
Abstract
Pinocembrin (PB), the most abundant flavonoid in propolis, has been proven to have neuroprotective property against neurotoxicity in vivo and in vitro. Our recent study demonstrated the neuroprotective effect of PB against Aβ25–35-induced SH-SY5Y neurotoxicity. However, the mechanism as how PB can induce neuroprotection is not known. In the present study, we demonstrate here that PB abrogates the effects of the neurotoxin 1-methyl-4-phenylpyridinium (MPP+) which mimics Parkinson’s disease (PD) with elevation of intracellular reactive oxygen species (ROS) level and apoptotic death. We found that pretreatment of SH-SY5Y cells with PB significantly reduced the MPP+-induced loss of cell viability, the generation of intracellular ROS, apoptotic rate, and the cleavage of caspase-3. PB strikingly inhibited MPP+-induced mitochondrial dysfunctions, including lowered membrane potential, decreased Bcl-2/Bax ratio, and the release of cytochrome c. Overall, these results suggest that PB is intimately involved in inhibiting MPP+-induced loss of mitochondrial function and induction of apoptosis that contributes toward neuronal survival. These data indicated that PB might provide a valuable therapeutic strategy for the treatment of PD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD), the second most frequent cause of dementia after Alzheimer’s disease, is characterized by a loss of dopaminergic neurons in the substantia nigra. Even though the etiology of PD remains unclear, several lines of evidence strongly support the involvement of oxidative stress and mitochondrial dysfunction (Pieczenik and Neustadt 2007).
Support for oxidative stress mechanisms in dopaminergic degeneration in the substantia nigra in Parkinson’s disease (Jenner et al. 1992) comes from a growing body of evidence, indicating that this region has a high propensity for oxidative stress and is also deficient in protective mechanisms (Dickson 2007). Dopaminergic neurons are characteristically vulnerable to oxidative damage due to the high probability of dopamine auto-oxidation and the high lipid and iron contents (Greenamyre et al. 1999). Insights into PD pathogenesis, the well-known neurotoxin 1-methyl-4-phenylpyridinium (MPP+), have long been used to establish experimental models of PD. MPP+, the active metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), is selectively neurotoxic to DA containing nigral cells and able to produce both cell death and Parkinson-like syndromes in primates. This neurotoxic metabolite accumulates in nigrostriatal neurons via the dopamine transporter (Chiba et al. 1985) and is subsequently transported into the mitochondria by the membrane potential (Ramsay et al. 1986). MPP+ potently inhibits complex I of the mitochondrial electron transport chain (Singer and Ramsay 1990) and induces a syndrome closely resembling PD in cellular and animal models (Przedborski and Jackson-Lewis 1998; Eberhardt and Schulz 2003).
MPP+ stimulates the production of the superoxide radical in vitro and induces cell death in PC12 cells (Itano et al. 1994). Moreover, MPTP generates hydroxyl radicals which cause membrane lipid peroxidation or DNA damage, and this is thought to be the mechanism of degeneration of dopaminergic neurons (Sriram et al. 1997; Chiueh and Rauhala 1998). Excessive free radical formation or antioxidant deficiency can result in oxidative stress, a possible mechanism of the toxicity of MPTP (Lotharius and O’Malley 2000). Hence, drugs that reduce the oxidative stress induced by MPP+ may prove to be neuroprotective in Parkinson’s disease.
Given the role of oxidative stress in the etiology of PD, one promising preventive or therapeutic intervention in PD may attenuate or suppress the oxidative stress-dependent cytotoxicity. Recently, there have been considerable efforts to search for natural substances for the neuroprotective potential, and attention has been focused on a wide array of natural antioxidants that can scavenge free radicals and protect cells from oxidative damage.
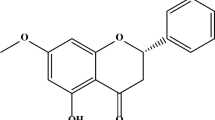
Pinocembrin (PB; 5,7-dihydroxyflavanone) is one of the primary flavonoids isolated from the variety of plants, mainly from Pinus heartwood, Eucalyptus, Populus, Euphorbia, and Sparattosperma leucanthum, in the diverse flora and purified by various chromatographic techniques. PB is a major flavonoid molecule incorporated as multifunctional in the pharmaceutical industry. Its vast range of pharmacological activities includes antimicrobial, anti-inflammatory, antioxidant, and anticancer activities. In addition, PB can be used as neuroprotective against cerebral ischemic injury (Rasul et al. 2013). Recently, the studies of Du’s groups showed that PB protected the human neuroblastoma SH-SY5Y cells overexpressing the Swedish mutant form of human APP (APPsw) against copper (Liu et al. 2012), and we also showed that PB protected SH-SY5Y cells against Aβ-induced neurotoxicity through a apoptotic pathway (submitted). Although PB exhibited neuroprotective activity, the protective effect of PB against other oxidative stress-induced cytotoxicity remained to be explored. The aim of the present study was therefore to assess the neuroprotective effect of PB against the toxicity of the parkinsonian neurotoxins MPP+ in relation to the mitochondria-mediated cell death process and role of oxidative stress. Our study verified that PB exerts neuroprotective effects against MPP+-induced cell injury through an apoptotic pathway.
