
Abstract
Morphine is a potent agonist of μ-opioid receptor and is widely used to relieve severe pain, including cancer pain. Some chemokines, for example, CX3CL1 and CCL2, participate in the regulation of opioid santinociception. In our previous study, we found overexpression of chemokine CXCL10/CXCR3 in spinal cord participated in the development of cancer-induced bone pain, so we supposed that CXCL10 may have influence in morphine analgesia in cancer pain relief. In this study, we found that a single dose of morphine could transiently increase the expression of CXCL10 in spinal cord. Blocking the function of CXCL10 enhanced morphine antinociception in cancer-induced bone pain rats. However, overexpression of CXCL10 induced acute algesia and decreased the analgesic effect of morphine in normal mice. The algesic effect of CXCL10 was blocked by inhibition of CXCR3 and Gi protein. These results suggested that CXCL10 in spinal cord serves as a novel negative regulator of morphine analgesia and provided evidence that activation of CXCL10/CXCR3 in spinal cord may attenuate antinociceptive potency of morphine in cancer pain relief.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Morphine, an agonist of μ opioid receptor (MOR), is widely used in clinical pain management, especially in the treatment of cancer pain. When morphine binds to μ opioid receptor, GDP dissociates from the Gα subunit and is placed by GTP. The Gα subunit bounding to GTP dissociates from the Gβ and Gγ subunits and interacts with the intracellular system. Subsequently, the release of nociceptive neurotransmitters (for example, substance P and glutamate) from the primary afferent terminals in spinal cord was blocked, and then the analgesic effect was produced (Bovill 1997).
Chemokines are a family of small cytokines which have the ability to induce directed chemotaxis in nearby responsive cells. They are involved in a wide range of biological functions including neuroinflammation, tumor metastasis, allergic diseases, and other neuropathies (Nguyen et al. 2009; Sano et al. 2005; Yoon et al. 2007). Recent studies suggest that chemokines are novel regulators of pain. In these chemokines, the roles of CX3CL1 (Fractalkine) and CCL2 (MCP-1) in pain are widely accepted, which implicate in the maintenance of hypersensitivity in pain conditions (Dauvergne et al. 2013; Zhuang et al. 2007). Chemokines also participate in the regulation of opioid analgesia. Similar to opioid receptors, chemokine receptors belong to the superfamily of G-protein coupling receptors (GPCRs). Until now, CX3CL1, CCL5 (RANTES), and CXCL12 (SDF-1) are known to interfere with the antinociceptive effect induced by opioid agonists (Chen et al. 2007; Szabo et al. 2002). These chemokines block opioid analgesia by rapid heterologous desensitization between opioid receptors and chemokine receptors. CXCL10 (IP-10), a ligand of CXC chemokine receptor CXCR3, is a potent chemoattractant for various immune cells, including CD8+ T cells, mast cells, natural killer cell, etc. (Harris et al. 2012; Petreaca et al. 2012). Fentanyl, but not morphine, could inhibit TNF-α-induced CXCL10 expression in human astroglial cells (Davis et al. 2007). This suggests that opioids may regulate the expression of CXCL10 in the central nervous system. In our previous studies, we found that chemokine CXCL10 and CXCR3 in spinal cord were up-regulated in the process of bone cancer and that activation of CXCL10/CXCR3 participated in the development of cancer-induced bone pain (CIBP) (Bu et al. 2013). These demonstrated that CXCL10 was an important regulator in pain mechanism. However, whether CXCL10 could affect morphine antinociception in cancer pain relief is still unknown. In this study, we investigated the effect of morphine on CXCL10 expression and the role of CXCL10 in the regulation of morphine analgesia in CIBP rats and normal mice.
Materials and Methods
Animals
Female Sprague–Dawley (SD) rats weighing 180–220 g and male C57BL/6 mice weighing 18–22 g were used. The animals were kept under controlled conditions (22 ±0.5 °C, relative humidity 40–60 %, alternate light–dark cycles, food and water ad libitum). All experimental procedures were reviewed and approved by our Institutional Animal Care and Use Committee and carried out in accordance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals.
Surgical Procedure of Bone Cancer Models
CIBP modeling was prepared following previous report (Cao et al. 2010). Briefly, rats were deeply anesthetized with pentobarbital sodium (50 mg/kg, intraperitoneal injection). The right tibia was carefully exposed. Walker 256 cells (10 μL, 4 × 106/mL) or D-Hanks solution (10 μL) were slowly injected into the bone cavity using a 50-μL Hamilton microsyringe. The injection site was closed using bone wax as soon as the syringe was removed. Then, the wound was closed after being carefully disinfected. Animals were placed in a warm pad until they had regained consciousness and were returned to their home cages.
