
1. Parkinson’s disease (PD) is considered to be an aging-related neurodegeneration of catecholamine (CA) systems [typically A9 dopamine (DA) neurons in the substantia nigra and A6 noradrenaline (NA) neurons in the locus coeruleus]. The main symptom is movement disorder caused by a DA deficiency at the nerve terminals of fibers that project from the substantia nigra to the striatum. Most PD is sporadic (sPD) without any hereditary history. sPD is speculated to be caused by some exogenous or endogenous substances that are neurotoxic toward CA neurons, which toxicity leads to mitochondrial dysfunction and subsequent oxidative stress resulting in the programmed cell death (apoptosis or autophagy) of DA neurons.
2. Recent studies on the causative genes of rare familial PD (fPD) cases, such as alpha–synuclein and parkin, suggest that dysfunction of the ubiquitin–proteasome system (UPS) and the resultant accumulation of misfolded proteins and endoplasmic reticulum stress may cause the death of DA neurons.
3. Activated microglia, which accompany an inflammatory process, are present in the nigro-striatum of the PD brain; and they produce protective or toxic substances, such as cytokines, neurotrophins, and reactive oxygen or nitrogen species. These activated microglia may be neuroprotective at first in the initial stage, and later may become neurotoxic owing to toxic change to promote the progression toward the death of CA neurons.
4. All of these accumulating evidences on sPD and fPD points to a hypothesis that multiple primary causes of PD may be ultimately linked to a final common signal-transduction pathway leading to programmed cell death, i.e., apoptosis or autophagy, of the CA neurons.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Parkinson’s disease (PD) is assumed to be a systemic neurodegenerative disease of the catecholamine (CA) neurons. The neurons mainly affected are the A9 dopamine (DA) neurons in the substantia nigra and the A6 noradrenaline (NA) neurons in the locus coeruleus, both of which contain neuromelanin. PD is the second most common aging-related neurodegenerative disease after Alzheimer’s disease (AD). The main symptom of PD is a movement disorder called parkinsonism, i.e., muscle rigidity, akinesia, and resting tremor, symptoms which are caused by a DA deficiency in the striatum due to degeneration of the A9 DA neurons in the substantia nigra. A small percentage of PD is familial (fPD), but most PD is sporadic (sPD) without any hereditary history and is related to aging. A deficiency of DA in the striatum in PD patients was predicted by Carlsson (1959) when he discovered DA as a new neurotransmitter, and this prediction was confirmed in postmortem brains by Ehringer and Hornykiewicz (1960). DA supplementation therapy by L-DOPA was also predicted by Carlsson from the results of animal experiments (1959); and after many clinical trials by several groups in the world, L-DOPA is now established as the gold standard of drug therapy for PD. sPD and some cases of fPD are characterized by the presence of intracytoplasmic eosinophilic inclusions called Lewy bodies in the neurons and glial cells of the affected individuals. Lewy bodies consist mainly of alpha-synuclein protein, which is encoded by the causative gene of fPD/PARK1 (familial Parkinson’s disease 1), and are observed not only in PD but also in dementia with Lewy bodies (DLB), a disorder also called diffuse Lewy body disease (DLBD; Kosaka, 2000), and in other neurodegenerative diseases. Thus, such neurodegenerative diseases with Lewy bodies are generally referred to synucleinopathies. Lewy bodies are frequently observed in the surviving DA and NA neurons of the substantia nigra and locus coeruleus of the PD brain. Braak et al. (2003) recently proposed, based on anatomical investigation of Lewy bodies, that the pathological process of sPD starts from the lower brain stem and spreads throughout the brain to the midbrain, limbic brain, and cerebral cortex and that the main symptom of PD, i.e., movement disorder, appears at the late stage when the nigro-striatal DA neurons in the midbrain become involved. In this theory, movement disorder parkinsonism in PD is speculated to be a late symptom. Lewy bodies are also observed not only in CA neurons but also in non-CA neurons such as acetylcholine, amino acid, or peptide neurons in the central and peripheral nervous systems, i.e., in neurons of the anterior olfactory nucleus, dorsal nucleus of the vagus nerve, peripheral autonomic nervous system including sympathetic ganglia, adrenal medulla, and parasympathetic ganglia, and intestinal Auerbach’s plexus. In relation to this wide distribution of Lewy bodies in sPD, various non-DA symptoms, e.g., REM sleep behavior disorder (RBD) (Abott, 2005), olfaction disturbance, neurocircuitry abnormalities due to cardiac sympathetic denervation (Goldstein et al., 2005), or constipation, have been noticed as early signs of sPD before the appearance of parkinsonism.
The pathogenesis of sPD is still enigmatic (Foley and Riederer, 1999), but free radicals produced by mitochondrial dysfunction and oxidative stress are thought to play an important role (Youdim and Riederer, 1997; Mizuno et al., 1998; Schapira et al., 1998). Mitochondrial dysfunction in sPD is supported by the findings on complex I (NADH-ubiquinone reductase complex, one of the five enzyme complexes of the inner mitochondrial membrane involved in oxidative phosphorylation) deficiency in the nigro-striatum of the postmortem brain from sPD patients and of complex I inhibition in the substantia nigra of animal PD models produced by treatment with neurotoxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP; Langston et al., 1983) or the insecticide rotenone (Betarbet et al., 2000). The discovery of MPTP causing PD in humans suggests the neurotoxin hypothesis, in which MPTP-like exogenous or endogenous neurotoxins acting together with presumed PD-susceptibility genes are assumed to be the cause of sPD.
On the other hand, recent molecular genetical studies on mutations of the causative genes of autosomal dominant or recessive fPD, especially alpha-synuclein for autosomal dominant and Lewy body-positive PARK1 (Polymeropoulos et al., 1997) and parkin for autosomal recessive and Lewy body-negative juvenile PARK2 (Kitada et al., 1998), have led to a new hypothesis, that fPD is caused by the accumulation and/or aggregation of misfolded proteins due to dysfunction of the ubiquitin–proteasome system (UPS; Table I). The discovery of the causative genes of fPD may also give important clues for elucidating the signaling pathway of cell death in sPD (for reviews, see Bonifati et al., 2004; Feany, 2004; Forman et al., 2004; Cookson, 2005; Krueger, 2004; Selko, 2004; Vila and Przedborski, 2004; Grandhi and Wood, 2005; Lozano and Kalia, 2005; Mizuno, 2006).
As another mechanism of neuronal death in PD, an inflammatory process in the brain called neuroinflammation, which is accompanied by changes in the levels of neuroprotective or neurotoxic cytokines and neurotrophins and the presence of activated microglia, has recently gained much attention with respect to not only PD, but also AD and other neurodegenerative diseases (for reviews, see McGeer and McGeer, 1995; Anglade et al., 1997; Hirsch et al., 1999; Mogi and Nagatsu, 1999; Nagatsu et al., 1999, 2000a,b; Hartmann et al., 2000; Jellinger, 2000; Nagatsu, 2002a; Hayley, 2005; Herrera et al., 2005; Nagatsu and Sawada, 2005). The brain is considered to be an immune privileged site, i.e., one free from immune reactions, since it is protected by the blood–brain-barrier. However, accumulating findings have revealed that immune responses may occur in the brain, especially due to activation of the microglia, which cells are known to produce pro-inflammatory cytokines. This inflammatory process is now thought to be fundamental to, if not at first the initiator of, the progression of PD pathogenesis. Not only DA neurons but also other non-DA neurons may be affected by this process, whose dysfunction may negatively impact DA and non-DA pathways in the PD patients.
Herein, we will review advances in our understanding of the cellular and molecular pathogenesis of PD by separately focusing on these three groups of causative factors, i.e., neurotoxins, causative genes, and inflammatory cytokines.
NEUROTOXIC SUBSTANCES PRODUCING PD IN HUMANS AND ANIMALS
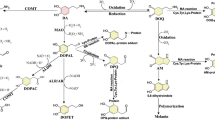
The cell death of A9 DA neurons in sPD may be caused by both genetic factors and environmental factors. As to the latter, certain neurotoxins are speculated to cause sPD. This neurotoxin hypothesis of PD started in the 1980s from the discovery of MPTP, which produces PD in humans (Davis et al., 1979; Langston et al., 1983). Since then, environmental or endogenous neurotoxins similar to MPTP in the brain and/or cerebrospinal fluid (CSF) of PD patients have been investigated, and two groups of amine-related compounds, i.e., isoquinolines and beta-carbolines, have been suggested as candidates of such neurotoxins (for review, see Nagatsu, 1997; Nagatsu, 2002b).
