
Abstract
Microglia cells are the primary mediators of the CNS immune defense system and crucial for the outcome of shaping inflammatory responses. They are highly dynamic, moving constantly, and become activated by neuronal signaling under pathological conditions. They fulfill a dual role by not only regulating local neuroinflammation but also conferring neuronal protection. Gonadal steroids are known to exert anti-inflammatory effects in the CNS. Recently, we have shown that the microglial-like cell line BV-2 is hypoxia-sensitive and regulated by gonadal steroids. The present study used primary rat cerebral cortex-derived microglia to analyze whether this cell type directly perceive and respond to acute hypoxia. Second, we investigated whether 17β-estradiol (E2) and progesterone (P) interfere with hypoxia-induced changes. Short-term hypoxia increased the expression of a subset of pro-inflammatory (TNFa, IL1b) and oxidative stress-related (Hif1a) genes. The induction of TNFa and IL1b was counteracted by P. Hypoxia shifted the primary microglia to the pro-inflammatory M1 phenotype. The administration of E2 and P favored the neuroprotective M2 phenotype. Our findings extend previous data obtained with BV-2 cells and show that the primary microglia directly perceive hypoxia which increase their inflammatory activity. Both steroid hormones directly and indirectly interact with the microglia cells by reducing the inflammatory scenario and stimulating neuroprotection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microglia cells belong to the mononuclear phagocyte lineage and represent the resident immune cells of the central nervous system (CNS) which mediate the brain's innate immune responses to acute injury and disease. They are constantly surveying the brain microenvironment and remove cellular debris by phagocytosis. Thereby, they maintain normal brain homeostasis and preserve healthy tissue (Loane and Byrnes 2010). After tissue damage, the microglia cells become activated in different ways. Besides recognizing pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), the microglia expresses receptors for different factors released by injured nerve cells and neighboring reactive astrocytes, thus being under control of circulating and paracrine-acting cytokines and chemokines (Block and Hong 2005; Bajramovic 2011). The local cross talk between the astrocytes and microglia appears essential for proper immune responses in the brain (Ransohoff and Brown 2012). Although they are regarded as key regulators of detrimental inflammation after trauma by releasing pro-inflammatory cytokines and other mediators such as prostaglandin E2 (PGE2), reactive oxygen species (ROS), and others, microglia activation may additionally have beneficial effects (Block and Hong 2005; Gelinas and McLaurin 2005; Li et al. 2007). For better differentiation of subsets of the microglia cells, the classification into an activated pro-inflammatory type M1 and a less inflammatory and neuroprotective type M2 involved in tissue repair and modeling of the extracellular matrix was launched (Kigerl et al. 2009).
The gonadal steroid hormones 17β-estradiol (E2) and progesterone (P) have been demonstrated as neuroprotective factors in acute traumatic brain injury and ischemic stroke (Stein 2008; Dang et al. 2011; Spence et al. 2011; Kipp et al. 2012). Both hormones interfere with a multitude of devastating pathophysiological processes initiated by brain damage under hypoxia such as ROS formation, failure of mitochondrial ATP synthesis, and glutamate overflow metabolism under hypoxic conditions (Pawlak et al. 2005; Arnold and Beyer 2009; Suzuki et al. 2009). There is also good evidence that both steroids regulate immune functions mediated in particular by the microglia in the brain and thereby modulate inflammatory responses (Mor et al. 1999; Baker et al. 2004; Loram et al. 2012). The microglia cells are known to better bear hypoxic challenges in the CNS similar to the astrocytes compared to the nerve cells. Not only the regulation of both glial cell types on their own but also the extensive cross talk between astroglia and microglia is an integral part of steroid-dependent detoxification and neuroprotection in the brain (Kipp and Beyer 2009; Arevalo et al. 2010; Liu et al. 2011; Johann and Beyer 2012).
We have recently demonstrated using a temporary middle cerebral artery occlusion rat model (tMCAO) that E2 and P are both individually capable of reducing the infarct volume and restoring behavioral function which is paralleled by a dampening of local inflammation in the penumbra (Dang et al. 2011; Ulbrich et al. 2012). In a further attempt to identify the responsive target cells and physiological reactions, we used an in vitro hypoxia system and defined ischemic conditions. Thereby, we have shown that astroglia sense hypoxia and directly respond by increasing the expression of pro-inflammatory genes (Habib P et al. Hypoxia-induced expression of aquaporin-4, cyclooxygenase 2 and hypoxia inducible factor 1alpha in mouse cortical astroglia is inhibited by sex steroids, in revision). Similar findings were obtained when the immortalized murine microglia-like cell line BV-2 was used (Habib et al. 2013). In both cases, E2 and P reduced the inflammatory responses and modulated the phagocytotic activity.