Intrathecal Injections
For rats, intrathecal drug administration was performed by a PE-10 catheter. Under the same surgical conditions to CIBP modeling, rats were intrathecally implanted with PE-10 catheter to the subarachnoid cavity at the level of lumbar enlargement, according to the method described previously (Yaksh and Rudy 1976). Proper intrathecal location was confirmed by a temporary motor block of both hind limbs after the injection of 10 μL 2 % lidocaine. The animals were allowed a 7-day recovery period before used in experiments.
For mice, intrathecal drug administration was performed in unanesthetized mice at the L5 and L6 intervertebral space as described by Hylden and Wilcox (1980). Briefly, after the mouse was lightly restrained to maintain the position of the needle, a volume of 3 μL of drug was intrathecally (i.t.) injected with a 50-μL Hamilton microsyringe. Puncture of the dura was indicated by a slight flick of the tail.
Behavioral Experiments
The changes of pain threshold in animals were assessed by measuring paw withdrawal thresholds (PWTs) as described previously (Liu et al. 2012). Briefly, animals were allowed to acclimatize to the boxes for 15 min before testing. Von Frey filaments, with ascending order of forces (0.04, 0.07, 0.16, 0.4, 0.6, 1, 2, 4, 6, 8, 10, 15, 26, 60, 100, and 300 g), were applied to the region between foot pads in the plantar aspect of the right hind paw. Abrupt paw withdrawal, licking, and shaking were considered as positive responses. Once a withdrawal response was established, the test was repeated starting with the next descending filament until no response occurred. The lowest amount of force which could elicit a response was recorded as the PWT (in grams).
Experiment 1. Effect of CXCL10 on Morphine Analgesia in CIBP Models
To detect the effect of CXCL10 on morphine analgesia in CIBP models, on the 14th day after modeling, CIBP rats received intrathecal (i.t.) injection of 100 ng neutralizing rabbit anti-CXCL10 antibody (anti-CXCL10, Peprotech, USA), followed by i.t. injection of 10 μg morphine (NEPG, China). Saline or normal rabbit IgG was injected as control. At 30 min after the last drug injection, the PWTs of rats in each group were measured, respectively.
Experiment 2. The Effect of CXCL10 on the PWTs of Normal Mice
To evaluate if overexpression of CXCL10 has influence on the pain threshold, mice were i.t. injected with 30 ng recombinant murine CXCL10 protein (rmCXCL10, PeproTech, USA) or saline (3 μL). Then, PWTs were measured at 15, 30, 60, 90, and 120 min after drug injection.
Experiment 3. The Role of CXCR3 in CXCL10-Induced Algesia
To evaluate if CXCL10 induced algesia via its receptor CXCR3, mice were i.t. injected with lentivirus encoding siRNA sequence targeting mouse CXCR3 (CXCR3i, NM_009910, GeneChem, China) ornon-targeting RNA (GeneChem, China). At 7 days after CXCR3i injection, mice were i.t. administrated with rmCXCL10 (30 ng). The PWTs were measured as experiment 2.
Experiment 4. The Role of CXCL10 in Morphine Analgesia in Normal Mice
To detect if overexpression of CXCL10 could influence the antinociceptive effect of morphine, mice were i.t. administrated with 30 ng rmCXCL10, followed by s.c. injection of 10 mg/kg morphine. The PWTs were measured as experiment 2.
Experiment 5. The role of Gi Protein in CXCL10-Induced Algesia
As CXCR3 is a member of GPCRs, we detected if Gi protein plays roles in CXCL10-induced algesia. Mice were i.t. injected with 100 ng pertussis toxin (PTX, a special inhibitor of Gi protein; Sigma, USA), followed by injection with rmCXCL10 (30 ng, i.t.) 30 min later. The PWTs were measured as experiment 2.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA extraction of spinal cord and the reverse transcription procedure were performed using RNAiso Plus (Takara, Shiga, Japan) according to the manufacturer’s instructions. One microgram of total RNA from each sample was added into a 20-μL reaction of reverse transcription, respectively. Specific primers for mouse CXCL10 and for the endogenous control mouse GAPDH were obtained from qPrimerDepot (Table 1). StepOne Real-Time PCR System (Applied Biosystems, USA) was used to conduct the qRT-PCR. Relative quantification of mRNA was performed by 2−∆∆Ct method. Data were presented as fold changes normalized to control group.