MPTP, an analogue of meperidine (a synthetic heroin) is a highly lipophilic precursor neurotoxin. After its systemic administration, it rapidly crosses the blood–brain barrier to enter the brain. Once in the brain, MPTP, which is a proneurotoxin, is metabolized to N-methyl-4-pheny-2,3-dihydropyridinium (MPDP+) by the enzyme monoamine oxidase B (MAO B), which is localized in the outer membrane of mitochondria within non-DA cells such as glial cells and serotonin neurons. MPDP+ is then probably spontaneously oxidized to 1-methyl-4-phenylpyridinium (MPP+), the active neurotoxin. MPP+ is taken up across the cell membrane to enter into the A9 nigro-striatal DA neurons via DA transporters, which are mostly localized at the nerve terminals in the striatum. MPP+ then passes from the cytoplasm of these neurons into synaptic vesicles by the action of vesicular monoamine transporter type 2 (VMAT2). MPP+ also accumulates within the inner mitochondrial membrane, where it inhibits complex I; interrupts electron transport; releases reactive oxygen species (ROS); and depletes ATP. Inhibition of the mitochondrial complex I also results in the opening of mitochondrial permeability transition pores, allowing the release of cytochrome c, which may trigger the signal transduction pathway culminating in apoptotic cell death. As a consequence of these actions, MPP+ decreases the DA level in the striatum, resulting in the appearance of PD-like symptoms. MPTP produces cell death specifically in A9 nigro-striatal DA neurons. Selegiline (l-deprenyl), which is a specific inhibitor of MAO B, completely blocks the neurotoxicity of MPTP by preventing the conversion of MPTP to MPP+ in glial cells. This may suggest that MAO B allele activity could play an important genetic component of sPD. Humans and monkeys are the most susceptible to MPTP, but mice and other non-primate animals also produce parkinsonism-like movement disorder in response to MPTP. In MPTP-induced animal models of PD, typical cytoplasmic inclusions such as the Lewy bodies found in sPD are not observed. However, chronic infusion of mice with MPTP via an osmotic minipump caused the appearance of alpha-synuclein- and ubiquitin-positive aggregates that looked like Lewy bodies (Fornai et al., 2005).
As a group of MPTP-like neurotoxins, the following isoquinolines have been identified by gas chromatography–mass spectrometric analysis of postmortem brain specimens and/or CSF from PD patients and normal controls: tetrahydroisoquinoline (TIQ), 1-methyl(Me)-TIQ, 2-Me-TIQ, N-Me-6,7-(OH)2-TIQ (N-Me-norsalsolinol), R-1,N-(Me)2-6,7-(OH)2-TIQ (R-N-Me-salsolinol), 1-phenyl-TIQ, N-Me-1-phenyl-TIQ, and 1-benzyl-TIQ (Kajita et al., 2002; Kotake et al., 1995; Moser and Koempf, 1992; Naoi et al., 1996; Niwa et al., 1987; Ohta et al., 1987). TIQs are also found in various foods in small concentrations. Exogenously administered TIQ easily crosses the blood–brain barrier and passes into the brain, although TIQs are metabolized in the liver to 4-OH-TIQs by the action of debrisoquine hydroxylase (P-450 CYP2D6). On the other hand, TIQs in the brain are also speculated to be synthesized from endogenous amines such as phenylethylamine or DA by various enzymes. However, the synthetic pathway and the enzymes for the biosynthesis of TIQs (versus beta-carbolines described below which arise from tryptophan metabolism) have not yet determined. Among these TIQs in the brain, 2-Me-TIQ (Fukuda, 1994), R-N-Me-salsolinol (Naoi et al., 1996), N-Me-norsalsolinol (Moser and Koempf, 1992), and 1-Bn-TIQ (Kotake et al., 1995) have gained much attention as neurotoxins that probably cause PD. R-N-Me-salsolinol, after its stereotaxic injection into the striatum of rats, induces behavioral changes similar to those seen in PD and is metabolized to 1,N-(Me)2-6,7-(OH)2-isoquinolinium in the substantia nigra, similar to the MPTP/MPP+ conversion (Naoi et al., 1996). Among TIQs, 1-Me-TIQ is unique in that its concentration is not increased but actually decreased in the striatum of PD patients and that it prevented PD-like behavior abnormalities produced by MPTP, TIQ, and 1-Bn-TIQ (Tasaki et al., 1991).
Beta-carbolines have structures similar to those of MPTP/MPP+, and may be synthesized in vivo from tryptophan via tryptamine (Collins and Neafsey, 2000; Matsubara, 2000). Like MPTP, beta-carbolines may be precursor neurotoxins that are N-methylated and oxidized by MAO B to form, in their case, beta-carbolinium ions, which may trigger apoptotic neuronal death and PD symptoms. Norharman, harman, 2-Me-norharmanium, and 2,9-(Me)2-norharmanium were found in the human brain and/or the CSF. A neurotoxic 2,9-dimethylated beta-carbolinium ion (2,9-(Me)2- norharmanium) was found in half of the PD, but not at all in non-PD, patients examined (Matsubara et al., 1995; Kuhnet al., 1996). These results suggest that beta-carbolinium compounds as well as isoquinolinium compounds may also be candidate neurotoxins that produce PD.
Endogenous isoquinolines and beta-carbolines in the brain, as in the case of MPTP, are speculated to be first N-methylated and then oxidized by MAO to the corresponding isoquinolinium ions or beta-carbolinium ions in glial cells. The mechanisms of the DA cell death caused by PD-producing neurotoxins, i.e., MPTP/MPP+, isoquinoline/isoquinolinium, or beta-carboline/beta-carbolinium, may be similar, acting to inhibit mitochondrial complex I, to induce ROS formation, and ATP depletion, and finally to cause apoptotic cell death of DA neurons.
Rotenone is a naturally occurring, lipophilic compound isolated from roots of certain plants (Derris species), and is used as the main component of many insecticides. Rotenone is not structurally related to amines, but is a specific inhibitor of mitochondrial complex I. Betarbet et al. (2000) reported that in Lewis rats, chronic, systemic inhibition of complex I by rotenone caused highly selective degeneration of the A9 nigro-striatal DA neurons with behavioral PD symptoms of hypokinesia and rigidity. Important morphological findings in these rotenone-treated rats were that the nigro-striatal DA neurons had accumulated fibrillar cytoplasmic inclusions containing ubiquitin and alpha-synuclein, similar to the Lewy bodies seen in human sPD. It was reported that the primary mechanism underlying the toxic effect of rotenone was a significant increase in O2 − generation that causes damage to mitochondrial complexes I and II, presumably at the level of the 4Fe–4S clusters (Panov et al., 2005).
All the properties of the afore-mentioned neurotoxins indicate that inhibition of complex I, resulting in ROS formation, oxidative stress, and ATP depletion in DA neurons may trigger the pathway of apoptotic cell death in these cells. In agreement with this neurotoxin-based hypothesis, a selective defect in complex I of the mitochondria in the nigro-striatal DA neurons from the postmortem brain of sPD patients was reported by several laboratories (Youdim and Riederer, 1997; Mizuno et al., 1998; Shapira et al., 1998). However, it should be noted that the development of PD has been confirmed in humans only in the case of MPTP, a synthetic neurotoxin. That is, the toxicity of the afore-mentioned endogenously identified neurotoxin candidates has been proved only in animals.
6-Hydroxydopamine (6-OHDA) is a neurotoxin specific for DA neurons in vitro and in vivo (Kostrzewa and Jacobowitz, 1974). It is a very unstable compound and is easily oxidized to produce ROS, which may kill DA neurons via apoptosis. Although 6-OHDA is formed from DA in vitro, it is believed not to be formed in vivo. 6-OHDA does not cross the blood–brain barrier. Thus, it is widely used to produce hemiparkinsonian animal models by stereotaxic injection of it directly into the nigro-striatal region.
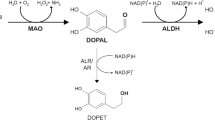
Since DA (or NA) is easily oxidized to form DA quinones and other reactive oxygen species, toxic metabolites of DA such as DA quinones (in the case of NA, NA quinones), which are intermediates of neuromelanin biosynthesis, or 3,4-dihydroxyphenylacetaldehyde (DOPAL; in the case of noradrenaline, 3,4-dihydroxyphenylethyleneglycolaldehyde, DOPEGAL) formed by MAO are postulated to play a role as endogenous neurotoxin candidates in PD (Eisenhofer et al, 2002). DA quinones may be formed either non-enzymatically or by the action of tyrosinase. Tyrosinase has been implicated in DA quinone and neuromelanin formation in the brain. However, we did not detect tyrosinase protein in the A9 nigral DA neurons in the human brain by using antibodies against human tyrosinase (Ikemoto et al., 1998). DOPAL was identified in the PD brain, but not in the normal brain, by gas chromatography–mass spectrometry (Mattammal et al., 1993), and was suggested to be toxic for DA neurons in vitro (Mattammal et al., 1995). However, DA depletion in DA-deficient mice did not protect against acute MPTP toxicity in vivo, suggesting that DA does not contribute to this toxicity in vivo (Hasbani et al., 2005).
Since iron exists in a high concentration in the basal ganglia and interacts with neuromelanin and DA, it is suggested to play a role in the formation of ROS and in the initiation of neurodegeneration of DA neurons by oxidative stress (Mochizuki et al., 1993; Gerlach et al., 1994).
All the effects of exogenous or endogenous neurotoxin candidates suggest that mitochondrial dysfunction and oxidative stress are important for the pathogenesis of sPD. Inhibition of complex I results in enhanced production of ROS, which in turn inhibits complex I. Thus, the vicious cycle resulting from even partial inhibition of complex I in DA neurons may lead to excessive stress and an ATP deficit that ends in cell death (Tretter et al., 2004).