Materials and Methods
Materials
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), rhodamine 123 (Rh123), pinocembrin, and MPP+ were purchased from Sigma-Aldrich Inc. Hoechst 33258 and ECL detection kit were purchased from Beyotime (Beijing, China). The fluorescent dyes 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) were purchased from Invitrogen. Complete Protease Inhibitor was from Roche Diagnostics GmbH (Penzberg, Germany). Modified Dulbecco’s Eagle’s medium (DMEM) supplement was obtained from Gibco Invitrogen Corporation. Anti-Bcl-2, anti-Bax antibodies, and antibodies against cytochrome c were from Cell Signaling (Beverly, MA, USA). Antibodies against cleaved caspase-3 were from Calbiochem (San Diego, CA, USA). All the other chemicals used were of the high-grade available commercially.
Cell Culture and Treatment
Human neuroblastoma SH-SY5Y cells were cultured in DMEM supplemented with 10 % (v/v) fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C and 5 % CO2 (Lee et al. 2008; Ahn et al. 2009). PB and MPP+ were dissolved in dimethyl sulfoxide (DMSO) and stored frozen at −20 °C until use. To induce cell injury, cells were incubated with 1 mM MPP+ for 24 h. To study the effects of PB, cells were pre-incubated with PB for 4 h, and then PB was added to the medium for an additional 24 h.
Determination of Cell Viability
Cell viability was assessed using conventional MTT reduction assay. The cultured cells in 96-well plates were pre-incubated with PB for 4 h and exposed to 50 μM PB for 24 h, and then 20 μl of MTT stock solution (5 mg/ml) was added to the culture medium for treating another 4 h at 37 °C. The resulted MTT formazan was extracted with 150 μl DMSO, and the absorbance was recorded with a microtiter plate reader.
Measurement of Apoptotic Cell Death
Apoptosis of SH-SY5Y cells was analyzed by Hoechst staining. After exposed to 1 mM MPP+ with or without PB as described above, the cells on coverslips were fixed in 4 % paraformaldehyde for 20 min and then stained with Hoechst 33258 for 15 min (Lee et al. 2008). Nuclear morphology was viewed using a fluorescence microscope. The number of cells with apoptotic morphology appearing condensed or fragmented nuclei was counted.
Intracellular ROS Generation Detection
Intracellular ROS was monitored by using the fluorescent probe 2,7-dichlorofluorescein diacetate (DCFH-DA) (Ahn et al. 2009). SH-SY5Y cells were seeded in 96-well plates and were incubated with increasing concentrations of MPP+ and/or PB for 24 h. Cells were incubated with 10 μM DCFH-DA at 37 °C for 30 min, then washed twice with PBS, and finally the fluorescence intensity of DCF was measured in a microplate reader (Wang et al. 2010). Data were analyzed and expressed as a percentage of the control.
Measurement of Mitochondrial Membrane Potential
Mitochondrial membrane potential (MMP) was monitored using the Rh123 (Wang et al. 2010). Rh123 was added to media (at a final concentration of 10 μg/ml) after the cells exposed to MPP+ with or without PB pretreatment. After incubation at 37 °C for 30 min, cells were washed and then measured by fluorescence microplate reader.
Cytochrome c Assay
For measurement of cytochrome c release, the cytosol fractions were prepared as previously reported (Wang et al. 2009). Briefly, cells were washed twice with 10 ml of cold PBS, resuspended in 500×g of fresh cytosolic extract buffer (250 mM sucrose, 20 mM Hepes pH 7.4, 10 mM KCl, 1 mM EGTA, 1 mM EDTA, 1 mM MgCl2, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 1 mM pepstatin A, 10 mg/ml leupeptin, and 2 mg/ml aprotinin), and incubated for 30 min on ice with frequent tube tapping. Cells were lysed with 50 strokes of a Dounce Homogenizer (2 ml, tight pestle) on ice, and then nuclei, unbroken cells, and cell debris were pelleted at 2,500 rpm for 10 min at 4 °C. The supernatant was spun again at 13,000 rpm for 20 min at 4 °C. The supernatant (now containing the cytosolic extract) was carefully transferred and the supernatants were stored at −80 °C, and the final pellet was used as the mitochondrial fraction. The cytochrome c levels were determined using a monoclonal antibody to cytochrome c by western blotting as described below.
Western Blot Analysis
Changes in the expression levels of indicated proteins were assessed by western blotting as described previously (An et al. 2010).