Double-Immunofluorescent Staining
At 14 days after modeling, CIBP rats were perfused with saline, followed by 4 % paraformaldehyde. The L3–L5 spinal cord segments were removed and post-fixed for 24 h at 4 °C and then dehydrated in 30 % sucrose solution. After increasing membrane permeability and blocking nonspecific binding, 30-μm-thick sections were incubated overnight at 4 °C with a mixture of the following primary antibodies: rabbit anti-MOR antibody (1:400, Millipore, USA) and goat anti-CXCL10 antibody (1:50, Santa Cruz, CA, USA). Then, sections were incubated with NorthernLights 557 Fluorochrome-labeled donkey anti-goat IgG (1:100, R&D, USA) and NorthernLights 493 Fluorochrome-labeled donkey anti-rabbit IgG (1:100, R&D, USA) for 3 h at room temperature (RT) and lastly stained with 4,6-diamidino-2-phenylindole (DAPI, Sigma, USA) for 10 min. Fluorescent images were captured using a fluorescence microscope (Leica, German).
Western Blots
The proteins of spinal cord tissue from each group were extracted using RIPA lysis buffer according to the instruction (Beyotime, China). After being denatured by boiling, 30 μg protein from each sample was loaded on 10 % SDS polyacrylamide gel. Electrophoresis was conducted at 200-mA constant current for each gel. After that, the proteins were electro-transferred (200 mA, 25 or 90 min) to PVDF membranes. The membranes were blocked non-specific binding with 5 % non-fat milk in Tris-buffered saline for 2 h at room temperature and incubated with rabbit anti-CXCR3 (1:200, Abnova), rabbit anti-CXCL10 (1:200, Peprotech, USA), or rabbit anti-GAPDH (1:1,000, Boster, China) overnight at 4 °C. After having been washed, the membranes were incubated with HRP-conjugated goat anti-rabbit IgG (1:5,000, Boster, China) for 2 h at RT. Finally, proteins were detected by ECL reagents (Beyotime, China) and visualized by exposure to Kodak film. The density was quantified by densitometric scanning. The level of CXCL10 and CXCR3 was exhibited as density relative to the density of GAPDH.
Enzyme-Linked Immunosorbent Assay
The proteins of spinal cord tissue were extracted as detailed earlier. Total unboiled proteins of each sample were diluted to the same concentration of 500 μg/mL. The levels of CXCL10 protein were measured using a mouse CXCL10 ELISA kit (Boster, China). Briefly, 100 μL of protein samples and standards was added to the plate and incubated for 90 min at 37 °C. Then, proteins were incubated with antibody, ABC solution, and TMB substrate solution in sequence. Finally, 100 μL stop solution was added to each well and the absorbance of optical density was determined at 450 nm using a plate reader. The concentration of CXCL10 was calculated by CurveExpert. The levels of CXCL10 protein were presented as pg/g total protein.
Statistical Analysis
All data were presented as mean ± SEM. Statistical significance (P < 0.05) was determined using a two-way ANOVA (treatment group × min) to detect the changes to PWTs after drug injection over time. Individual comparisons were made with unpaired t-test. The change of CXCL10 expression and PWTs in CIBP rats was tested using one-way ANOVA.
Results
The Co-localization of MOR and CXCL10 in Spinal Cord
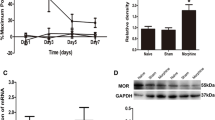
The co-localization of MOR and CXCL10 was detected by double-immunofluorescent staining. Results showed that MOR was distributed in the lamina II of spinal cord with several pericellular ring-like structures (Fig. 1. b, f, h), which suggested that the MOR was distributed as a membrane protein. CXCL10 was also distributed in the lamina II,with punctiform structures (Fig. 1. a, e). The merged pictures showed that MOR and CXCL10 were co-localizated in the lamina II of spinal cord (Fig. 1d, h).

The co-localization of MOR and CXCL10 in the spinal cord of rat as detected by immunofluorescence. MOR was distributed in the lamina II with ring-like structures (white arrowheads; b, f). CXCL10 was distributed in the lamina II with punctiform structures (a, e). The yellow spots in the merged pictures show the co-localization of MOR and CXCL10 (d, h). The nuclei were stained with DAPI (c, g). Scale bar a–d, 200 μm; e–h, 50 μm
Single Injection of Morphine Transiently Increased the Expression of CXCL10 in Mice
The effect of morphine on expression of CXCL10 was measured by real-time PCR and ELISA. Mice were treated with single morphine (10 mg/kg, s.c.). Then, the expression of CXCL10 in spinal cord was measured at 1, 2, 3, 6, and 12 h after drug administration, respectively. Results showed that a single dose of morphine could increase the level of CXCL10 mRNA in spinal cord from 1 h after drug administration (7.18 ± 1.70 vs. 1.13 ± 0.37, compared with saline-treated mice, P < 0.01) (Fig. 2a). The upregulation of CXCL10 mRNA lasted to 3 h after drug administration (3.23 ± 1.49). A single dose of morphine could also increase the level of CXCL10 protein in spinal cord. CXCL10 protein in morphine-treated mice was significantly increased at 3 h after drug administration (5,112.21 ± 607.95 vs. 3,202.01 ± 322.94 pg/g, compared with saline-treated mice, P < 0.01) (Fig. 2b). The expression of CXCL10 was returned to the base level at 6 and 12 h.