CAUSATIVE GENES OF FAMILIAL PD (PARK)
A small percentage (approximately 5%) of PD cases are familial with a hereditary history (fPD; Table I). Several causative genes of fPD, which mutations produce parkinsonism, and their chromosomal localization have recently been identified: PARK1 (alpha-synuclein), PARK2 (parkin), PARK4 (triplication of alpha-synuclein), PARK5 (UCHL1), PARK6 (PINK1), PARK7 (DJ-1), and PARK8 (LRRK2) (for reviews, see Chiba-Falek and Nussbaum, 2003; Feany, 2004; Forman et al., 2004; Selkoe, 2004; Cookson, 2005; Grandhi and Wood, 2005; Lozano and Kalia, 2005; Mizuno, 2006).
In 1997, a causative mutation of fPD was first identified in the PARK1 gene, encoding the protein alpha-synuclein in autosomal dominant Italian and Greek families (Polimeropoulos et al., 1997). Alpha-synuclein is a small (144 amino acids), presynaptic protein and probably plays a role in signaling between neurons (Goedert, 2001). As a finding on the physiological role of alpha-synuclein, Chandra et al. (2005) reported that alpha-synuclein in conjunction with CSP-alpha (cystein-string protein alpha) has a powerful in vivo activity in protecting nerve terminals against injury in mice. CSP-alpha is a synaptic vesicle protein, essential for neural survival, has a cochaperon function, and may prevent the accumulation of nonnative, potentially toxic molecules during the continuous operation of a nerve terminal.
Not only mutations in alpha-synuclein such as a single amino acid substitution A30P or A53P in PARK1, but also triplication of the wild-type alpha-synuclein gene also causes autosomal dominant PARK4. Alpha-synuclein is a major protein component of Lewy bodies (Spillantini et al., 1998), and thus may play an important role in both fPD and sPD. Alpha-synuclein is a natively unfolded soluble protein and has a central hydrophobic region and a high potency to aggregate to form oligomers or protofibrils and ultimately insoluble polymers or fibrils under certain conditions. The protofibrillar intermediates are toxic in neurons (Conway et al., 2000). Alpha-synuclein fibrils cause mitochondrial complex I deficiency (Sherer et al., 2003) and oxidative stress (Ischiropoulos and Beckman, 2003). Thus, alpha-synuclein may elicit a pathogenetic mechanism similar to that acting in sPD.
Drosophila (fruit fly) models based on overexpression of normal and mutant forms of the alpha-synuclein gene show selective loss of DA neurons and the formation of alpha-synuclein inclusions (Feany and Bender, 2000). Experiments using this model confirmed that phosphorylation at the Ser 129 residue is crucial to the toxicity of alpha-synuclein and that mutations of this serine residue abolishes the toxicity. The reduction in toxicity in this model is associated with increased inclusion body formation, which suggests that inclusion bodies may protect neurons by reducing the amount of diffusible toxic protein by sequestering it in inert bodies (Chen and Feany, 2005). All these findings on alpha-synuclein suggest that the misfolding of the protein is the key steps in mediating degeneration of DA neurons in both fPD and sPD. Alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration, which also suggests a close correlation between alpha-synuclein and the pathogenesis of sPD (Martin et al., 2006).
In PARK2 the gene encoding parkin protein, which was discovered in a Japanese family, is the most common mutant gene in fPD (Kitada et al., 1998). The parkin gene is very large, about 1.4 Mb; and mutations of it are responsible for most of the cases of autosomal recessive juvenile (young-onset) PD. The parkin protein is an E3 ubiquitin ligase with two characteristic RING finger domains separated by an IBR (in-between ring) domain (Shimura et al., 2000). There is a notable absence of Lewy bodies in patients with the homozygous deletion of parkin, although these bodies are present in patients with compound heterozygous parkin mutations. These findings suggest that parkin plays a significant role in Lewy body formation and that nigral cell loss and parkinsonism can occur in the absence of Lewy bodies. The identification of mutations of the parkin gene in PARK2 suggests that dysfunction of the UPS due to loss of function has an important role in PD and that the parkin gene may play a protective role. Ubiquitin is added to proteins by the action of E3 ubiquitin ligase to target them to the proteasome, a large multiprotein complex that functions to degrade most ubiquitin-marked cellular proteins. Parkin mutations cause the accumulation of parkin substrates, which probably contributes to the death of DA neurons. Many putative parkin substrates have been identified, including synphilin-1, 22-kDa O-glycosilated form of alpha-synuclein (alphaSp22; Shimura et al., 2001), Pael-R (parkin-associated endothelin receptor-like receptor; Imai et al., 2001), CHIP, Cdc-Rel1A, cyclin E, and synaptotagmin XI. Overexpression of the parkin substrate Pael-R produces DA cell death in vitro, which cells can be rescued by parkin overexpression (Yang et al., 2003).
Parkin has been shown to be S-nitrosylated in vitro and in vivo in the MPTP mouse model of PD and in brains of patients with PD or LBD, and both neuron- and microglia-derived NO contributes to the S-nitrosylation of parkin in a biphasic fashion after MPTP intoxication. S-Nitrosylation inhibits the E3 ubiquitin ligase activity of parkin and thus its protective function (Chung et al., 2004).
Synphilin-1, a substrate of parkin, was shown to interact with alpha-synuclein and to promote the formation of cytosolic inclusions. Synphilin-1 also interacts with E3 ubiquitin ligase SIAH (seven in absentia homologues)-1 and SIAH-2. SIAH proteins ubiquitinate synphilin-1, promoting its degradation by UPS, and may play a role in inclusion formation, since SIAH immunoreactivity was demonstrated in Lewy bodies in PD patients (Liani et al., 2004). MAO inhibitor deprenyl (selegiline) was reported to act for protection of DA neurons by binding to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Tatton et al., 2003). Snyder (2005) recently reported that an apoptotic stimulus turns on inducible NO synthase with its product NO causing S-nitrosylation of GAPDH in DA neurons. This modification allows GAPDH to bind to the E3 ubiquitin ligase SIAH, which transports GAPDH to the nucleus, leading to apoptotic cell death. They also showed that MAO B inhibitor deprenyl binds to SIAH protein, thereby preventing translocation of the GAPDH–SIAH complex from the cytoplasm to the nucleus and thus preventing apoptotic cell death.
PARK5 is another autosomal dominant fPD. The gene encodes UCHL1 (ubiquitin C-terminal hydrolase L1), which generates free ubiquitin and aids the recycling of polyubiquitin chains back to monomeric ubiquitin (Leroy et al., 1998). UCLH1 can also exert a ubiquitin ligase activity. The discovery of the UCHL1 gene further supports the importance of the role of UPS in the pathogenesis of PD.
The UPS may be important not only in fPD (PARK1, PARK2, PARK4, and PARK5) but also in sPD. Postmortem brain tissues from sPD patients show functional deficits in their 20S proteasome activity (Chung et al., 2001). Also, systemic administration of inhibitors of the UPS to rodents produces selective nigral cell loss and Lewy body-like inclusions, which are accompanied by clinical signs of parkisonism (McNaught et al., 2004).
PARK6 is another autosomal recessive fPD, and is caused by a mutation in a mitochondrial protein kinase called PINK1 [PTEN (phosphatase and tensin Romolog deleted on chromosome ten)-induced kinase-1; Valente et al., 2004)]. PINK1 is the first nucleus-encoded mitochondrial protein to be implicated in the pathogenesis of fPD. The PINK1 gene encodes a serine/threonine protein kinase with significant homology to the calcium/calmodulin-dependent protein kinase. Neuroblastoma cells transiently transfected with either wild-type or mutant PINK1 do not show any detectable alterations in viability. In contrast, when these cells are challenged with a proteasome inhibitor, MG132, overexpression of the wild-type PINK1 mitigates cell death; whereas, overexpression of mutant PINK1 neither attenuates nor enhances MG132-mediated cytotoxicity (Valente et al., 2004). These results suggest that the loss of PINK1 function renders DA neurons more vulnerable to injury. Transient knockdown of PINK1 renders cells susceptible to apoptosis on exposure to taxol (MacKeigan et al., 2005). This neuroprotective function and the mitochondrial localization of PINK1 suggest its probable important role in mitochondria also in sPD.
DJ-1 is the causative gene of autosomal recessive early-onset PARK7 (Bonifati et al., 2003). DJ-1 is a homodimeric and multifunctional protein, ubiquitously expressed in human tissues; and it plays essential roles in tissues with higher-order biological functions such as the testis and brain. DJ-1 is related to male fertility, and its level in sperm is decreased in response to exposure to sperm toxicants. DJ-1 was discovered as a novel mitogen-dependent oncogene product involved in a Ras-related signal transduction pathway (Nagakubo et al., 1997). DJ-1 is up-regulated after oxidative stress and may play a role as an antioxidant protein and a sensor for oxidative stress. The crystal structure of DJ-1 indicates that the protein is structurally similar to a cysteine protease and may induce conformational changes to acquire its catalytic activity in response to oxidative stress (Honbou et al., 2003). The function of DJ-1 as an anti-oxidant protein again suggests its pathogenetic role also in sPD.
