were present in a homozygous pattern, consistent with the pattern of autosomal recessive inheritance of MLD and the high consanguineous rate among the Egyptian population (Regis et al. 2004). Interestingly, the six unreported missense variants p.Arg60Pro, p.Val112Asp, p.Gly124Asp, p.Thr276Lys, p.Pro193Ser, p.Arg498His, and p.Gly311Arg (Table 1) were suggested to have a pathological nature on protein feature. In silico tools revealed that it might involve an alteration of metal binding. This was supported by the involvement of conserved residues in the ARSA protein and their absence in more than 100 chromosomes of the Egyptian population, confirming the absence of any spurious changes. The enzyme levels of arylsulfatase in these patients were severely affected compared to controls, suggesting that the resulting mutant protein primarily affects its folding and decreases its stability, which is concordant with previous reports (Dorboz et al. 2009; Cesani et al. 2016). On the other hand, the characterized missense variants p.Pro150Leu, p.Trp195Cys, p.Thr276Met, p.Asp283Tyr, p.Asn284Ser, p.Gly295Ser, and p.Trp320Cys in our patients were previously detected in different ethnic groups (Harvey et al. 1993; Tsuda and Hasegawa 1996; Halsall et al. 1999; Qu et al. 1999; Berná et al. 2004; Grossi et al. 2008; Önder et al. 2009; Dehghan Manshadi et al. 2017). Assessment of the phenotypic presentation of this group was marked by variable severity and varying age at onset, with the possible presence of a genotype–phenotype correlation, as a result of enzyme deficiency and altered protein production.
Some of the identified mutations, such as p.Pro150, p.Gly295, p.Asp283, and p.Asn284, exhibited their effect on ARSA activity either through their default binding interactions with other neighboring proteins, or by functionally disrupting the protein folding. They were also found to have an effect on the correction of the folding in the ER or instability of the lysosomes and consequently their hydrolysis. In addition, their presence in cis with the pseudo-allele would ultimately inactivate the enzyme (Halsall et al. 1999; Kay et al. 2000; Berná et al. 2004; Grossi et al. 2008). Furthermore, the unreported nonsense mutations p.Gln238* found in patients 21–26, p.Gln456* in patient 43 (Table 1), and p.Arg116* in two carrier parents screened for carrier detection (Table 2) produce a premature termination codon that could lead to either nonsense-mediated mRNA decay (NMD) or a truncating protein escaping NMD, with dominant negative activity. In the latter process, a more severe phenotype is produced as a result of the dominant negative effects of the translated faulty protein (Inoue et al., 2004). Further functional studies are needed to determine whether the detected nonsense mutations may cause NMD.
This study also reported a new splice variant, c.685-1G > A, in patient 18, in addition to the reported donor splice c.465 + 1G > A in patients 10 and 11, which are the most frequent mutations detected in Caucasians and Italian patients as described by Biffi et al. (2008). The effect of these two mutations that occurred in splice-site consensus sequences is mis-splicing that does not alter the reading frame but would disrupt the amino acid sequence via either the loss or addition of amino acid residues in c.685-1G > A and c.465 + 1G > A, respectively. Both of these alterations in the primary structure of the protein would likely interfere with folding and lead to protein instability, and mRNA functional analysis is needed to confirm their effect.
Furthermore, our study detected a new in-frame deletion 3bpc.954_956delCTT located in exon 5 in one patient (patient 40), in addition to the previously reported in-frame deletion c.583delT found in patients 16 and 17. This new in-frame deletion was found to be deleterious, with a score of −9.89, using PROVEAN [Protein Variation Effect Analyzer] pathogenicity prediction software (cutoff value of −2.5). Our cohort suggests a genotype/phenotype correlation, as patients with either deletions or nonsense mutations were associated with severe deficiency of ARSA activity and presented with severe clinical manifestations and extremely rapid disease progression. These findings are concordant with previous reports (Biffi et al. 2008; Hettiarachchi and Dissanayake 2020).
In this study, three families underwent prenatal testing of the targeted causative variants and were given genetic counseling. The obvious variability in the clinical phenotype and the correlation with the characterized genotype for predicting the patient’s prognosis represents a challenge, especially in those who harbor heteroallelic mutations, one identified allele and one new allele, as in patients 9, 10, 18, 19, and 27. These results suggest that Egyptian MLD patients have a different distribution of ARSA mutations than those found in Caucasian or other Arab populations (Dorboz et al. 2009; Luzi et al. 2013).
Although there is no curative treatment currently available, hematopoietic stem cell transplantation has slowed disease progression in some patients. Future novel therapies, such as enzyme replacement and gene editing, are promising (Böhringer et al. 2017).