Changes of CXCL10 mRNA and protein expression as induced by a single dose of morphine in the spinal cord. CXCL10 levels were measured at 1, 2, 3, 6, and 12 h after morphine injection (10 mg/kg, s.c.). a CXCL10 mRNA was increased at 1, 2, and 3 h after morphine treatment, measured by real-time PCR. **P < 0.01, vs. saline-treated mice; n = 6 in each group. b CXCL10 protein was increased at 3 h after morphine injection, measured by ELISA. **P < 0.01, vs. saline-treated mice; n = 6 in each group
Blocking of CXCL10 Function Enhanced Morphine Analgesia in CIBP Rats
To detect if blocking CXCL10 function has influence on morphine analgesia in CIBP rats, on the 14th day after modeling, CIBP rats were treated with anti-CXCL10 (100 ng, i.t.) and (or) morphine (10 μg, i.t.). Results showed that the expression of CXCL10 was increased in CIBP rats compared with sham rats (34.68 ± 1.92 vs. 20.97 ± 2.92 %, P < 0.05; Fig. 3a). The CXCR3 level was also higher in CIBP rats than that in sham rats (21.27 ± 1.09 vs. 14.51 ± 0.80 %, P < 0.05; Fig. 3a). On postoperative day 14, the PWTs of saline-treated CIBP rats were decreased compared with sham rats (0.93 ± 0.52 vs. 11.33 ± 2.69 g, P < 0.01). Both anti-CXCL10 and morphine treatment increased the PWTs in CIBP rats (9.5 ± 2.81 vs. 0.93 ± 0.52 g; 17 ± 6.68 vs. 0.93 ± 0.52 g, compared with saline-treated CIBP rats, P < 0.01; Fig. 3b). While blocking the function of CXCL10 by pre-treatment with anti-CXCL10, the analgesic effect of morphine in CIBP rats was enhanced compared with that in rats which were treated by morphine only (41.17 ± 19.19 vs. 17 ± 6.68 g, P < 0.01; Fig. 3b).

The effect of blocking CXCL10 on morphine analgesia in CIBP rats. a On day 14 post-modeling, CXCL10 and CXCR3 levels in each group were measured by western blots. CXCL10 and CXCR3 protein levels in spinal cord were higher in CIBP rats than those in sham rats (P < 0.05). *P < 0.05; n = 3 in each group. b On day 14, CIBP rats were i.t. injected with anti-CXCL10 and (or) morphine. Anti-CXCL10 was injected 30 min before morphine. Behavioral tests were accessed 30 min after the last drug injection. The PWTs of CIBP rats were significantly decreased (P < 0.01, vs. sham rats). Morphine- or anti-CXCL10-treated rats showed a significant increase of PWTs (P < 0.01, vs. saline-treated CIBP rats). Rats receiving a combination of morphine and anti-CXCL10 showed higher PWTs compared with rats receiving morphine treatment only (P < 0.01). &&P < 0.01, compared with sham rats; ** P < 0.01, compared with saline-treated CIBP rats; ## P < 0.01, compared with morphine-treated CIBP rats (n = 6 in each group)
Overexpression of CXCL10 Induced Algesia in Mice Via Receptor CXCR3
To detect if overexpression of CXCL10 in normal mice affects pain threshold, mice were i.t. injected with 30 ng rmCXCL10. The PWTs of rmCXCL10-treated mice decreased from 15 min (0.35 ± 0.17 vs. 0.60 ± 0.22 g, compared with saline-treated mice, P < 0.01). This decrease of PWTs lasted up to 120 min until the test was finished (0.32 ± 0.12 vs. 0.73 ± 0.21 g, compared with saline-treated mice, P < 0.01) (Fig. 4). These results suggested that rmCXCL10 induced an algesic effect in mice. To detect if CXCR3 participated in CXCL10-induced algesia, mice were i.t. injected with lentiviral vectors carrying siRNA-targeting murine CXCR3 (CXCR3i, 4 μL, 1 E + 8 TU) or non-targeting RNA (LV-con, 4 μL, 1 E + 8 TU). At 7 days after infection, mice were i.t. administrated with 30 ng rmCXCL10. While rmCXCL10-treated LV-con-treated mice showed a significant decrease of PWTs by 0.10 ± 0.05 g from 30 min after injection, the PWTs of rmCXCL10-treated CXCR3i-mice still maintained a high level of 0.53 ± 0.10 g at 30 min (Fig. 5a). The PWTs of rmCXCL10-treated CXCR3i-mice had no significant changes during the experiment (P > 0.05, compared with PWTs at time 0; Fig. 5a). To verify the downregulation of gene induced by CXCR3i, the CXCR3 level was measured by western blots. Results showed that CXCR3 siRNA downregulated CXCR3 protein level in spinal cord (Fig. 5b). These results suggested that CXCL10 induced algesia via coupling receptor CXCR3.