The gene for the LRRK2 (leucine-rich repeat kinase 2) was identified as the PARK8 locus (Paisan-Ruiz et al., 2004; Zimprich et al., 2004). Mutations in LRRK2 cause autosomal dominant PD with a broad spectrum of neuropathological features, such as neuronal loss in the substantia nigra either in the absence or in the widespread presence of Lewy bodies or in the presence of neurofibrillary tangles. The affected families originated from Italy, Portugal, and Brazil, indicating the presence of this mutation in different populations. The associated phenotype is broad, including early and late disease onset (Di Fonzo et al., 2005). The LRRK2 gene encodes a 286-kDa protein that is a member of a novel family of protein kinases called “dardarin” (meaning “tremor” in the Basque region, from where some of the affected patients came). Dardarin contains leucine-rich repeats and a Ras/small GTPase superfamily domain, a tyrosine kinase-like domain, and the WD40 domains with sequence similarity to both tyrosine and serine/threonine kinases (Shen, 2004). The presence of these novel domains in dardarin suggests a unique and new function for this kinase in the survival of nigral DA neurons.
Mutations in NR4A2 have also been found to be significantly associated with fPD (Le et al., 2003). The NR4A2 gene (also called Nurr1; i.e., nuclear receptor-related 1) encodes a transcription factor that belongs to the steroid/thyroid hormone receptor superfamily. Alternative splicing and selective use of transcription initiation sites control the expression of the human Nurr1 gene (Ichinose et al., 1999a,b). Interestingly, Nurr1 is essential for the differentiation of the nigral DA neurons and is closely related to the expression and function of the DA system. The Nurr1 gene activates expression of tyrosine hydroxylase (TH; Iwawaki et al., 2000), and also enhances transcription of the DA transporter (Sacchetti et al., 2001). NR4A2 (+/−) mice have a parkinsonian-like phenotype and are more susceptible than the wild type to MPTP (Warbt et al., 2003). Although the mechanism is not yet clear, NR4A2 is thought to be a susceptibility gene for sPD.
DA deficiency due to mutations of the genes of the enzymes involved in DA biosynthesis, i.e., DA-synthesizing-enzyme TH or its cofactor tetrahydrobiopterin (BH4)-synthesizing enzyme GTP cyclohydrolase I (GCH), causes DOPA-responsive dystonia, parkinsonism in infancy or progressive infantile encephalopathy with L-DOPA-nonresponsive dystonia, depending upon the degree of DA deficiency (Hoffmann et al., 2003; Segawa et al., 2003; Kobayashi and Nagatsu, 2005). Autosomal dominant GCH deficiency, which was first described by Segawa and thus called Segawa’s disease, is a DOPA-responsive dystonia caused by a partial decrease of the activity of GCH due to a mutation of one of its alleles (Ichinose et al., 1994, 1995, 1999; Nagatsu and Ichinose, 1999; Segawa et al., 2003). Segawa’s disease is a partial DA deficiency without any DA cell death and the symptom is completely controllable by L-DOPA administration. In contrast, PARK2 or autosomal recessive juvenile PD is initially similar to DOPA-responsive dystonia but progresses to parkinsonism, and is accompanied by DA cell death.
fPDs indicate the importance of the dysfunction of UPS and protein misfolding in the pathogenesis of PD. Overexpression of alpha-synuclein in mice and rats leads to the development of mitochondrial degeneration and produces DA cell death similarly as in sPD (Yamada et al., 2004; Martin et al., 2006). On the other hand, mitochondrial dysfunction in sPD also causes the dysfunction of UPS due to ATP deficiency. The above genes involved in fPD may be susceptibility genes for sPD. It has been reported that DA covalen1tly modifies and functionally inactivates parkin, suggesting a vulnerability of parkin to modification by DA and a mechanism for the progressive loss of the neuroprotective parkin function in DA neurons during aging and sPD (LaVoie et al., 2005). Thus, fPD and sPD are different in their primary causes, but may ultimately produce nigral DA cell death by a final common pathway.
CHANGES IN CYTOKINES PRODUCED BY ACTIVATED MICROGLIA DURING THE NEUROINFLAMMATORY PROCESS IN PD
Elevated Pro-Inflammatory Cytokine Expression in the Presence of Activated Microglia in the Nigro-Striatal Region in sPD
We and others have reported increases and decreases in the levels of pro-inflammatory cytokines and neurotrophins, along with the appearance of activated microglia, in the brain of sPD patients, thus suggesting the presence of an inflammatory process (McGeer and McGeer, 1995; Anglade et al., 1997; Hirsch et al., 1999; Mogi and Nagatsu, 1999; Nagatsu et al., 1999; Jellinger, 2000; Hartmann et al., 2000; Nagatsu et al., 2000a,b; Nagatsu, 2002a; Hayley, 2005; Herrera et al., 2005; Nagatsu and Sawada, 2005). Based on the results of enzyme-linked immunosolvent assay (ELISAs; for a review, see Nagatsu, 2002a), we reported the changes in the levels of the following cytokines and neurotrophins in the postmortem brain (striatum) and/or ventricular or lumbar cerebrospinal fluid (CSF) in sPD patients as compared with their normal levels: (1) increased levels of TNF-alpha (Mogi et al., 1994a), IL-1beta, IL-6 (Mogi et al., 1994b), IL-2, IL-4, EGF, TGF-alpha, TGF-beta1, TGF-beta2, Bcl-2 (Mogi et al., 1996), soluble FAS, TNF R1 (p55), caspase 1, and caspase 3 (Mogi et al., 2000); and (2) decreased levels of neurotrophins BDNT and NGF. These data on changes in the levels of cytokines in human PD brains were also supported by the results obtained from animal models of PD. For example, MPTP-treated mice show an increased level of IL-1beta and a decreased level of NGF specifically in their striatum (Mogi et al., 1998). As another model of PD, in hemiparkinsonian rats produced by injecting 6-OHDA into one side of the ventrotegmental bundle without or with L-DOPA treatment, the levels of TNF-alpha were significantly increased only in the substantia nigra and striatum of the injected side. L-DOPA administration did not produce any significant changes in TNF-alpha levels in either 6-OHDA-treated or control side of any of the brains (Mogi and Nagatsu, 1999). These results agree with the changes seen in the TNF-alpha levels in the striatum and lumbar CSF in PD patients and also suggest that the increased cytokine levels may not be due to the secondary effects of L-DOPA therapy in PD patients.
The increased levels of pro-inflammatory cytokines such as TNF-alpha, IL-6, and IL-1beta and the decreased levels of neurotrophins such as BDNF and NGF, which changes are known to trigger the process of apoptosis, strongly suggest a pro-apoptotic environment in the striatum in PD. In fact, the levels of apoptosis-related factors such as Bcl-2 (Mogi et al., 1996), soluble FAS, TNF R1 (p55), caspase 1 (IL-1-beta converting enzyme), and caspase 3 are increased in the PD brain (Mogi et al., 2000). Fas antigen and 2 TNF receptors, p55 and p75, are implicated in triggering cell death upon stimulation by their natural ligands, i.e., TNF-alpha and Fas ligands (Nagata and Goldstein, 1995). Since TNF R1 and caspases 1 and 3 have been implicated as mediators of apoptotic cell death (Kumar, 1995), their increased levels support the presence of pro-apoptotic environment in the striatum in the PD brain. The increased levels of Bcl-2 (Mogi et al., 1996) and sFAS, which are anti-apoptotic factors, may suggest their compensatory production to cope with apoptosis. Marshall et al. (1997) also reported up-regulation of Bcl-2 in the basal ganglia in PD patients. We also found that the levels of two other factors trophic toward DA neurons, i.e., GDNF (glial cell line-derived neurotrophic factor) and bFGF (basic fibroblast growth factor) were not decreased, although their concentrations were high in the striatum in control or PD brains. This is in contrast to the markedly reduced levels of BDNF or NGF specifically in that region in PD. The unchanged level of GDNF in PD could be due to compensatory production in glial cells, which occurs with neither BDNF nor NGF. In agreement with our results obtained by ELISA, Boka et al. (1994) found TNF-alpha immunoreactive glial cells in the substantia nigra in the PD brain, and other workers also reported increased cytokine levels in de novo PD without L-DOPA treatment: IL-1beta and IL-6 in lumbar CSF (Blum-Degan et al., 1995) and TGF-beta1 and TGF-beta2 in ventricular CSF (Vawter et al., 1996). Activated caspase 3 was also detected immunohistochemically and was proposed to be the final effector in the apoptotic cell death of DA neurons in PD (Hartmann et al., 2002).
Pro-Inflammatory Cytokines in the PD Brain are Produced from Activated Microglia
The origin of pro-inflammatory cytokines in the PD brain is speculated to be activated microglia. McGeer et al. (1988) were the first to report an increase in the number of major histocompatibility complex class II antigen [human leukocyte antigen-DR (HLA-DR)]-positive reactive microglia in the substantia nigra in PD patients. We also speculated that activated microglia are present in the PD brain to produce pro-apoptotic cytokines and neuroinflammation, ultimately promoting death of DA neurons in the substantia nigra. Imamura et al. (2003) proved that increased amounts of cytokines are produced by activated microglia in the putamen of sPD patients. Mogi et al. (1994a,b) had previously shown, by enzyme immunoassay, increased levels of TNF-alpha and IL-6 in the striatum in sPD. Imamura et al. (2003) identified by Western blot analysis TNF-alpha protein and IL-6 protein, along with MHC class II (CR3/43) protein, in homogenates of the putamen from sPD patients; and they further proved by an immunofluorescence technique the coexistence of TNF-alpha and IL-6 proteins in ICAM-I and LFA-1-positive MHC class II-bearing activated microglia in the putamen from sPD patients. These results confirmed that TNF-alpha and IL-6 proteins are produced from activated microglia in the putamen in sPD.