Statistical Analysis
All data were presented as the mean ± SEM. Data were subjected to statistical analysis via one-way ANOVA followed by Student’s t test with GraphPad Prism 4.0 software (GraphPad Software, Inc., San Diego, CA, USA). Mean values were considered to be statistically significant at P < 0.05.
Results
PB Ameliorated MPP+-Induced Loss of Neuronal Cell Viability
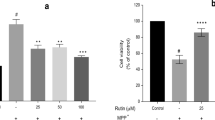
In the current experiment, we first examined the toxic effect of MPP+ at various concentrations (0.1–1 mM) on cell viability of SH-SY5Y cells. The incidences of cell death after exposure to 1 mM MPP+ for 24 and 48 h were about 75 and 65 %, respectively (Fig. 1a). Therefore, 1 mM MPP+ for treatment 24 h was used to further study the mechanisms. First, we tested the toxicity of PB to SH-SY5Y cells. As shown in Fig. 1b, treatment with PB alone did not cause any cytotoxic effect on the cell viability up to the highest concentration (50 μM). However, when the cells were treated with 1 mM MPP+ for 24 h, their viability decreased to 74 % (Fig. 1c). The MPP+-induced viability loss was attenuated by PB in a concentration-dependent manner (Fig. 1c). Furthermore, the results of the phase-contrast microscope were in line with the results measured by MTT assay (Fig. 1d).

Effects of PB on SH-SY5Y cell viability. The viability of SH-SY5Y cells was measured by MTT assay. a Cells were exposed to MPP+ with indicated concentrations for 24 or 48 h. b Cells were incubated with various concentrations of PB for 24 h. c SH-SY5Y cells were pre-incubated with vehicle or various concentrations of PB for 4 h, followed by incubation with 1 mM MPP+ for another 24 h. After this incubation, cell viability was determined with the MTT assay. d Morphological changes of SH-SY5Y cells under the phase-contrast microscope. Results are shown as the mean ± SEM and represent three independent experiments. *P < 0.05; **P < 0.01
PB Depressed MPP+-Induced Formation of ROS
To determine whether the MPP+-induced cell death in SH-SY5Y cells is mediated by oxidative stress, we investigated the formation of ROS within cells by monitoring a conversion of DCFH2-DA to DCF. Exposure of cells to MPP+ for 24 h caused a significant increase in DCF fluorescence, whose response was significantly depressed by the pretreatment with PB (Fig. 2). These results indicated that PB has the ability to scavenge ROS induced by MPP+.

PB inhibits MPP+-induced ROS generation. ROS formation, evaluated by the oxidation of H2DCF to DCF, was assessed 24 h after incubation with MPP+. PB was pretreated before MPP+ for 4.0 h. Intracellular ROS was determined by fluorescent spectrometer. Relative fluorescence intensity was obtained from three independent experiments, and the amount of intracellular ROS is expressed relative to that in controls. *P < 0.05; **P < 0.01
PB Inhibited MPP+-Triggered Nuclear Change and Apoptosis
To further study the protective effect of PB on the MPP+-treated SH-SY5Y cells, Hoechst 33258 staining was used. The specific DNA stain Hoechst 33258 was used to assess changes in DNA and nuclear structure following different treatments. As shown in Fig. 3a, MPP+-treated SH-SY5Y cells showed presence of aggregated and condensed nuclei as compared to normal nuclei in control cells. Pretreatment with 20 μm PB decreased the number of aggregated and condensed nuclei compared to cells treated with MPP+ alone. The results of inhibition of caspase-3 activation confirmed apoptosis inhibiting of PB. As shown in Fig. 3c, d, a marked increase of activated caspase-3 in MPP+ treatment was observed. However, MPP+-induced activation of caspase-3 was inhibited by PB pretreatment in a dose-dependent manner. This result indicated that PB can inhibit the induction of caspase-3. These results indicated that PB suppresses MPP+-induced apoptosis in SH-SY5Y cells.

Involvement in anti-apoptosis in the neuroprotective effects of PB. a SH-SY5Y cells, with or without pretreated PB for 4.0 h, were exposed to MPP+ for 24 h and then were subjected to Hoechst 33258 staining and viewed under a fluorescence microscope. Arrows indicated condensed nuclei, and arrowheads indicated fragmented nuclei. b Quantification of abnormal nuclei after exposure to MPP+ in the presence or absence of PB. Results are taken from three independent experiments. c Representative image of immunoblots for cleaved caspase-3. The cells were pretreated with PB for 4 h prior to exposure to MPP+ for 24 h. The amount of cleaved caspase-3 was determined using western blot analysis. d Densitometric analysis of changes in levels of cleaved caspase-3. Data are means ± SEM for three independent experiments. *P < 0.05; **P < 0.01
Neuroprotective Effects of PB Against MPP+-Elicited SH-SY5Y Cell Apoptosis in Mitochondria-Dependent Manner
Previous studies suggested that the mitochondrion was a critical effector of MPP+-induced cell apoptosis. Disruption of the MMP is one of the earliest intracellular events that occur following induction of apoptosis. To evaluate the role of mitochondria in MPP+-induced apoptosis, we investigated its ability to induce alterations in MMP. The drop in fluorescence, indicative of MMP disruption, was measured by fluorescent spectrometer. When cells were treated with 1 mM MPP+ for 24 h, an increase in the percentage of depolarized cells was observed (Fig. 4a). PB dose-dependently attenuated the MPP+-induced collapse of MMP in SH-SY5Y cells. Figure 4b showed that MPP+ markedly decreased Rh123 staining imaged on fluorescence microscope, indicating a drop in MMP which is related to mitochondrial dysfunction. PB (20 μM) pretreatment significantly improved MPP+-induced impairments in MMP.