Administration with rmCXCL10 induced algesic effect in normal mice. RmCXCL10 (30 ng, i.t.) or saline (3 μL) was given to the mice. The mice that received rmCXCL10 treatment showed a significant decrease of PWTs than the saline-treated mice (P < 0.01). **P < 0.01, vs. saline-treated mice; n = 6 in each group

RmCXCL10-induced algesia involved CXCR3. a Mice that received i.t. injection of lentiviral vectors containing CXCR3siRNA or control RNA. At 7 days later, the mice were i.t. injected with 30 ng rmCXCL10 or 3 μL saline. The PWTs of rmCXCL10-treated LV-con mice were significantly decreased. However, in CXCR3i-rmCXCL10-treated mice, the PWTs had no statistical differences in the time course. *P < 0.05, **P < 0.01, vs. rmCXCL10-treated LV-con mice; n = 6 in each group. b Mice were i.t. injected with lentivirus encoding CXCR3i or control RNA. At 7 days later, the spinal cord of mice was removed to detect the level of CXCR3 protein by western blots. CXCR3 level was significantly decreased in CXCR3i-treated mice compared with naïve mice and LV-con-treated mice (P < 0.05). *P < 0.05, vs. naïve mice. # P < 0.05, vs. LV-con-treated mice; n = 3 in each group
Overexpression of CXCL10 Attenuated Morphine Analgesia in Normal Mice
As previous studies showed, some chemokine blocked morphine antinociception (Chen et al. 2007). Next, we investigated if overexpression of CXCL10 could change the antinociceptive effect of morphine. Mice were i.t. administrated with rmCXCL10 (30 ng), followed by morphine (10 mg/kg, s.c.). In morphine-treated mice, the PWTs were increased from 15 min and reached a peak of 14.17 ± 2.04 g at 60 min after drug administration. However, in rmCXCL10-pretreated mice, the increase of PWTs induced by morphine reached a lower level of 7.00 ± 2.76 g than that in morphine-treated mice at 60 min (P < 0.01; Fig. 6a). These results suggested that overexpression of CXCL10 in spinal cord partly inhibited morphine-induced analgesic effect.

RmCXCL10-attenuated morphine analgesia. a The mice were i.t. injected with 30 ng rmCXCL10 or saline and subsequently s.c. injected with 10 mg/kg morphine. The PWTs of mice that received saline–morphine treatment increased from 30 min after the last drug injection. Co-treatment of morphine and rmCXCL10 also induced increase of PWTs, but to a significantly lesser extent. **P < 0.01, vs. saline–morphine-treated mice; n = 6–7 in each group. b Mice were pre-treated with i.t. injection of 100 ng PTX or saline. At 30 min later, mice were i.t. injected with 30 ng rmCXCL10. The PWTs of saline–rmCXCL10-treated mice were significantly decreased. However, pre-treatment with PTX partly blocked the algesic effect induced by rmCXCL10. *P < 0.05, **P < 0.01, vs. saline–rmCXCL10-treated mice; n = 6 in each group
CXCL10-Induced Algesia Involved Gi Protein
CXCR3 is a member of GPCRs family. Gi protein plays an important role in the initiation of intracellular signal transduction induced by active GPCRs. PTX was used to detect the role of Gi protein in CXCL10-induced algesia. Mice were i.t. injected with 100 ng PTX. At 30 min later, mice were treated with rmCXCL10 (30 ng, i.t.) or saline. Results showed that PTX pretreatment partially blocked the decrease of PWTs induced by rmCXCL10 (PTX-rmCXCL10-treated mice vs. saline-rmCXCL10-treated mice, at 30 min with 0.32 ± 0.12 vs. 0.11 ± 0.05 g, P < 0.01, and at 90 min with 0.43 ± 0.08 vs. 0.19 ± 0.15 g, P < 0.05) (Fig. 6b). These results demonstrated that the algesic effect induced by CXCL10 was mediated by Gi proteins.
Discussion
Our study demonstrated the role of CXCL10 in morphine analgesia. Acute morphine administration increased the expression of CXCL10 in spinal cord in mice. Blocking the function of CXCL10 in spinal cord enhanced morphine antinociception in CIBP rats. However, overexpression of CXCL10 in spinal cord blocked the analgesic effect of morphine and produced algesic effect via activation of CXCR3 and Gi protein. This is a novel finding that CXCL10 plays a role in the regulation of pain management.