Activated Microglia may be Initially Non-Toxic/Neuroprotective and Then by a Toxic Change Become Neurotoxic to Cause Progression of PD
Activated microglia are known to produce either neuroprotective or neurotoxic factors. The question is whether these activated microglia are neuroprotective or neurotoxic toward the nigro-striatal DA neurons.
We aimed at elucidating the role of activated microglia in the postmortem brain of sPD at the cellular level. Activated microglia have multiple roles: (1) MHC class II-positive ones act in antigen presentation; (2) activated microglia phagocytose damaged cells; (3) they produce substances such as the pro-inflammatory cytokines TNF-alpha and IL-6, which are pleiotropic and act either as neurotoxins or as neuroprotective agents, as well as neurotoxic substances, i.e., ROS, nitric oxide (reactive nitrogen species, RNS), and glutamate; and (4) they also produce neurotrophic substances such as the neurotrophin BDNF and cytokines that act neuroprotectively.
In the normal brain, many Kp1-positive resting microglia, which are non-toxic, are seen in the substantia nigra and putamen. In the PD ones, however, a large number of MHC class II-positive ramified, activated microglia are found in these regions compared with their number in normal controls. Furthermore, the number of MHC class II-positive microglia in the putamen in PD increases as the stage of PD advances. In the early stages of PD, MHC class II-positive microglia in the putamen and substantia nigra are associated with intensively TH-positive DA neurites showing no signs of degeneration. In the advanced stages, however, MHC class II-positive microglia in these areas are found with damaged TH-positive neurons and neurites. These results suggest that activated microglia in the substantia nigra and putamen may be non-toxic/neuroprotective or neurotoxic, depending on the stage of PD.
Imamura et al. (2003) observed that the number of MHC class II-positive activated microglia was significantly higher not only in the substantia nigra and putamen but also in various other brain regions such as the hippocampus, transentorhinal cortex, cingulate cortex, and temporal cortex in PD brains than in the control ones. Imamura et al. (2005) also observed activated microglia in the nigro-striatum and hippocampus in dementia with Lewy bodies (DLB), and compared them with those in PD. Neuronal degeneration in the putamen was observed in both PD and DLB, whereas neuronal loss in the hippocampus was observed in DLB but not in PD without dementia. In normal controls, neuronal loss, activated microglia, and alpha-synyclein-positive cells were not observed in the hippocampus (CA2/3 region), and neurons were strongly BDNF positive. In the hippocampus (CA2/3 region) in PD, the number of MHC class II-positive microglia was increased, which cells were also positive for ICAM-I (CD54), LFA-1, TNF-alpha, and IL-6. Alpha-synuclein-positive cells were also observed. BDNF-stained neurons were only slightly decreased in number in PD compared with those in controls. In the hippocampus (CA2/3 region) in DLB, the numbers of MHC class II (CR3/43)-positive microglia and alpha-synuclein-positive microglia, and alpha-synuclein-positive neurons were greater than those in PD, and the neurons were very weakly stained with anti-BDNF. These immunohistochemical data on the hippocampus (CA 2/3 region) indicate that the number of activated microglia increases in both PD and DLB and that the content of neurotrophic BDNF protein is markedly decreased in DLB but not in PD. Furthermore, in the hippocampus, mRNA levels of IL-6 and TNF-alpha were increased in both PD and DLB compared with the control levels; whereas the mRNA level of BDNF was greatly decreased in DLB, as compared with that in PD or normal controls. These different changes in the levels of mRNA and protein of BDNF, IL-6, or TNF-alpha in the hippocampus and putamen between PD and DLB suggest that activated microglia in these brain regions in PD and DLB are different in their properties and may secrete different kinds and different amounts of cytokines and neurotrophins such as BDNF and IL-6.
As other evidence supporting this concept of the presence of non-toxic/neuroprotective and neurotoxic microglia, we (Sawada et al., 2006) separated two subsets of microglia from normal mouse brain by cell sorting based on profiles of intracellular ROS production induced by phorbol myristate acetate (PMA) stimulation: one subset of microglia producing a large amount of ROS and the other, just a minute amount of ROS. Furthermore, we obtained two cell lines of microglia, Ra2 cells and 6–3 cells, by spontaneous immortalization of mouse microglia in primary cultures. The Ra2 microglia cell line did not produce ROS upon PMA stimulation, whereas the 6–3 one produced ROS in large amounts in response to this stimulant. When co-cultured with N18 neuronal cells, Ra2 cells were neuroprotective, whereas 6–3 cells were neurotoxic. Furthermore, Sawada and co-workers found in a cell culture experiment a toxic change in activated microglia from neuroprotective to neurotoxic, caused by transduction of the cells with a lenti virus vector carrying HIV-1 Nef cDNA (Vilhardt et al., 2002). It is speculated that a similar toxic change in activated microglia may occur in vivo in the PD brain as the second step, one caused by other factors such as invasion of serum, viruses, toxic substances, or inflammatory cells in some of the neuroprotective microglia in a specific brain regions, i.e., the nigro-striatum in PD. As a result of this toxic change, large amounts of cytotoxic factors such as ROS, NO, and RNS produced by NADPH oxidase, myeloperoxidase, cyclooxygenase 2 (COX 2), or nitric oxide synthase may promote the observed neuronal loss. The presence of reactive microglia in the substantia nigra years after MPTP exposure was detected in experimental monkeys (McGeer et al., 2003) and in human patients (Langston et al., 1999). These reactive microglia might have been produced by a toxic change in response to the exposure to MPTP. These results also suggest that a variety of causative agents of sPD, disappearing after having instituted long-lasting inflammatory changes, might cause progression of the disease.
Based on the results described earlier, Sawada et al. (2006) recently proposed a hypothesis of two-step activation of microglia in vivo in the PD brain. The observation on activated microglia associated with non-degenerating neurons and neurites in various brain regions such as the hippocampus in the early stage of PD suggests that microglia activated by the initiating factors of PD may be at first non-toxic and act for neuroprotection by producing neurotrophins, neurotrophic cytokines, and antioxidant substances in the first step. However, a toxic change in the activated microglia may occur as the second step to promote progression of the disease.
CONTRIBUTION OF JULIE AXELROD TO PD RESEARCH
Aside from his other numerous accomplishments, Dr. Julie Axelrod has made many great contributions also to PD research. One great contribution was his discovery of the reuptake of neurotransmitter catecholamines into the pre-synaptic nerve endings via membrane transporters and then from the cytoplasm to the synaptic vesicles via vesicular transporters (Axelrod et al., 1959). This discovery provided a general principle for the termination of neurotransmission, and led to the identification of neurotransmitter transporters such as DA transporter (DAT) and NA transpoter (NAT), and vesicular monoamine transporters (VMAT), and to the development of innovative drugs such as serotonin noradrenaline reuptake inhibitors as anti-depression drugs and of new diagnostic methods such as molecular imaging by PET (positron emission tomography) or SPECT (single photon emission computed tomography) of synaptic function. Another great contribution is his discovery of catechol O-methyl transferase (COMT; Axelrod, 1957). Inhibitors of COMT are of great importance to the L-DOPA therapy of PD in combination with MAO B inhibitors such as deprenyl.
CONCLUSIONS AND FUTURE PROSPECTS
sPD is thought to be caused by the combination of a susceptible genetic background and various environmental factors. The biochemical analysis of postmortem brain from PD patients and neurotoxin-induced animal models indicates mitochondrial dysfunction and oxidative stress to be important. On the other hand, the causative genes of fPD indicate the accumulation of misfolded proteins due to UPS dysfunction to be important. It should be noted that both mitochondrial dysfunction and UPS dysfunction may be related to each other, and may trigger a common signal transduction pathway to programmed cell death. There are 2 types of programmed cell death, i.e., apoptosis and autophagy. Alpha-synuclein is degraded by both the ubiquitin–protesome pathway and the autophagy–lysosome pathway (Webb et al., 2003). Much data on the pathogenesis of PD support the programmed cell death mechanism by apoptosis. However, this still remains controversial. The process of neuroinflammation may also be important, especially for the progression of PD. Sawada et al. (2006) has proposed a hypothesis of two-step activation of microglia in the brain and their toxic change in PD patients. In order to confirm this hypothesis, the following points remain to be proved: (1) the first and second causative stimuli must be identified; (2) a toxic change should be confirmed to occur in vivo in the nigro-striatum in PD models. A stimulus such as MPTP toxicity may directly produce degeneration of DA cells, and some signal from degenerating DA neurons may trigger activation of microglia, which may, due to the toxic change for producing neurotoxic cytokines, promote cell death of DA neurons, perpetuating a vicious circle. It is also possible that in fPD damaged DA neurons may send unknown signals to microglia to activate them. Thus, activated microglia producing neurotoxic cytokines may promote the progression of the disease in both sPD and fPD.