Conclusion
Our findings expanded the spectrum of ARSA mutations with 12 novel variants, discovered in Egyptian families affected by MLD. Furthermore, it provided carrier detection, prenatal diagnosis, and proper genetic counseling. To our knowledge, this is the first detailed clinical, biochemical, and molecular testing report of MLD patients in Egypt. The metachromatic leukodystrophy diagnosis should be considered in cases with consanguineous marriages, developmental regression, and slowing of nerve conduction velocity. A genotype/phenotype association was observed with regard to enzyme activity and underlying molecular pathology. Further investigations of the novel mutations through in vitro expression experiments are needed to reveal their impact on protein function.
Declarations
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Berná L, Gieselmann V, Poupetová H, Hrebícek M, Elleder M, Ledvinová J (2004) Novel mutations associated with metachromatic leukodystrophy: phenotype and expression studies in nine Czech and Slovak patients. Am J Med Genet A 129A(3):277–281
Biffi A, Lucchini G, Rovelli A, Sessa M (2008) Review Metachromatic leukodystrophy: an overview of current and prospective treatments. Bone Marrow Transplant 42(Suppl 2):S2-6
Böhringer J, Santer R, Schumacher N, Gieseke F, Cornils K, Pechan M, Kustermann-Kuhn B, Handgretinger R, Schöls L, Harzer K, Krägeloh -Mann I, Müller I, (2017) Enzymatic characterization of novel arylsulfatase A variants using human arylsulfatase A deficient immortalized mesenchymal stromal cells. Hum Mutat 38(11):1511–1520
Cameron CL, Kang PB, Burns TM, Darras BT, Jones HR Jr (2004) Multifocal slowing of nerve conduction in metachromatic leukodystrophy. Muscle Nerve 29(4):531–536
Cesani M, Lorioli L, Grossi S, Amico G, Fumagalli F, Spiga I, Filocamo M, Biffi A (2016) Mutation Update of ARSA and PSAP Genes Causing Metachromatic Leukodystrophy. Hum Mutat 37(1):16–27
Dehghan Manshadi M, Kamalidehghan B, Aryani O, Khalili E, Dadgar S, Tondar M, Ahmadipour F, Yong Meng G, Houshmand M (2017) Four novel ARSA gene mutations with pathogenic impacts on metachromatic leukodystrophy: a bioinformatics approach to predict pathogenic mutations. Ther Clin Risk Manag 13:725–731
Dorboz I, Eymard-Pierre E, Kefi R, Abdelhak S, Miladi N, Boespflug-Tanguy O (2009) Identification of a new Arylsulfatase A (ARSA) gene mutation in Tunisian patients with metachromatic leukodystrophy (MLD). J Neurol Sci 287(1–2):278–280
Gieselmann V, Polten A, Kreysing J, von Figura K, Arylsulfatase A, pseudodeficiency, (1989) loss of a polyadenylation signal and N-glycosylation site. Proc Natl Acad Sci USA 86(23):9436–9440
Giugliani R (2012) Newborn screening for lysosomal diseases: current status and potential interface with population medical genetics in Latin America. J Inherit Metab Dis 35:871–877
Grossi S, Regis S, Rosano C, Corsolini F, Uziel G, Sessa M, Di Rocco M, Parenti G, Deodato F, Leuzzi V, Biancheri R, Filocamo M (2008) Molecular analysis of ARSA and PSAP genes in twenty-one Italian patients with metachromatic leukodystrophy: identification and functional characterization of 11 novel ARSA alleles. Hum Mutat 29(11):E220–E230
Halsall DJ, Halligan EP, Elsey TS, Cox TM (1999) Metachromatic leukodystrophy: a newly identified mutation in arylsulphatase A, D281Y, found as a compound heterozygote with I179L in an adult onset case. Hum Mutat 14:447
Harvey JS, Nelson PV, Carey WF, Robertson EF, Morris CP (1993) An arylsulfatase A (ARSA) missense mutation (T274M) causing late-infantile metachromatic leukodystrophy. Hum Mutat 2(4):261–267
Harvey JS, Carey WF, Morris CP (1998) Importance of the glycosylation and polyadenylation variants in metachromatic leukodystrophy pseudodeficiencyphenotype. Hum Mol Genet 7(8):1215-9
Hettiarachchi D, Dissanayake VHW (2020) Three novel variants in the arylsulfatase A (ARSA) gene in patients with metachromatic leukodystrophy (MLD). BMC Res Notes 13(1):40
Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD, Mancias P, Butler IJ, Wilkinson MF, Wegner M, Lupski JR (2004) Molecularmechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet 36:361–369