Previous study showed that 24 h of treatment with fentanyl could inhibit the increase of CXCL10 induced by TNF-α in vitro (Davis et al. 2007). Morphine can activate a pro-inflammatory response via increasing the production of cytokines or chemokines (Loram et al. 2012; Schwarz et al. 2013). In our study, a single dose of morphine increased CXCL10 level in spinal cord of mice in the first 3 h after drug injection. This suggested that the expression of CXCL10 induced by opioids may be complicated, which involves not only astroglia but also other cells. We also detected the co-localization of MOR and CXCL10 in spinal cord, which provided evidence that the upregulation of CXCL10 can be explained as the activation of neuroinflammation directly induced by single morphine administration.
In clinical treatment, morphine is widely used for cancer pain relief. However, chronic administration of morphine usually induced antinociceptive tolerance or hyperalgesia, which limited its use for pain relief. Furthermore, morphine antinociception could be blocked by many factors (Jackson and Damaj 2013; Weibel et al. 2013). Chemokines contributed to the development of morphine tolerance (Zhao et al. 2012) and also induced loss of acute morphine analgesia (Rivat et al. 2013). There are several changes in spinal cord associated with cancer-induced pain condition, including excitatory synaptogenesis (Ke et al. 2013), activation of glial cells (Mao-Ying et al. 2012), increasing of pro-hyperalgesic factors, and chemokine upregulation (Hu et al. 2012; Suzuki et al. 2012; Wang et al. 2012). In this study, we found that chemokine CXCL10 was upregulated in spinal cord of CIBP rats. When we blocked CXCL10 function with neutralizing antibody, the antinociceptive effect of morphine was enhanced. This suggested that the upregulation of CXCL10 induced by bone cancer may play a negative role to the analgesia induced by morphine. In the subsequent studies, we investigated the role of CXCL10 in morphine-induced analgesia in normal mice. After overexpression of CXCL10 by acute injection of rmCXCL10, mice exhibited hyperalgesia immediately. This algesic effect of CXCL10 was promised by coupling to CXCR3. Meanwhile, injection of rmCXCL10 attenuated morphine analgesia. These strongly support that CXCL10 is an algesic molecule and that activation of CXCL10/CXCR3 signaling plays a negative feedback role in the regulation of morphine analgesia.
Chemokine receptors are members of GPCR families. Gi protein coupling chemokine receptor could activate Erk and Akt signals and then mediated chemotaxis of T cells, proliferation, and survival of immune cells (Korniejewska et al. 2011; Sanchez-Sanchez et al. 2004; Smit et al. 2003; Willox et al. 2010). Moreover, chemokine-mediated tumor metastasis also involves Gi protein. Inhibitor of Gi protein, PTX, could inhibit SDF-1α-induced cancer cell adhesion and CXCL12-mediated chemotactic responses of cancer cells (Akekawatchai et al. 2005; Hidalgo et al. 2001). PTX-sensitive Gi protein also participated in chemokine CCL2-induced CGRP release in the DRG neurons and activation of primary sensory neurons (Belkouch et al. 2011; Qin et al. 2005). These suggest that Gi protein plays an important role in the chemokine-mediated regulation of chronic pain and other pathological conditions. However, there is still lack of direct evidence. In our study, pretreatment with PTX blocked CXCL10-induced algesia in mice. It can be concluded that CXCR3 coupling to Gi protein contributed to the regulation of the pain induced by CXCL10.
Based on these data, we presume that there may be a cross-talk between MOR and CXCR3. Activation of CXCR3 may interfere with the function of μ opioid receptor via rapid heterologous desensitization, which subsequently attenuates morphine analgesia. Another possible hypothesis is that activation of CXCR3 may block the downstream signals of μ opioid receptor, for example, activation of p38 MAPK-mediated nuclear NF-κB activation. However, these hypotheses need to be confirmed in further studies.
The findings of this study may have significant therapeutic implications. As blocking CXCL10 function enhanced morphine analgesia in CIBP rats and upregulation of CXCL10 induced algesia in normal mice, targeting at inhibiting the function of CXCL10/CXCR3 may prove to be an effective adjunctive strategy in cancer pain relief with morphine. However, further studies need to be done to investigate more details and the underlying mechanism of the cross-talk between chemokines and opioids.