In order to supplement these observations and to better picture the natural situation in vivo, we have isolated the primary rat microglia from the cerebral cortex and used these cell cultures for hypoxic studies. We aimed at demonstrating the expression pattern of E2 and P receptors in the microglia, their responsiveness towards transient sub-lethal hypoxia, and the regulative effects of both steroid hormones on these functional parameters and, in particular, the switching from a M1 to a M2 phenotype.
Materials and Methods
Primary Cell Cultures
The cerebral cortices were prepared from 1–3 day-old rat pups. The tissues were pooled, minced, and dispersed in Dulbecco's phosphate-buffered saline (PBS) containing 1 % trypsin and 0.02 % EDTA. Cell suspensions were filtered through a 50-μm nylon mesh and centrifuged at 400×g for 5 min. Cell pellets were re-suspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10 % heat-inactivated and charcoal-stripped fetal calf serum (PAA, Austria), streptomycin (0.5 %, Invitrogen, Germany), and fungizone (0.1 %, Invitrogen, Germany). Cell culturing was performed as recently described with some minor modifications (Habib et al. 2013). Briefly, cells were seeded in 75 cm3 cell culture flasks coated with poly-L-ornithine (Sigma, Germany) at a density of 1–2 × 10−6 cells/cm2. Cultures remained for 3–4 days at 37 °C in a humidified incubator with 5 % CO2. Medium was changed every second day. After 2 weeks, the microglia was detached from the confluent astroglia cell layer by mild shaking at 100–150 rpm for 2–4 h. The supernatant was collected and pelleted at 1,000 rpm for 5 min. Microglia cells were then seeded in 24-well plates as secondary cultures. This procedure yielded microglia cultures with a >95 % homogeneity and purity.
Hypoxic Stimulation and Steroid Hormone Treatment
As previously described, a cube-shaped hypoxia chamber was flooded with inert nitrogen gas to replace oxygen for 30 min and create hypoxia (Habib et al. 2013; Habib P et al. Hypoxia-induced expression of aquaporin-4, cyclooxygenase 2 and hypoxia inducible factor 1alpha in mouse cortical astroglia is inhibited by sex steroids, in revision). Oxygen levels were continuously monitored using an oxygen detector (Gox 100 T, Greisinger Electronic GmbH, Germany). Temperature was maintained at 37 °C. Before hypoxia, the medium was replaced. For steroid hormone treatment, E2 and P (both Sigma-Aldrich, Munich, Germany) were diluted in pro-analysis EtOH to yield a concentration of 10−2 M and then further diluted in the RPMI 1640 culture medium in the experiment to a final concentration of 10−7 M. Controls were treated with the same concentration of EtOH/medium. Cells were exposed to hypoxia for 3 h with or without hormone supplementation and subsequently processed.
Cell Viability Assay
Cell viability was determined by vital metabolic activity to reduce the non-fluorescent resazurin (7-hydroxy-3H-phenoxazin-3-one 10-oxide) into the fluorescence emitting dye resorufin at 590 nm (CellTiter-Blue® assay, Promega, Germany) and by the release of lactate dehydrogenase (LDH, CytoTox 96® Non-Radioactive Cytotoxicity Assay, Promega, Germany) into the cell supernatant measured at 490–520 nm. Fluorescence was measured using a microplate reader (Tecan GmbH, Switzerland). In both assays, lysed cells (Triton X-100) served as internal positive controls and cells without treatment as negative control.