Mitochondria dependence of neuroprotective effects of PB against MPP+-elicited SH-SY5Y cell apoptosis. a SH-SY5Y cells were treated with indicated concentrations of MPP+ with/without PB for 24 h, and then MMP alteration was measured by fluorescence microplate reader using Rh123 staining. b Representative micrographs of Rh123-derived fluorescence in MPP+ treated cells with or without 20 μM PB were imaged on fluorescence microscope. c Effect of PB on the expression of Bcl-2 and Bax in SH-SY5Y cells. Cells were with MPP+ with/without PB for 24 h, and then cell lysates were subject to western blot analysis. The levels of Bax and Bcl-2 were quantified by densitometric analysis (upper) and the Bcl-2/Bax ratio was determined (lower). d PB inhibits MPP+-induced cytochrome c release. The cells were pretreated with PB for 4 h prior to exposure to 1 mM MPP+ for 24 h. The amounts of cytosolic cytochrome c (C Cyto C) and mitochondrial cytochrome c (M Cyto C) were determined by western blot analyses. Results are shown as the mean ± SEM and represent three independent experiments. **P < 0.01
Next we also detected the expression of Bcl-2 and Bax in the mitochondria-dependent apoptosis. As shown in Fig. 4c, treatment of cells with MPP+ induced an increase in the protein level of Bax and robust decrease in the protein level of Bcl-2, and there was a significant decrease in the ratio of Bcl-2/Bax expression in MPP+ treatment compared with the control using western blot analysis, while PB pretreatment could prevent the MPP+-induced decrease of the Bcl-2/Bax ratio. The effect of PB against MPP+-induced apoptosis may be, at least in part, mediated by regulating of Bcl-2 and Bax expression.
The mitochondria-mediated cell death was assessed by measuring a release of cytochrome c into the cytosol and subsequent activation of caspase-3 (Bras et al. 2005). Next, we investigated the effect of PB on MPP+-induced cytochrome c release. As shown in Fig. 4d, MPP+ significantly increased the release of cytochrome c from mitochondria to the cytosol, and PB (20 μM) pretreatment inhibited the release of cytochrome c.
Discussion
PD is a chronic, progressive, neurodegenerative disease with no effective treatment. Thus, the development of effective neuroprotective drugs is urgently needed. A variety of medicinal plants have long been used in traditional Oriental medicine as crude extracts and mixtures, in order to prevent or alleviate neurological symptoms (Houghton and Howes 2005). These extracts are shown to relieve neurological symptoms in experimental animal models, as well as reducing in vitro activity. The diverse array of bioactive nutrients present in the natural products plays a pivotal role in prevention and cure of various neurodegenerative diseases, such as Alzheimer’s disease, PD, and other neuronal dysfunctions (Essa et al. 2012).
Pinocembrin (5,7-dihydroxyflavanone) is one of the primary flavonoids isolated from the variety of plants, mainly from P. heartwood, Eucalyptus, Populus, Euphorbia, and S. leucanthum, in the diverse flora and purified by various chromatographic techniques. Its vast range of pharmacological activities has been well researched including antimicrobial, anti-inflammatory, antioxidant, anticancer, and most recent neuroprotective activities. The present study demonstrates that PB protects human dopaminergic SH-SY5Y cells against MPP+-induced cytotoxicity in several aspects.