REFERENCES
Abott, A. (2005). While you are sleeping. Nature (News feature)437:1220–1222.
Anglade, P., Vyas, S., Javoy-Agid, F., Ilerreto, M. T., Michel, P. P., Marquez, J., Pouatt-Prigent, A., Ruberg, M., Hirsch, C., and Agid, Y. (1997). Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 12:25–31.
Axelrod, J., Weil-Malherbe, H., and Tomchick, R. (1959). The physiological distribution of 3H-epinephrine and its metabolite epinephrine. J. Pharm. Exp. Therap. 127:251–256.
Axelrod, J. (1957). O-Methylation of epinephrine and other catecholamines in vitro and in vivo. Science: 126:400–401.
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenmyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurobiol. 3:1301–1306.
Blum-Degan, D., Mueller, T., Kuhn, W., Gerlach, M., Przuntek, H., and Riederer, P. (1995). Interleukin 1-beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease. Neurosci. Lett. 202:17–20.
Boka, G., Anglade, P., Wallach, D., Javoy-Azid, F., Agid, Y., and Hirsch, E. C. (1994). Immunocytochemical analysis of tumor necrosis factor and its receptor in Parkinson’s disease. Neurosci. Lett 172:151– 154.
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O. J., Breedveld, G. J., Krieger, E., Dekker, M. C. J., Squitieri, F., Ibanez, P., Joosse, M., van Dongen, J. W., Vanacore, N., van Swieten, J. C., Brice, A., Meco, G., van Duijn, C. M., Oostra, B. A., and Heutink, P. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259.
Bonifati, V., Oostra, B. A., and Heutink, P. (2004). Unraveling the pathogenesis of Parkinson’s disease: the contribution of monogenic forms. Cell. Mol. Life Sci. 61:1729–1750.
Braak, H., DelTredici, K., Rub, U., deVos, R. A. I., Steur, E. N. H. J., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24:197–212.
Carlsson, A. (1959). The occurrence, distribution and physiological role of dopamine in the nervous system. `Pharmacol. Rev. 11:490–493.
Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schlueter, O. M., and Suedhof, T. C. (2005). Alpha-synuclein cooperates with CSP alpha in preventing neurodegeneration. Cell 123:383–396.
Chen, L., and Feany, M. B. (2005). Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson’s disease. Nat. Neurosci. 8:657–663.
Chiba-Falek, O., and Nussbaum, R. L. (2003). Regulation of alpha-synuclein expression: Implication for Parkinson’s disease. Cold Spring Harbor Sym. Quantit. Biol. LXVIII:409–415.
Chung, K. K., Dawson, V. L., and Dawson, T. M. (2001). The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci. 24(Suppl. 11):S7–S14.
Chung, K. K., Thomas, B., Li, X., Pletnikova, O., Troncosa, J. C., Marsh, L., Dawson, V. L., and Dawson, T. M. (2004). S-Nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 304:1328–1331.
Collins, M. A., and Neafsey, E. J. (2000). Beta-carboline analogues of MPP+ as environmental neurotoxins. In Storch, A., and Collins, M. A. (eds.), Neurotoxic Factors in Parkinson’s Disease and Related Disorders, Kluwer Academic Publishing/Plenum, New York, pp. 115–130.
Conway, K. A., Lee, S. J., Rochet, J. C., Ding, T. T., Williamson, R. E., and Lansbury, P. T., Jr. (2000). Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 97:571–576.
Cookson, M. R. (2005). The biochemistry of Parkinson’s disease. Ann. Rev. Biochem. 74:29–74.
Dawer, W., and Przedborski, S. (2003) Parkinson’s disease: Mechanisms and models, Neuron 39:889– 909.
Davis, G. C. B., Williams, A. C., Markey, S. P., Ebert, M. H., Caine, E. D., Reichert, C. M., and Kopin, I. J. (1979). Chronic parkinsonism secondary to intravenous injection of meperidine analogus. Psychiatry Res. 1:249–254.
Di Fonzo, A., Rohe, C. F., Ferreira, J., Chien, H. F., Vacca, L., Stocchi, F., Guedes, L., Fabrizio, E., Manfredi, M., Vanacore, N., Goldwurm, S., Breedveld, G., Sampaio, C., Meco, G., Barbosa, E., Oostra, B. A., and Bonifati, V. Italian Parkinson Genetics Network (2005). A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365:412–415.
Ehringer, H., and Hornykiewicz, O. (1960). Verteilung von Noradrenalin und Dopamin (3-Hydroxytyramin) im Gehirn des Menschen und ihr Verhalten bei Erkarankungen des extrapyramidalen Systems. Klin. Wschr. 38:1236–1239.
Eisenhofer, G., Lamensdorf, I., Kirk, K. L., Kawamura, M., and Sato, S. (2002). Oxidative deamination of monoamines and biogenic aldehydes in neurodegenetration. In Creveling, C. R. (ed.), Role of Catechol Quinone Species in Cellular Toxicity, F.P. Graham Publishing, Johnson City, pp. 147–167.
Feany, M. B. (2004). New genetic insights into Parkinson’s disease. New Engl. J. Med. 351:1937–1940.
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson’s disease. Nature 404:394–398.
Foley, P., and Riederer, P. (1999). Pathogenesis and preclinical course of Parkinson’s disease. J. Neural. Transm. Suppl. 56:31–74.
Forman, M. S., Trojanowski, J. Q., and Lee, V. M.-Y. (2004). Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nature Med. 10:1055–1063.
Fornai, F., Schlueter, O. M., Lenzi, P., Gesi, M., Ruffoli, R., Ferrucci, M., Lazzeri, G., Busceti, C. L., Pontarelli, F., Battaglia, G., Pellegrini, A., Nicoletti, F., Ruggieri, S., Paparelli, A., and Suedhof, T. C. (2005). Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. USA 102:3413–3418.
Fukuda, T. (1994). 1-Methyl-1,2,3,4-tetrahydroisoquinoline does dependently reduces the number of tyrosine hydroxylase-immunoactive cells in the substantia nigra and locus ceruleus of C57BL/6J mice. Brain Res. 639:325–328.
Gerlach, M., Ben-Shachar, D., Riederer, P., and Youdim, M. B. H. (1994). Altered brain metabolism of iron as a cause of neurodegenerative diseases? J. Neurochem. 63:793–807.
Goedert, M. (2001). Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2:492–501.
Goldstein, D. S., Eldadah, B. A., Holmes, C., Pechnik, S., Moak, J., Saleem, A., and Sharabi, Y. (2005). Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotention: Independence from Levodopa treatment. Hypertension 46:1333–1339.
Grandhi, S., and Wood, N. W. (2005). Molecular pathogenesis of Parkinson’s disease. Human Mol. Genet. 14:2749–2755.
Hartmann, A., Hunot, S., Michel, P. P., Muriel, M. P., Vyas, S., Faucheux, B. A., Mouatt-Prignet, A., Turmel, H., Srinivasan, A., Ruberg, M., Evan, G. I., Agid, Y., and Hirsch, E. C. (2000). Caspase-3: A vulnerable factor and a final effector in the apoptotic cell death of dopaminergic neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 97:2875–2880.
Hasbani, D. M., Perez, F. A., Palmiter, R. D., and O’Malley, K. L. (2005). Dopamine depletion does not protect against acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in vivo. J. Neurosci. 25:9428–9433.
Hayley, S. (2005). Multiple mechanisms of cytokine activation in neurodegenerative and psychiatric states: Neurochemical and molecular substrates. Curr. Pharmac Design 11:947–962.
Hirsch, E. C., Hunot, S., Faucheux, B. A., Agid, Y., Mizuno, Y., Mochizuki, H., Tatton, W. G., Tatton, N., and Olanow, W. C. (1999). Dopaminergic neurons degenerate by apoptosis in Parkinson’s disease. Mov. Disord. 14:383–385.
Hoffmann, G. F., Assmann, B., Braeutigam, C., Dionisi-Vici, C., Haeussler, M., and deKlerk, J. B. C., Neumann, M., Steenbergen-Spanjers, G. C. H., Strassburg, M.-H., and Wevers, R. A. (2003). Tyrosine hydroxylase deficiency causes progressive encephalopathy and dopa-non-responsive dystonia. Ann. Neurol. 54(Suppl. 6):S56–S65.
Honbou, K., Suzuki, N. N., Horiuchi, M., Niki, T., Taira, T., Ariga, H., and Inagaki, F. (2003). The crystal structure of DJ-1, a protein related to male fertility and`Parkinson’s disease. J. Biol. Chem. 278:31380–31384.
Ichinose, H., Ohye, T., Takahashi, E., Seki, N., Hori, T., Segawa, M., Nomura, Y., Endo, K., Tanaka, H., Tsuji, S., Fujita, K., and Nagatsu, T. (1994). Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydolase I gene. Nature Genet. 8:236–242.
Ichinose, H., Ohye, T., Matsuda, Y., Hori, T., Blau, N., Burlina, A., Rouse, B., Matalon, R., Fujita, K., and Nagatsu, T. (1995). Characterization of mouse and human GTP cyclohydrolase I genes. Mutations in patients with GTP cyclohydrolase I deficiency. J. Biol. Chem. 270:10062–10071.