Gene Expression Analysis
After hypoxia (3 h), RNA was isolated by phenol–chloroform extraction using peqGold RNA pure (PeqLab, Erlangen, Germany). Efficiency and quality of RNA extraction was measured using 260/280 ratios of optical density from each sample (Nanodrop 1000, Peqlab, Germany). Samples were approved pure if the 260/280 ratios were around 2.0 ± 0.1. Reverse transcription from mRNA to complementary DNA (cDNA) was conducted using the M-MLV reverse transcription (RT)-kit and random hexanucleotide primers (Invitrogen, Germany) using 1 μg/ml of mRNA. Measurement of expression levels of target genes was performed with a mixture consisting of 2 μl reversed-transcribed cDNA, 2 μl RNAse-free water (Invitrogen, Germany), 5 μl 2×Sensi mix plus SYBR & Fluoresceine (Quantace, USA), and 0.5 μl primers (10 pmol/μl). For assessment of sex hormone receptor expression, semi-quantitative PCR was performed using 0.75 μl of rt cDNA. The mixture for the amplification process contains 1.25 μl 10× PCR buffer, 0.38 μl MgCl, 0.25 μl dNTPs, 0.08 μl Taq-polymerase, and the respective sense (s) and anti-sense (as) primers (see Table 1) (Invitrogen, Germany). Primers were designed with the free-available software Primer3 input (Version 0.4.0, Whitehead Institute for Biomedical Research, Cambridge, USA). PCR reactions were conducted using a thermal cycler (Eppendorf, Germany) starting at 94 °C for 30 s, followed by 30 cycles of DNA amplification (30 s at 94 °C, 30 s at 58–63 °C, and 1 min at 68 °C) with final extension at 68 °C for 5 min and hold at 4 °C. PCR products were separated by electrophoresis using 2 % (w/v) agarose gels containing 0.0025 % ethidium bromide (Roth, Germany). Every experiment included negative and positive controls. For visualization of bands, an UV transluminator chamber E-Box VX2 (Peqlab, Germany) was used. Quantitative PCR analyses were performed using the MyIQ RT-qrtPCR detection system (Bio-Rad, Germany) in special 96-well plates (Bio-Rad, Germany) under the following configurations: 10 min enzyme activation at 95 °C, 15 s denaturation at 95 °C, 40 cycles of 30 s at primer-specific annealing temperatures, 30 s amplification at 72 °C, and 5 s fluorescence measurement at 72 °C. To calculate the expression levels of target genes, an external standard was generated by six-time repeated tenfold dilutions of total sample pooling. Note that each sample was also coevally diluted tenfold to adjust concentration within the range of the external standard. The target and the housekeeping gene HPRT were measured at cycle threshold (C t values) by the reverse transcription quantitative polymerase chain reaction (RT-qPCR). Expression levels of target genes were calculated by the ratios of C t values of target to housekeeping. Software-supported evaluation of melting curve (iQ5 System Software, Biorad, Germany) and electrophoretical analysis were routinely performed to qualify the specificity of the RT-qrt-PCR products. Annealing temperatures of used primers/genes are given in Table 1.
Statistics
Data are shown as arithmetic means ± SEM calculated from at least four independent experiments per readout parameter and treatment. Percentage data were arc/sin-transformed for statistical analysis. Data were statistically analyzed as indicated in the figure legends by one-way ANOVA followed by Tukey's post hoc test. The criterion for statistical significance was set at p ≤ 0.05.
Results
The duration and magnitude of the hypoxic condition in vitro was chosen in such a way that no substantial cell loss and pronounced cell damage occur but was sufficient to set and activate primary microglia to hypoxic challenges similar to acute stroke mechanisms. These experimental requirements have been proven in preliminary studies (Habib et al. 2013; Habib et al. Hypoxia-induced expression of aquaporin-4, cyclooxygenase 2 and hypoxia inducible factor 1alpha in mouse cortical astroglia is inhibited by sex steroids, in revision). Increasing hypoxic exposure times resulted in a rise of microglia cell death (not shown).
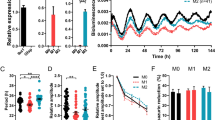
The release of LDH and metabolic activity was only marginally affected by hypoxia compared with normoxia. The administration of E2 and P did not modify these parameters (Fig. 1a). Primary microglia expressed several types of classical and non-classical steroid hormone receptors (Fig. 1b). We could demonstrate ample expression of ERb, GPR30 (E2) and Pgrmc1, and mPRa (P) receptors under both experimental conditions. There was no obvious variation in the mRNA levels of these receptors with respect to the chosen experimental conditions neither did the application of sex steroids change the expression profiles (not shown).
Hypoxic conditions influenced the morphology of primary microglia (Fig. 2a). Under normoxia, most of the cells had three or more branched processes. This morphological appearance was also seen in the presence of E2 and P. In contrast, hypoxia caused a rapid transformation of the cells (Fig. 2b). The number of multi-process-bearing cells was reduced, and only few microglia cells showed three or more processes. Again, sex steroids did not influence this parameter.