Issa AB, Feki FK, Jdila MB, Khabou B, Rhouma BB, Ammar-Keskes L, Triki C, Fakhfakh F (2018) Clinical, Molecular, and Computational Analysis Showed a Novel Homozygous Mutation Among the Substrate-Binding Site of ARSA Protein in Consanguineous Family with Late-Infantile MLD. J Mol Neurosci 66(1):17–25
Kay BK, Williamson MP, Sudol M (2000) The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J 14:231–241
Kehrer C, Groeschel S, Kustermann-Kuhn B, Bürger F, Köhler W, Kohlschütter A, Bley A, Steinfeld R, Gieselmann V, Krägeloh-Mann I, Leukonet G (2014) Language and cognition in children with metachromatic leukodystrophy: onset and natural course in a nationwide cohort. Orphanet J Rare Dis 9:18
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding nonsynonymous variants on protein function using the SIFT algorithm. Nat Protoc 4(7):1073–1081
Liaw HR, Lee HF, Chi CS, Tsai CR (2015) Late infantile metachromatic leukodystrophy: Clinical manifestations of five Taiwanese patients and Genetic features in Asia. Orphanet J Rare Dis 10:144
Lugowska A, Amaral O, Berger J, Berna L, Bosshard NU, Chabas A et al (2005) Mutations c.459 + 1G N A and p. P426L in the ARSA gene: prevalence in metachromatic leukodystrophy patients from European countries. Mol Genet Metab 86:353–359
Luzi P, Rafi MA, Rao HZ, Wenger DA (2013) Sixteen novel mutations in the arylsulfatase A gene causing metachromatic leukodystrophy. Gene 530(2):323–328
Lynch S, Wade C, de Paiva ARB, John N, Kinsella JA, Merwick Á, Ahmed RM et al (2019) Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era. J Neurol Neurosurg Psychiatry 90:543–554
Önder E, Sinici I, Sönmez FM, Topçu M, Özkara HA (2009) Identification of two novel arylsulfatase A mutation with a polymorphism as a cause of metachromatic leukodystrophy. Neurol Res 31:60–66
Ozkan A1, Ozkara HA (2016) Metachromatic leukodystrophy: Biochemical characterization of two (p.307Glu→Lys, p.318Trp→Cys) arylsulfatase A mutations. Intractable Rare Dis Res 5(4):280–283
Qu Y, Shapira E, Desnick RJ (1999) Metachromatic leukodystrophy: subtype genotype/phenotype correlations and identification of novel missense mutations (P148L and P191T) causing the juvenile-onset disease. Mol Genet Metab 67(3):206–212
Regis S, Corsolini F, Ricci V, Di Duca M, Filocamo M (2004) An unusual arylsulfatase A pseudodeficiency allele carrying a splice site mutation in a metachromatic leukodystrophy patient. Eur J Hum Genet 12:150–154
Tsuda T, Hasegawa Y (1996) Eto Y (1996) Two novel mutations in a Japanese patient with the late-infantile form of metachromatic leukodystrophy. Brain Dev 18:400–403
Wang Y, Chen X, Liu C, Wu S, Xie Q, Hu Q, Chen S, Liu Y (2019) Metachromatic leukodystrophy: Characterization of two (p.Leu433Val, p.Gly449Arg) arylsulfatase A mutations. Exp Ther Med 18(3):1738–1744
Wang Z, Lin Y, Zheng D, Yan A, Tu X, Lin J, Lan F (2016) Whole-exome sequencing identifies compound heterozygous mutations in ARSA of twosiblings presented with atypical onset of metachromatic leukodystrophy from a Chinese pedigree. Clin Chim Acta 460:135-7
Wenger DA, Williams C (1991) Screening for lysosomal disorders. In: Hommes FA (ed) Techniques in Diagnostic Human Bio-chemical Genetics. Wiley-Liss, New York, pp 587–617
Zhi-Hong C, Yu-xiong G, Mu-qing Z, Yu-xin Z, Chun W, Lin-Gan W, Qiong-xiang Z (2016) A novel ARSA gene mutation c.302 delG in a Chinese patient with metachromatic leukodystrophy. Int J Clin Exp Pathol 9(2):1800–1804
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare no conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration. Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Amr, K., Fateen, E., Mansour, L. et al. Clinical, Biochemical, and Molecular Characterization of Metachromatic Leukodystrophy Among Egyptian Pediatric Patients: Expansion of the ARSA Mutational Spectrum. J Mol Neurosci 71, 1112–1130 (2021). https://doi.org/10.1007/s12031-020-01734-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-020-01734-1