References
Akekawatchai C, Holland JD, Kochetkova M, Wallace JC, McColl SR (2005) Transactivation of CXCR4 by the insulin-like growth factor-1 receptor (IGF-1R) in human MDA-MB-231 breast cancer epithelial cells. J Biol Chem 280:39701–39708
Belkouch M, Dansereau MA, Reaux-Le Goazigo A, Van Steenwinckel J, Beaudet N, Chraibi A, Melik-Parsadaniantz S, Sarret P (2011) The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gbetagamma-dependent mechanism. J Neurosci 31:18381–18390
Bovill JG (1997) Mechanisms of actions of opioids and non-steroidal anti-inflammatory drugs. Eur J Anaesthesiol Suppl 15:9–15
Bu H, Shu B, Gao F, Liu C, Guan X, Ke C, Cao F, Hinton AO, Jr., Xiang H, Yang H, Tian X, Tian Y (2013) Spinal IFN-gamma-induced protein-10 (CXCL10) mediates metastatic breast cancer-induced bone pain by activation of microglia in rat models. Breast Cancer Res Treat. doi:10.1007/s10549-013-2807-4
Cao F, Gao F, Xu AJ, Chen ZJ, Chen SS, Yang H, Yu HH, Mei W, Liu XJ, Xiao XP, Yang SB, Tian XB, Wang XR, Tian YK (2010) Regulation of spinal neuroimmune responses by prolonged morphine treatment in a rat model of cancer induced bone pain. Brain Res 1326:162–173
Chen X, Geller EB, Rogers TJ, Adler MW (2007) The chemokine CX3CL1/fractalkine interferes with the antinociceptive effect induced by opioid agonists in the periaqueductal grey of rats. Brain Res 1153:52–57
Dauvergne C, Molet J, Reaux-Le Goazigo A, Mauborgne A, Melik-Parsadaniantz S, Boucher Y, Pohl M (2013) Implication of the chemokine CCL2 in trigeminal nociception and traumatic neuropathic orofacial pain. Eur J Pain. doi:10.1002/j.1532-2149.2013.00377.x
Davis RL, Buck DJ, Saffarian N, Stevens CW (2007) The opioid antagonist, beta-funaltrexamine, inhibits chemokine expression in human astroglial cells. J Neuroimmunol 186:141–149
Harris TH, Banigan EJ, Christian DA, Konradt C, Tait Wojno ED, Norose K, Wilson EH, John B, Weninger W, Luster AD, Liu AJ, Hunter CA (2012) Generalized Levy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 486:545–548
Hidalgo A, Sanz-Rodriguez F, Rodriguez-Fernandez JL, Albella B, Blaya C, Wright N, Cabanas C, Prosper F, Gutierrez-Ramos JC, Teixido J (2001) Chemokine stromal cell-derived factor-1alpha modulates VLA-4 integrin-dependent adhesion to fibronectin and VCAM-1 on bone marrow hematopoietic progenitor cells. Exp Hematol 29:345–355
Hu JH, Yang JP, Liu L, Li CF, Wang LN, Ji FH, Cheng H (2012) Involvement of CX3CR1 in bone cancer pain through the activation of microglia p38 MAPK pathway in the spinal cord. Brain Res 1465:1–9
Hylden JL, Wilcox GL (1980) Intrathecal morphine in mice: a new technique. Eur J Pharmacol 67:313–316
Jackson KJ, Damaj MI (2013) Calcium/calmodulin-dependent protein kinase IV mediates acute nicotine-induced antinociception in acute thermal pain tests. Behav Pharmacol 24:689–692
Ke C, Li C, Huang X, Cao F, Shi D, He W, Bu H, Gao F, Cai T, Hinton AO Jr, Tian Y (2013) Protocadherin20 promotes excitatory synaptogenesis in dorsal horn and contributes to bone cancer pain. Neuropharmacology 75C:181–190
Korniejewska A, McKnight AJ, Johnson Z, Watson ML, Ward SG (2011) Expression and agonist responsiveness of CXCR3 variants in human T lymphocytes. Immunology 132:503–515
Liu X, Bu H, Liu C, Gao F, Yang H, Tian X, Xu A, Chen Z, Cao F, Tian Y (2012) Inhibition of glial activation in rostral ventromedial medulla attenuates mechanical allodynia in a rat model of cancer-induced bone pain. J Huazhong Univ Sci Technolog Med Sci 32:291–298
Loram LC, Grace PM, Strand KA, Taylor FR, Ellis A, Berkelhammer D, Bowlin M, Skarda B, Maier SF, Watkins LR (2012) Prior exposure to repeated morphine potentiates mechanical allodynia induced by peripheral inflammation and neuropathy. Brain Behav Immun 26:1256–1264
Mao-Ying QL, Wang XW, Yang CJ, Li X, Mi WL, Wu GC, Wang YQ (2012) Robust spinal neuroinflammation mediates mechanical allodynia in Walker 256 induced bone cancer rats. Mol Brain 5:16
Nguyen KD, Vanichsarn C, Fohner A, Nadeau KC (2009) Selective deregulation in chemokine signaling pathways of CD4 + CD25(hi)CD127(lo)/(−) regulatory T cells in human allergic asthma. J Allergy Clin Immunol 123(933–9):e10