Effect of hypoxia on LDH release and metabolic activity in cultured primary rat microglia. (a) Hypoxia did not affect LDH release or metabolic activity compared to normoxia. Steroid hormones had no influence on LDH release and metabolic activity. Lysis increased LDH release to 100 % and decreased metabolic activity to zero. Data represent means ± SEM from four independent experiments. (b) Semi-quantitative rt-PCR showed expression of ERb, GPR30, Pgrmc1, and mPRa in primary rat microglia. Hypoxia did not change expression of steroid receptors

Effect of hypoxia on cell morphology of cultured primary rat microglia. (a) Under normoxia conditions, a large fraction of cells had three or more primary processes with ramifications, and only few cells showed two or less processes. Steroid hormones did not modify the appearance and ratio. (b) Hypoxia changed the cell morphology. Most of the cells had two or less processes and only few cells revealed a multi-process bearing phenotype. Again, steroid hormones showed no influence
As a consequence of hypoxia, not only the oxygen-sensing Hif1a is significantly increased but also the expression of a subset of pro-inflammatory and oxidative stress-related genes in microglial cells (Figs. 3a–f). We found that TNFa, Hif1a, and IL1b transcript levels are all elevated in the absence of O2, whereas VEGFa, CCL5, and IL10 expression was not significantly affected. P but not E2 antagonized the induction of TNFa (Fig. 3a) and IL1b (Fig. 3e), whereas E2 slightly increased Hif1a compared to hypoxia, whereas P had no effect on Hif1a induction (Fig. 3b). In addition, IL10 expression which remained unchanged after hypoxia was slightly but not significantly down-regulated by P (Fig. 3f).

Effect of hypoxia and steroid hormone treatment on gene expression levels in cultured primary rat microglia. Hypoxia increased TNFa (a), Hif1a (b), and IL1b (e) mRNA expression. These effects were partially antagonized by P (TNFa and IL1b). In addition, P reduced IL10 mRNA expression (f). Asterisk (*) indicates p ≤ 0.05 (compared to N) and triple asterisks (***) indicate p ≤ 0.01 (compared to N). a p ≤ 0.05 (compared to P). Data represent means ± SEM from four independent experiments
In order to evaluate the influence of hypoxia and steroid hormone treatment on the different functional activation states of microglia cells, the classical M1 and alternative M2 profile, we have analyzed different subtype-specific genes such as iNOS as a M1 marker and Trem2 as well as Arg1 as M2 markers (Fig. 4). Hypoxia increased iNOS and decreased Trem2 and Arg1 mRNA levels. E2 and P antagonized hypoxia effects on Trem2 and Arg1, and E2 even increased Arg1 compared to normoxia (Figs. 4b–d). Ratios of M1 vs. M2 markers delineate that hypoxia shifts the microglia phenotype towards M1, whereas the simultaneous treatment with E2 and P returned these effects (Figs. 4d–e).

Effect of hypoxia and steroid hormone treatment on gene expression related to the M1 and M2 phenotype in cultured primary rat microglia. (a) Hypoxia increased iNOS mRNA levels. This effect was not changed by E2 but inhibited by P. Hypoxia reduced Trem2 (b) and Arg1 (c) mRNA levels. P and E2 antagonized these effects, and E2 further boosted Arg1 compared to normoxia and hypoxia; asterisk (*) indicates p ≤ 0.05 (compared to N), double asterisks (**) indicate p ≤ 0.01 (compared to N), and triple asterisks (***) indicate p ≤ 0.001 (compared to N). a p ≤ 0.05 (E2 or P compared to H), b p ≤ 0.01 (E2 compared to H). Data represent means ± SEM from three to five independent experiments
Discussion
Consistent with an earlier report on the microglial-like cell line BV-2, we report here that primary cultures of microglia cells isolated from the cerebral cortex of neonatal rat pups have the ability to individually recognize transient sub-lethal hypoxia by a yet unknown intracellular perceiving mechanism. The resulting cell stress provokes changes in their morphological appearance which is accompanied by an up-regulation of TNFa, IL1b, and Hif1a and a switch from a balanced phenotype towards an activated and secretion M1 status. The concomitant application of E2 and P at different degrees counteracted these functional responses and favored the formation of a less inflammatory and neuroprotective M2 phenotype which is associated with tissue repair and remodeling of the extracellular matrix components.