Oxidative damage occurs in the parkinsonian brain (Nagatsu and Sawada 2006), and overproduction of ROS can cause severe impairment of cellular functions, is also involved in apoptotic mechanisms, and may contribute to the apoptotic process found in PD. Both exogenous and endogenous neurotoxic substances, which induced a parkinsonian symptom in vivo and in vitro, are known to provide partial explanation for chronic neuronal degeneration in PD. The early chemical agents used for this purpose, such as 6-hydroxydopamine, were then replaced by MPTP (Kopin 1987; Kopin and Markey 1988). In order to function, MPTP must be converted into the MPP+ ion, which is an inhibitor of complex I of the mitochondrial electron transport chain (Beal 2003). MPP+ is concentrated by the dopamine transporter into dopamine neurons via selective uptake (Pifl et al. 1993). In simplified in vitro systems, MPP+ is used directly. Incubation of neural cells with MPP+ increases oxidative stress (Cassarino et al. 1997) and induces apoptosis (Fall and Bennett Jr 1999). This treatment results in the rapid simultaneous activation of both cell survival and death-promoting signaling pathways (Halvorsen et al. 2002). Recently, the endogenous damaging agent, salsolinol (1-methyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline, SALS), was also shown as toxic to dopaminergic neurons in vitro as well as in vivo. SALS, found for the first time in the urine of patients treated with l-DOPA (Sandler et al. 1973) and subsequently in human brain (Moser et al. 1995; Maruyama et al. 1997), is an endogenous dopamine metabolite that has structural similarity to MPTP. SALS is a more realistic model for selective toxicity to nigral dopaminergic neurons (Storch et al. 2002; Maruyama et al. 2004; Naoi et al. 2004) and mimics the natural course of PD that develops slowly, allowing the brain to adapt to progressive damage (Antkiewicz-Michaluk et al. 2000). SALS leads to neurotoxicity in dopaminergic cells by inhibiting mitochondrial electron transport chain (Storch et al. 2000) and can trigger typical apoptotic dopaminergic cell death (Copeland Jr et al. 2007) through a reactive oxygen species activated cascade (Chun et al. 2001). Therefore, numerous studies have used the SALS-treated cells to investigate the protective effect of some antioxidants to inhibit SALS-induced neurotoxicity (Copeland Jr et al. 2005, 2007; Das and Tizabi 2009; Arshad et al. 2014; Brown et al. 2013).
In the present study, we used MPP+-treated SH-SY5Y cells to investigate the protective effect of PB against MPP+-induced neurotoxicity. MPP+-treated SH-SY5Y cells were chosen as a model system to investigate cell death because these cells exhibit (1) DA neuron characteristics including dopamine synthesis, (2) expression of dopamine receptors, (3) specific uptake and sequestration of dopamine consistent with expression of the dopamine transporter (Biedler et al. 1978; Farooqui 1994; Takahashi et al. 1994), and (4) they can be differentiated into a neuronal phenotype by incubation with retinoic acid (Kaplan et al. 1993). MPP+ has been widely used as a neurotoxin because it elicits a severe Parkinson’s disease-like syndrome with elevation of intracellular ROS level and apoptotic death. Our present results show that SH-SY5Y cells exposed to 1 mM MPP+ significantly increase their production of ROS and that PB treatments suppress the MPP+-induced accumulation of ROS dose-dependently and attenuate MPP+-induced SH-SY5Y death.
Neuronal cell death due to MPP+ is mediated by opening of the mitochondrial permeability transition pore, releases of Ca2+ and cytochrome c, and activation of caspases (Cassarino et al. 1999; Lotharius et al. 1999). Formation of the mitochondrial permeability transition due to exposure of MPP+ seems to be associated with increased oxidative stress (Leist et al. 1998). It is thought that neuronal cell death induced by MPP+ is mediated by the collapse of the MMP and the opening of mitochondrial permeability transition (MPT) pore (Seaton et al. 1997). With the MMP depletion by ROS, the permeability transition pore opened, and intermembrane proteins were released out of the mitochondrial, which in turn activated a downstream executive caspase-3 and cell death. The present data show that PB treatment significantly improved MPP+-induced impairments in MMP (Fig. 4a). After the disruption of MMP, mitochondrial cytochrome c was released, which ultimately cleave pro-caspase-3 to form active caspase-3(Bras et al. 2005). The interplay between pro- and anti-apoptotic Bcl-2 family members may determine the fate of cells by regulating the permeability of mitochondrial membrane and controlling the release of cytochrome c from mitochondria (Yang et al. 1997). Certain lines of evidence suggest that the Bcl-2 plays a significant role in MPP+-induced apoptotic cell death (Blum et al. 2001; Kim et al. 2009). Bcl-2 family members are intimately involved in cell death processes caused by MPP+. Next, we investigated the effect of PB on MPP+-induced Bcl-2/Bax expression. Our results showed that treatment of cells with MPP+ induced an increase in the protein level of Bax and decrease in the protein level of Bcl-2, and there was a significant decrease in the ratio of Bcl-2/Bax expression in MPP+ treatment compared with the control. It ameliorates the MPP+-induced Bcl-2/Bax ratio decrease in SH-SY5Y cells. Therefore, the effect of PB on MPP+-induced apoptosis may be, at least in part, mediated by regulation of Bax and Bcl-2 expression and regulation of Bax and Bcl-2 in an anti-apoptotic mechanism (Fig. 4c).