Ichinose, H., Suzuki, T., Inagaki, H., Ohye, T., and Nagatsu, T. (1999). Molecular genetics of dopa-responsive dystonia. Biol. Chem. 380:1355–1364.
Ichinose, H., Ohye, T., Suzuki, T., Sumi-Ichinose, C., Nomura, T., Hagino, Y., and Nagatsu, T. (1999). Molecular cloning of the human Nurr1 gene: Characterization of the human gene and cDNA. Gene 230:233–239.
Ikemoto, K., Nagatsu, I., Ito, S., King, R., Nishimura, A., and Nagatsu, T. (1998). Does tyrosinase exist in neuromelanin-pigmented neurons in the human substantia nigra? Neurosci. Lett. 253:198–200.
Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y., and Takahashi, R. (2001). An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell 105:891–902.
Imamura, K., Hishikawa, N., Sawada, M., Nagatsu, T., Yosida, M., and Hashizume, Y. (2003). Distribution of major histocompatibility complex II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 106:518–526.
Imamura, K., Hishikawa, N., Ono, K., Suzuki, H., Sawasa, M., Nagatsu, T., Yoshida, M., and Hashizume, Y. (2005). Cytokine production of activated microglia and decrease on neurotrophic factors of neurons in the hippocampus of Lewy body disease brain. Acta Neuropathol. 109:141–150.
Ischiropoulos, H., and Beckman, J. S. (2003). Oxidative stress and nitration in neurodegeneration: Cause, effect, or association? J. Clin. Invest. 111:163–169.
Iwawaki, T., Kohno, K., and Kobayashi, K. (2000). Identification of a potential Nurr1 response element that activates the tyrosine hydroxylase gene promoter in cultured cells. Biochem. Biophys. Res. Cpmmun. 274:590–595.
Kajita, M., Niwa, T., and Nagatsu, T. (2002). Tetrahydroisoquinolines (TIQ) and neurodegeneration. In Creveling, C. R. (ed.), Role of Quinone Species in Cellular Toxicity, F. P. Graham Publishing, Johnson City, TN, pp. 169–190.
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y., and Shimizu, N. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608.
Kobayashi, K., and Nagatsu, T. (2005). Molecular genetics of tyrosine 3-monooxygenase and inherited diseases. Biochem. Biophy. Res. Commun. 338:267–270.
Kosaka, K. (2000). Lewy body disease. Neuropathol. Suppl. 20:73–78.
Kostrzewa, R. M., and Jacobowitz, D. M. (1974). Pharmacological actions of 6-hydroxydopamine. Pharm. Rev. 26:199–288.
Kotake, Y., Tasaki, Y., Makino, S., Hirobe, M., and Ohta, S. (1995). 1-Benzyl-1,2,3,4-tetrahydroisoquinoline as a parkinsonism-inducing agent: A novel endogenous amine in mouse brain and parkinsonian CSF. J. Neurochem. 65:2633–2638.
Krueger, R. (2004). Genes in familial parkinsonism and their role in sporadic Parkinson’s disease. J. Neurol. 251(Supl. 6 ):VI/2–VI/6.
Kuhn, W., Mueller, Th., Grosse, H., and Rommelspacher, H. (1996). Elevated levels of harman and norharman in cerebrospinal fluid of Parkinsonian patients. J. Neural. Transm. 103:1435–1440.
Kumar, S. (1995). ICE-like proteases in apoptosis. Trends Biochem. Sci. 20:198–202.
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (1983). Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–980.
LaVoie, M. J., Ostaszewski, B. L., Weihofen, A., Schlossmacher, M. G., and Selkoe, D. J. (2005). Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 11:1214–1221.
Leroy, E., Boyer, R., Auburger, G., Leube, B., Ulm, G., Mezey, E., Harta, G., Brownstein, M. J., Jonnalagada, S., Chernova, T., Dehejia, A., Lavedan, C., Gasser, T., Steinbach, P. J., Wilkinson, K. D., and Polymeropoulos, M. H. (1998). The ubiquitin pathway in Parkinson’s disease. Nature 395:451–452.
Le, W., Xu, P., Jankovic, J., Jiang, H., Appel, S. H., Smith, R. G., and Vassilatis, K. (2003). Mutations in Nr4A2 associated with familial Parkinson’s disease. Nat. Genet. 33:85–89.
Liani, E., Eyal, A., Avraham, E., Shemer, R., Szargel, R., Berg, D., Bornemann, A., Riess, O., Ross, C. A., Rott, R., and Engelender, S. (2004). Ubiquitination of synphilin-1 and alpha-synyclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Nat. Acad. Sci. USA 101:5500–5505.
Lozano, A. M., and Kalia, S. K. (2005). New movement in Parkinson’s. Scientific American, pp. 68–75.
MacKeigan, J. P., Murphy, L. O., and Blenis, J. (2005). Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nature Cell Biol. 7:591–600.
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., Price, D. L., and Lee, M. K. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26:41–50.
Marshall, K. A., Daniel, S. E., Cairns, N., Jenner, P., and Halliwell, B. (1997). Upregulation of the anti-apoptotic protein Bcl-2 may be an early event in neurodegeneration: Studies on Parkinson’s and incidental Lewy body disease. Biochem. Biophys. Res. Commun. 240:84–87.
Matsubara, K. (2000). N-Methyl-beta-carbolinium neurotoxins in Parkinson’s disease. In Storch, A., and Collins, M. A. (eds.) Neurotoxic Factors in Parkinson’s Disease and Related Disorders, Kluwer Academic Publishing/Plenum, New York, pp. 131–143.
Matsubara, K., Kobayashi, S., Kobayashi, Y., Yamashita, K., Koide, H., Hatta, M, Iwamoto, K., Tanaka, O., and Kimura, K. (1995). Beta-carbolinium cations, endogenous MPP+ analogs in the lumbar cerebrospinal fluid of parkinsonian patients. Neurology 45:2240–2245.
Mattammal, M. B., Chung, H. D., and Strong, R. (1993). Confirmation of a dopamine metabolite in parkinsonian brain tissue by gas-chromatography-mass spectrometry. J. Chromatogr. B 614:205–212.
Mattammal, M. B., Haring, J. H., Chung, H. D., Raghu, G., and Strong, R. (1995). An endogenous dopaminergic neurotoxin: Implication for Parkinson’s disease. Neurodegeneration 4:271–281.
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s disease and Alzheimer’s disease brain. Neurology 38:1285–1291.
McGeer, P. L., and McGeer, E. G. (1995). The inflammatory response system of brain, implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 21:195–218.
McNaught, K. S., Perl, D. P., Brownell, A. L., and Olanow, C. W. (2004). Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 56:149–162.
Mizuno, Y., Yoshino, H., Ikebe, S., Hattori, N., Kobayashi, T., Shimoda-Matsubayashi, S., Matsumine, H., and Kondo, T. (1998). Mitochondrial dysfunction in Parkinson’s disease. Ann. Neurol. 44(Suppl 1):S99–S109.
Mizuno, Y. (2006). Progress in familial Parkinson’s disease. In Riederer, P. (eds.), Proceedings of the 16th International Congress on Parkinson’s Disease and Related Disorders. J. Neural Transm, in press.
Mochizuki, H., Nishi, K., and Mizuno, Y. (1993). Iron-melanin complex is toxic to dopaminergic neurons in a nigrostriatal co-culture. Neurodegeneration 2:1–7.
Mogi, M., Harada, M., Riederer, P., Narabayashi, H., Fujita, K., and Nagatsu, T. (1994a). Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 165:208–210.
Mogi, M., Harada, M., Kondo, T., Riderer, P., Inagaki, H., Miura, M., and Nagatsu, T. (1994b). Interleukin 1-beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 180:147–150.
Mogi, M., Harada, M., Kondo, T., Mizuno, Y., Narabayashi, H., Riederer, P., and Nagatsu, T. (1996). bcl-2 Protein is increased in the brain from parkinsonian patients. Neurosci. Lett. 215:137–139.
Mogi, M., Togari, A., Ogawa, M., Ikeguchi, K., Shizuma, N., Fan, D.-S., Nakano, I., and Nagatsu, T. (1998). Effects of repeated administration of 1-methyl-4-phenyl-1, 2, 3, 6- tetrahydropyridine (MPTP) to mice on interleukin-1beta and nerve growth factor in the striatum. Neusci. Lett. 250:25–28.
Mogi, M., and Nagatsu, T. (1999). Neurotrophins and cytokines in Parkinson’s disease. Adv. Neurol. 80:135–139.
Mogi, M., Togari, A., Kondo, T., Mizuno, Y., Komure, O., Kuno, S., Ichinose, H., and Nagatsu, T. (2000). Caspase activities and tumor necrosis factor receptor R1 level are elevated in the substantia nigra in Parkinson’s disease. J. Neural. Transm. 107:335–341.
Moser, A., and Koempf, D. (1992). Presence of methyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinolines, derivatives of the neurotoxin isoquinoline, in parkinsonian lumbar CSF. Life Sci. 50:1885–1891.
Nagakubo, D., Taira, T., Kitaura, H., Ikeda, M., Tamai, K., Iguchi-Ariga, S. M. M., and Ariga, H. (1997). DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 231:509–513.
Nagata, S., and Goldstein, P. (1995). The Fas death factor. Science 267:1449–1456.