Petreaca ML, Do D, Dhall S, McLelland D, Serafino A, Lyubovitsky J, Schiller N, Martins-Green MM (2012) Deletion of a tumor necrosis superfamily gene in mice leads to impaired healing that mimics chronic wounds in humans. Wound Repair Regen 20:353–366
Qin X, Wan Y, Wang X (2005) CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. J Neurosci Res 82:51–62
Rivat C, Sebaihi S, Steenwinckel JV, Fouquet S, Kitabgi P, Pohl M, Parsadaniantz SM, Goazigo AR (2013) Src family kinases involved in CXCL12-induced loss of acute morphine analgesia. Brain Behav Immun. doi:10.1016/j.bbi.2013.11.010
Sanchez-Sanchez N, Riol-Blanco L, de la Rosa G, Puig-Kroger A, Garcia-Bordas J, Martin D, Longo N, Cuadrado A, Cabanas C, Corbi AL, Sanchez-Mateos P, Rodriguez-Fernandez JL (2004) Chemokine receptor CCR7 induces intracellular signaling that inhibits apoptosis of mature dendritic cells. Blood 104:619–625
Sano R, Tessitore A, Ingrassia A, d'Azzo A (2005) Chemokine-induced recruitment of genetically modified bone marrow cells into the CNS of GM1-gangliosidosis mice corrects neuronal pathology. Blood 106:2259–2268
Schwarz JM, Smith SH, Bilbo SD (2013) FACS analysis of neuronal–glial interactions in the nucleus accumbens following morphine administration. Psychopharmacology (Berl). doi:10.1007/s00213-013-3180-z
Smit MJ, Verdijk P, van der Raaij-Helmer EM, Navis M, Hensbergen PJ, Leurs R, Tensen CP (2003) CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood 102:1959–1965
Suzuki M, Narita M, Hasegawa M, Furuta S, Kawamata T, Ashikawa M, Miyano K, Yanagihara K, Chiwaki F, Ochiya T, Suzuki T, Matoba M, Sasaki H, Uezono Y (2012) Sensation of abdominal pain induced by peritoneal carcinomatosis is accompanied by changes in the expression of substance P and mu-opioid receptors in the spinal cord of mice. Anesthesiology 117:847–856
Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ (2002) Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci U S A 99:10276–10281
Wang LN, Yang JP, Ji FH, Zhan Y, Jin XH, Xu QN, Wang XY, Zuo JL (2012) Brain-derived neurotrophic factor modulates N-methyl-d-aspartate receptor activation in a rat model of cancer-induced bone pain. J Neurosci Res 90:1249–1260
Weibel R, Reiss D, Karchewski L, Gardon O, Matifas A, Filliol D, Becker JA, Wood JN, Kieffer BL, Gaveriaux-Ruff C (2013) Mu opioid receptors on primary afferent nav1.8 neurons contribute to opiate-induced analgesia: insight from conditional knockout mice. PLoS One 8:e74706
Willox I, Mirkina I, Westwick J, Ward SG (2010) Evidence for PI3K-dependent CXCR3 agonist-induced degranulation of human cord blood-derived mast cells. Mol Immunol 47:2367–2377
Yaksh TL, Rudy TA (1976) Chronic catheterization of the spinal subarachnoid space. Physiol Behav 17:1031–1036
Yoon Y, Liang Z, Zhang X, Choe M, Zhu A, Cho HT, Shin DM, Goodman MM, Chen ZG, Shim H (2007) CXC chemokine receptor-4 antagonist blocks both growth of primary tumor and metastasis of head and neck cancer in xenograft mouse models. Cancer Res 67:7518–7524
Zhao CM, Guo RX, Hu F, Meng JL, Mo LQ, Chen PX, Liao XX, Cui Y, Feng JQ (2012) Spinal MCP-1 contributes to the development of morphine antinociceptive tolerance in rats. Am J Med Sci 344:473–479
Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR (2007) Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun 21:642–651
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of the People's Republic of China (No. 81171259, No. 81371250, and No. 81070890) and also supported by 2010 Clinical Key Disciplines Construction Grant from the Ministry of Health of China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dawei Ye and Huilian Bu contributed equally to this work.
Rights and permissions
About this article
Cite this article
Ye, D., Bu, H., Guo, G. et al. Activation of CXCL10/CXCR3 Signaling Attenuates Morphine Analgesia: Involvement of Gi Protein. J Mol Neurosci 53, 571–579 (2014). https://doi.org/10.1007/s12031-013-0223-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-013-0223-1