The opening of the MPT pores causes a release of apoptogenic substances such as cytochrome c from mitochondria into the cytosol (Nicholls and Budd 2000). Cytochrome c release from mitochondria was proven to play a critical role in apoptosis. Using western blot analysis, we investigated the possible effect of PB on MPP+-induced cytochrome c release from mitochondria. Our results showed that an increase in cytochrome c release correlates well with a decrease in the Bcl-2/Bax ratio, as pro-apoptotic Bax is thought to be upstream of cytochrome c release in the mitochondria-mediated apoptosis pathway.
After the disruption of MMP, mitochondrial cytochrome c was released, which ultimately cleave pro-caspase-3 to form active caspase-3. Caspase-3 activation led to DNA breakage, nuclear chromatin condensation, and cell apoptosis. The activation of caspase-3 is also believed to be important for commitment to or execution of neuronal apoptosis. The suppressive effect of PB on caspase-3 activity further suggests that the neuroprotective effect of PB against MPP+-induced cell death is related to its antioxidant effects.
In summary, we document the neuroprotective effects of PB in attenuating MPP+-induced neurotoxicity. One of the most salient features of our present study is that we demonstrate a new pharmacological activity of PB, i.e., neuroprotective activity against MPP+-induced neurotoxicity. PB reduces the toxicity MPP+-induced neurotoxicity in SH-SY5Y cells through an apoptotic pathway by limiting the induction of death signaling proteins, Bax, Bcl-2, and caspase-3 enzymes (Fig. 5).

A proposed pathway for pinocembrin-mediated anti-apoptosis against MPP+. Pinocembrin could inhibit MPP+-induced elevation of intracellular ROS level, inhibit MPP+-induced apoptosis through downregulation of pro-apoptotic protein Bax and the upregulation of anti-apoptotic protein Bcl-2, and inhibit MPP+-induced cytochrome c release, thus to stabilize mitochondrial membrane potential and suppress the induction of caspase-3, thereby inhibiting the MPP+-induced neurotoxicity
References
Ahn KH, Kim YS, Kim SY, Huh Y, Park C, Jeong JW (2009) Okadaic acid protects human neuroblastoma SH-SY5Y cells from 1-methyl-4-phenylpyridinium ion-induced apoptosis. Neurosci Lett 449:93–97
An H, Kim IS, Koppula S, Kim BW, Park PJ, Lim BO, Choi WS, Lee KH, Choi DK (2010) Protective effects of Gastrodia elata Blume on MPP+-induced cytotoxicity in human dopaminergic SH-SY5Y cells. J Ethnopharmacol 130:290–298
Antkiewicz-Michaluk L, Romanska I, Papla I, Michaluk J, Bakalarz M, Vetulani J, Krygowska-Wajs A, Szczudlik A (2000) Neurochemical changes induced by acute and chronic administration of 1,2,3,4-tetrahydroisoquinoline and salsolinol in dopaminergic structures of rat brain. Neuroscience 96:59–64
Arshad A, Chen X, Cong Z, Qing H, Deng Y (2014) TRPC1 protects dopaminergic SH-SY5Y cells from MPP+, salsolinol, and N-methyl-(R)-salsolinol-induced cytotoxicity. Acta Biochim Biophys Sin (Shanghai) 46:22–30
Beal MF (2003) Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci 991:120–131
Biedler JL, Roffler-Tarlov S, Schachner M, Freedman LS (1978) Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res 38:3751–3757
Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol 65:135–172
Bras M, Queenan B, Susin SA (2005) Programmed cell death via mitochondria: different modes of dying. Biochemistry (Mosc) 70:231–239
Brown D, Tamas A, Reglodi D, Tizabi Y (2013) PACAP protects against salsolinol-induced toxicity in dopaminergic SH-SY5Y cells: implication for Parkinson’s disease. J Mol Neurosci 50:600–607
Cassarino DS, Fall CP, Swerdlow RH, Smith TS, Halvorsen EM, Miller SW, Parks JP, Parker WD Jr, Bennett JP Jr (1997) Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson’s disease. Biochim Biophys Acta 1362:77–86
Cassarino DS, Parks JK, Parker WD Jr, Bennett JP Jr (1999) The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim Biophys Acta 1453:49–62
Chiba K, Trevor AJ, Castagnoli N Jr (1985) Active uptake of MPP+, a metabolite of MPTP, by brain synaptosomes. Biochem Biophys Res Commun 128:1228–1232
Chiueh CC, Rauhala P (1998) Free radicals and MPTP-induced selective destruction of substantia nigra compacta neurons. Adv Pharmacol 42:796–800