Nagatsu, T. (1997). Isoquinoline neurotoxins and Parkinson’s disease. Neurosci. Res. 29:99–111.
Nagatsu, T. (2002a). Parkinson’s disease: Changes in apoptosis-related facors suggesting possible gene therapy. J. Neural Transm. 109:731–745.
Nagatsu, T. (2002b). Amine-related neurotoxins in Parkinson’s disease. Past, present, and future. Neurotoxicol Teratol 24:565–569.
Nagatsu, T., and Ichinose, H. (1999). Molecular biology of catecholamine-related enzymes in relation to Parkinson’s disease. Cell. Mol. Neurobiol. 19:57–66.
Nagatsu, T., Mogi, M., Ichinose, H., Togari, A., and Riederer, P. (1999). Cytokines in Parkinson’s disease. NeuroSci. News 2:88–90.
Nagatsu, T., Mogi, M., Ichinose, H., and Togari, H. (2000a). Cytokines in Parkinson’s disease. J. Neural. Transm. Suppl 58:143–151.
Nagatsu, T., Mogi, M., Ichinose, H., and Togari, A. (2000b). Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural Transm. Suppl. 60:277–290.
Nagatsu, T., and Sawada, M. (2005). Inflammatory process in Parkinson’s disease: Role for cytokines. Curr. Pharmac. Design 11:999–1016.
Naoi, M., Maruyama, W., Dostert, P., Hashizume, Y., Nakahara, D., Takahashi, T., and Ota, M. (1996). Dopamine-derived endogenous 1(R), 2(N)-dimethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline, N-methyl-(R)-salsolonol, induced parkinsonism in rats: Biochemical, pathological and behavioral studies. Brain Res. 709:285–295.
Niwa, T., Takeda, N., Kaneda, N., Hashizume, Y., and Nagatsu, T. (1987). Presence of tetrahydroisoquinoline and 2-methyl-tetrahydroisoquinoline in parkinsonian and normal human brains. Biochem. Biophys. Res. Commun. 144:1084–1089.
Norris, E. H., Giasson, B. I., and Lee, V. M. (2004). Alpha-synuclein: normal function and role in neurodegenerative diseases. Curr. Top. Dev. Biol. 60:17–54.
Ohta, S., Kohno, M., Makino, Y., Tachikawa, O., and Hirobe, O. (1997). Tetrahydroisoquinoline and 1-methyl-tetrahydroisoquinoline are present in the human brain. Biomed. Res. 8:453–456.
Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., de Munain, A. L., Aparicio, S., Gil, A. M., Khan, N., Johnson, J., Martinez, J. R., Nicholl, D., Carrera, I. M., Pena, A. S., de Silva, R., Lees, A., Marti-Masso, J. F., Perez-Tur, J., Wood, N. W., and Singleton, A. B. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44:595– 600.
Panov, A., Dikalov, S., Shalbuyeva, N., Taylor, G., Sherer, T., and Greenamyre, J. T. (2005). Rotenone model of Parkinson’s disease: Multiple brain mitochondria dysfunctions after short-term systemic rotenone intoxication. J. Biol. Chem. 280:42026–42035.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Deheijia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenrous, E. S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoiosin, R. C., DiIorio, G., Golbe, L. I., and Nussbaum, R. L. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047.
Sacchetti, P., Mitchell, T. R., Grameman, J. G., and Bannon, M. J. (2001). Nurr1 enhances transcription of the human dopamine transporter gene through a novel mechanism. J. Neurochem. 76:1565–1572.
Sawada, M., Imamura, K., and Nagatsu, T. (2006). Role of cytokines in inflammatory process in Parkinson’s disease. In Riederer, P. (ed.) Proceedings of the 16th International Congress on Parkinson’s disease and Related Disorders. J. Neural Transm. in press.
Schapira, A. H. V., Gu, M., Taanman, J.-W., Tabrizi, S. J., Seaton, T., Cleeter, M., and Cooper, J. M. (1998). Mitochondria in the etiology and pathogenesis of Parkinson’s disease. Ann. Neurol. 44(Suppl 1):S89–S98.
Segawa, M., Nomura, Y., and Nishiyama, N. (2003). Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann. Neurol. 54(Suppl. 6 ):S32–S45.
Selkoe, D. (2004). Cell biology of protein misfolding: The example of Alzheimer’s and Parkinson’s disease. Nat. Cell Biol 6:1054–1061.
Shen, J. (2004). Protein kinases linked to the pathogenesis of Parkinson’s disease. Neuron 44:575–577.
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., Shimizu, N., Imai, K., Chiba, T., Tanaka, K., and Suzuki, T. (2000). Familial Parkinson gene product, parkin, is a ubiquitin-protein ligase. Nature Genet 25:302–305.
Shimura, H., Schlossmacher, M. G., Hattori, N., Frosch, M. P., Trockenbacher, A., Schneider, R., Mizuno, Y., Kosik, K. S., and Selko, D. J. (2001). Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 293:263–269.
Sherer, T. B., Kim, J. H., Batarbet, R., and Greenamyre, J. T. (2003). Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 179:9–16.
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., and Nussbaum, R. (2003). Alpha-synuclein locus triplication causes Parkinson’s disease. Science 302:841.
Snyder, S. H. (2005). Messengers of life and death. Society for Neuroscience 2005: Program No. 467.
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 95:6469–6473.
Tasaki, Y., Makino, Y., Ohta, S., and Hirobe, M. (1991). 1-Methyl-1.2.3.4-tetrahydroisoquinoline, decreased in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mouse, prevents parkinsonism-like behavior abnormalities. J. Neurochem. 57:1940–1943.
Tatton, W., Chalmers-Redman, R., and Tatton, N. (2003). Neuroprotection by deprenyl and other propargylamines: Glyceraldehyde-3-phosphate dehydrogenase rather than monoamine oxidase. J. Neural Transm. 110:509–515.
Tretter, L., Sipos, I., and Adam-Vizi, V. (2004). Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson’s disease. Neurochem. Res. 29:569–577.
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M. K., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A. R., Healy, D. G., Albanese, A., Nussbaum, R., Gonzalez-Maldonado, R., Deller, T., Salvi, S., Cortelli, P., Gilks, W. P., Latchman, D. S., Harvey, R. J., Dallapiccola, B., Auburger, G., and Wood, N. W. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160.
Vawter, M. P., Dillon-Carter, O., Tourtellotte, W. W., Carvey, P., and Freed, W. J. (1996). TGF beta1 and TGF beta2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp. Neurol. 142:313–322.
Vila, M., and Przedborski, S. (2004). Genetic clues to the pathogenesis of Parkinson’s disease. Nat. Med. S58–S62.
Vilhardt, F., Plastre, O., Sawada, M., Suzuki, K., Wiznerowicz, M., Kiyokawa, E., Trono, D., and Krause, K.-H. (2002). The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J. Biol. Chem. 277:42136–42143.
Warbt, S., MacDonald, M. L. E., and Abrahams, B. S. (2003). New mutations, new etiologies for Parkinson disease. Clin. Genet. 63:352–357.
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinstein, D. C. (2003). Alpha-synclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278:25009–25013.
Wood, P. L. (2003). Microglia: Role of microglia in chronic neurodegeneration. In Wood, P. L. (ed.) Neuroinflammation. Humana Press, Totowa, NJ, pp. 3–27.
Yamada, M., Iwatsubo, T., Mizuno, Y., and Mochizuki, H. (2004). Overexpression of alpha-synuclein in rat substantia nigra and activation of caspase-9: Resemblance to pathogenetic changes in Parkinson’s disease. J. Neurochem. 91:451–461.
Yang, Y., Nishimura, I., Imai, Y., Takahashi, R., and Lu, B. (2003). Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 37:911–924.
Youdim, M. B. H., and Riederer, P. (1997). Understanding Parkinson’s disease. Scientific American, pp. 82–89.
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti, R. J., Calne, D. B., Stoessel, A. J., Pfeiffer, R. F., Patenge, N., Carbajal, I. C., Vieregge, P., Asmus, F., Mueller-Myhsok, B., Dickson, D. W., Meitinger, T., Strom, T. M., Wszolek, Z. K., and Gasser, T. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607.
ACKNOWLEDGMENTS
Toshi Nagatsu dedicates this paper to the late Dr. Julie Axelrod with great admiration for him for his outstanding scientific achievements as Nobel Laureate and for his extremely warm personality and humanitarian efforts. This work was supported by grants-in-aid for scientific research from the Ministry of Labor and Welfare of Japan (MS), from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MS), and from the Japan Health Sciences Foundation (MS). We are thankful to all of our collaborators, especially Drs. K. Imamura, K. Ono, H. Suzuki, Y. Hashizume, and M. Mogi and to Drs. P. Riederer, Y. Mizuno, T. Kondo, and S. Kuno for their collaboration in supplying us post mortem brain samples from their brain banks.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue dedicated to Dr. Julie Axelrod
Rights and permissions
About this article
Cite this article
Nagatsu, T., Sawada, M. Cellular and Molecular Mechanisms of Parkinson’s Disease: Neurotoxins, Causative Genes, and Inflammatory Cytokines. Cell Mol Neurobiol 26, 779–800 (2006). https://doi.org/10.1007/s10571-006-9061-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-006-9061-9