Chun HS, Gibson GE, DeGiorgio LA, Zhang H, Kidd VJ, Son JH (2001) Dopaminergic cell death induced by MPP(+), oxidant and specific neurotoxicants shares the common molecular mechanism. J Neurochem 76:1010–1021
Copeland RL Jr, Leggett YA, Kanaan YM, Taylor RE, Tizabi Y (2005) Neuroprotective effects of nicotine against salsolinol-induced cytotoxicity: implications for Parkinson’s disease. Neurotox Res 8:289–293
Copeland RL Jr, Das JR, Kanaan YM, Taylor RE, Tizabi Y (2007) Antiapoptotic effects of nicotine in its protection against salsolinol-induced cytotoxicity. Neurotox Res 12:61–69
Das JR, Tizabi Y (2009) Additive protective effects of donepezil and nicotine against salsolinol-induced cytotoxicity in SH-SY5Y cells. Neurotox Res 16:194–204
Dickson DW (2007) Linking selective vulnerability to cell death mechanisms in Parkinson’s disease. Am J Pathol 170:16–19
Eberhardt O, Schulz JB (2003) Apoptotic mechanisms and antiapoptotic therapy in the MPTP model of Parkinson’s disease. Toxicol Lett 139:135–151
Essa MM, Vijayan RK, Castellano-Gonzalez G, Memon MA, Braidy N, Guillemin GJ (2012) Neuroprotective effect of natural products against Alzheimer’s disease. Neurochem Res 37:1829–1842
Fall CP, Bennett JP Jr (1999) Characterization and time course of MPP+-induced apoptosis in human SH-SY5Y neuroblastoma cells. J Neurosci Res 55:620–628
Farooqui SM (1994) Induction of adenylate cyclase sensitive dopamine D2-receptors in retinoic acid induced differentiated human neuroblastoma SHSY-5Y cells. Life Sci 55:1887–1893
Greenamyre JT, MacKenzie G, Peng TI, Stephans SE (1999) Mitochondrial dysfunction in Parkinson’s disease. Biochem Soc Symp 66:85–97
Halvorsen EM, Dennis J, Keeney P, Sturgill TW, Tuttle JB, Bennett JB Jr (2002) Methylpyridinium (MPP(+))- and nerve growth factor-induced changes in pro- and anti-apoptotic signaling pathways in SH-SY5Y neuroblastoma cells. Brain Res 952:98–110
Houghton PJ, Howes MJ (2005) Natural products and derivatives affecting neurotransmission relevant to Alzheimer’s and Parkinson’s disease. Neurosignals 14:6–22
Itano Y, Kitamura Y, Nomura Y (1994) 1-Methyl-4-phenylpyridinium (MPP+)-induced cell death in PC12 cells: inhibitory effects of several drugs. Neurochem Int 25:419–424
Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD (1992) Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann Neurol 32(Suppl):S82–S87
Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ (1993) Induction of TrkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Eukaryotic Signal Transduction Group. Neuron 11:321–331
Kim IS, Koppula S, Park PJ, Kim EH, Kim CG, Choi WS, Lee KH, Choi DK (2009) Chrysanthemum morifolium Ramat (CM) extract protects human neuroblastoma SH-SY5Y cells against MPP+-induced cytotoxicity. J Ethnopharmacol 126:447–454
Kopin IJ (1987) Toxins and Parkinson’s disease: MPTP parkinsonism in humans and animals. Adv Neurol 45:137–144
Kopin IJ, Markey SP (1988) MPTP toxicity: implications for research in Parkinson’s disease. Annu Rev Neurosci 11:81–96
Lee DY, Lee KS, Lee HJ, Noh YH, Kim DH, Lee JY, Cho SH, Yoon OJ, Lee WB, Kim KY, Chung YH, Kim SS (2008) Kynurenic acid attenuates MPP(+)-induced dopaminergic neuronal cell death via a Bax-mediated mitochondrial pathway. Eur J Cell Biol 87:389–397
Leist M, Volbracht C, Fava E, Nicotera P (1998) 1-Methyl-4-phenylpyridinium induces autocrine excitotoxicity, protease activation, and neuronal apoptosis. Mol Pharmacol 54:789–801
Liu R, Wu CX, Zhou D, Yang F, Tian S, Zhang L, Zhang TT, Du GH (2012) Pinocembrin protects against beta-amyloid-induced toxicity in neurons through inhibiting receptor for advanced glycation end products (RAGE)-independent signaling pathways and regulating mitochondrion-mediated apoptosis. BMC Med 10:105
Lotharius J, O’Malley KL (2000) The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J Biol Chem 275:38581–38588
Lotharius J, Dugan LL, O’Malley KL (1999) Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci Off J Soc Neurosci 19:1284–1293
Maruyama W, Sobue G, Matsubara K, Hashizume Y, Dostert P, Naoi M (1997) A dopaminergic neurotoxin, 1(R), 2(N)-dimethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline, N-methyl(R)salsolinol, and its oxidation product, 1,2(N)-dimethyl-6,7-dihydroxyisoquinolinium ion, accumulate in the nigro-striatal system of the human brain. Neurosci Lett 223:61–64
Maruyama W, Yi H, Takahashi T, Shimazu S, Ohde H, Yoneda F, Iwasa K, Naoi M (2004) Neuroprotective function of R-(−)-1-(benzofuran-2-yl)-2-propylaminopentane, [R-(−)-BPAP], against apoptosis induced by N-methyl(R)salsolinol, an endogenous dopaminergic neurotoxin, in human dopaminergic neuroblastoma SH-SY5Y cells. Life Sci 75:107–117
Moser A, Scholz J, Nobbe F, Vieregge P, Bohme V, Bamberg H (1995) Presence of N-methyl-norsalsolinol in the CSF: correlations with dopamine metabolites of patients with Parkinson’s disease. J Neurol Sci 131:183–189
Nagatsu T, Sawada M (2006) Cellular and molecular mechanisms of Parkinson’s disease: neurotoxins, causative genes, and inflammatory cytokines. Cell Mol Neurobiol 26:781–802
Naoi M, Maruyama W, Nagy GM (2004) Dopamine-derived salsolinol derivatives as endogenous monoamine oxidase inhibitors: occurrence, metabolism and function in human brains. Neurotoxicology 25:193–204
Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80:315–360
Pieczenik SR, Neustadt J (2007) Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol 83:84–92
Pifl C, Giros B, Caron MG (1993) Dopamine transporter expression confers cytotoxicity to low doses of the parkinsonism-inducing neurotoxin 1-methyl-4-phenylpyridinium. J Neurosci 13:4246–4253
Przedborski S, Jackson-Lewis V (1998) Mechanisms of MPTP toxicity. Mov Disord 13(Suppl 1):35–38
Ramsay RR, Salach JI, Dadgar J, Singer TP (1986) Inhibition of mitochondrial NADH dehydrogenase by pyridine derivatives and its possible relation to experimental and idiopathic parkinsonism. Biochem Biophys Res Commun 135:269–275
Rasul A, Millimouno FM, Ali EW, Ali M, Li J, Li X (2013) Pinocembrin: a novel natural compound with versatile pharmacological and biological activities. Biomed Res Int 2013:379850
Sandler M, Carter SB, Hunter KR, Stern GM (1973) Tetrahydroisoquinoline alkaloids: in vivo metabolites of l-dopa in man. Nature 241:439–443
Seaton TA, Cooper JM, Schapira AH (1997) Free radical scavengers protect dopaminergic cell lines from apoptosis induced by complex I inhibitors. Brain Res 777:110–118
Singer TP, Ramsay RR (1990) Mechanism of the neurotoxicity of MPTP. An update. FEBS Lett 274:1–8
Sriram K, Pai KS, Boyd MR, Ravindranath V (1997) Evidence for generation of oxidative stress in brain by MPTP: in vitro and in vivo studies in mice. Brain Res 749:44–52
Storch A, Kaftan A, Burkhardt K, Schwarz J (2000) 1-Methyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (salsolinol) is toxic to dopaminergic neuroblastoma SH-SY5Y cells via impairment of cellular energy metabolism. Brain Res 855:67–75
Storch A, Ott S, Hwang YI, Ortmann R, Hein A, Frenzel S, Matsubara K, Ohta S, Wolf HU, Schwarz J (2002) Selective dopaminergic neurotoxicity of isoquinoline derivatives related to Parkinson’s disease: studies using heterologous expression systems of the dopamine transporter. Biochem Pharmacol 63:909–920
Takahashi T, Deng Y, Maruyama W, Dostert P, Kawai M, Naoi M (1994) Uptake of a neurotoxin-candidate, (R)-1,2-dimethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline into human dopaminergic neuroblastoma SH-SY5Y cells by dopamine transport system. J Neural Transm Gen Sect 98:107–118
Wang H, Xu Y, Yan J, Zhao X, Sun X, Zhang Y, Guo J, Zhu C (2009) Acteoside protects human neuroblastoma SH-SY5Y cells against beta-amyloid-induced cell injury. Brain Res 1283:139–147
Wang HQ, Sun XB, Xu YX, Zhao H, Zhu QY, Zhu CQ (2010) Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against beta-amyloid-induced cytotoxicity in SH-SY5Y cells. Brain Res 1360:159–167
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275:1129–1132
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (81260196 to HW; 81201844 to JZ), Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (NJYT-13-B20) to HW, Scientific Research Projects in Universities of Inner Mongolia Autonomous Region (NJSZ12306) to HW, and Beijing Natural Science Foundation Proposed Program (7132137) to JG.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Y., Gao, J., Miao, Y. et al. Pinocembrin Protects SH-SY5Y Cells Against MPP+-Induced Neurotoxicity Through the Mitochondrial Apoptotic Pathway. J Mol Neurosci 53, 537–545 (2014). https://doi.org/10.1007/s12031-013-0219-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-013-0219-x