It has been widely accepted that gonadal sex steroid hormones play an explicit role as neuroprotective factors in the CNS guarding neurons from cell dysfunction, degeneration, and death (Stein 2008; Sherwin 2009; Arevalo et al. 2010; Spence et al. 2011). Besides controlling detrimental pathophysiological mechanisms which are described and listed in the introduction, the regulation and fine-tuning of local and systemic inflammatory responses appears to be a key event in the transmission of steroid-mediated defense strategies (Morale et al. 2006; Kipp and Beyer 2009; Girard et al. 2012; Kipp et al. 2012). The expression of different subtypes of E2 and P receptors in microglia and microglia-like cell lines, either classical nuclear or non-classical membrane-associated and cytoplasmic receptors, support the concept that these steroid hormones directly alter the function and characteristics of this innate and brain resident immune cell type (Sierra et al. 2008; Spence et al. 2011; Wood 2011; Habib et al. 2013). Among those receptors, the presence and signaling through microglial ERb and Pgrmc1 as well as mPRa receptors has been demonstrated to be beneficial in acute traumatic brain injury (Bali et al. 2013; Meffre et al. 2013), chronic experimental autoimmune encephalomyelitis (EAE) (Saijo et al. 2011; Wu et al. 2013), and lipopolysaccharide-induced brain inflammation (Brown et al. 2010; Loram et al. 2012). It is important to note that neuroprotection by E2 and P also comprises the firsthand activation and functional regulation of astroglial cell function as shown by numerous experimental studies (Dhandapani and Brann 2007; Kipp and Beyer 2009; Sayeed and Stein 2009; Arevalo et al. 2010). Interestingly, ERa-dependent signal transduction in astroglia seems to be more decisive to transmit neuroprotection than ERb in the EAE and spinal cord injury model (Spence et al. 2011; Hu et al. 2012a; Spence et al. 2013). With respect to hypoxic brain damage, the expression of both ER receptor subtypes is elevated in the penumbra (Takahashi et al. 2004; Dang et al. 2011). Concordantly, both steroid receptor subsets, most likely by affecting activated astroglia and microglia at the same time, take part in the propagation of E2-mediated nerve cell protection (Cimarosti et al. 2006; Shin et al. 2013). Although not explicitly mentioned here, this also applies for P, since this gestagen also targets both glia types and mediates neuroprotection under hypoxic conditions (Sayeed and Stein 2009; Liu et al. 2012; Habib et al. 2013; Habib P et al. Hypoxia-induced expression of aquaporin-4, cyclooxygenase 2 and hypoxia inducible factor 1alpha in mouse cortical astroglia is inhibited by sex steroids, in revision).
From the above cited studies, the question arises what is the detailed function of E2 and P to dampen or even prevent the detrimental local inflammatory scenario? In the initial reperfusion phase after stroke, mediators such as iNOS, TNFa, IL1β, and CCL5 are massively induced and implicated in the fine-tuning of the inflammatory cascade as well as in the context of inflammatory cell–cell communication (Neumann et al. 2006; Bajramovic 2011; Woodruff et al. 2011). This clearly defines this early period as anti-inflammatory and protective therapeutic time window with respect to microglia regulation. Microglia-triggered inflammatory action on nerve cells is equally regulated by their competence to detect damaged neurons by transmembrane receptors and their receptiveness for soluble and paracrine-acting mediators arising from neighboring activated astroglia (Liu et al. 2011; Linnartz and Neumann 2013). Under ischemic conditions, Hif1a and Toll-like receptor-4 appear to be important determinants in microglia within the lesioned brain area and implicated in microglia activation (Weinstein et al. 2010). Hif1a is a transcriptional regulator of O2 homeostasis, typically boosted in different cell types and tissues under hypoxia and modulates adaptive cell responses resulting from hypoxia such as angiogenesis, erythropoiesis, energy metabolism, and survival (Freeman and Barone 2005; Ong and Hausenloy 2012). VEGF is also characteristically up-regulated in the lesioned core and penumbra region after stroke and implicated in the restoration of the disrupted blood–brain barrier, but this effect seems to be mainly involve activated local astroglia (Kaur and Ling 2008; Narantuya et al. 2010; Weinstein et al. 2010; Dang et al. 2011). In vivo and in vitro, gonadal sex steroids have been demonstrated to control the transcription levels of the abovementioned mediators in the ischemic cerebral cortex and down-regulate hypoxia-induced expression (Gibson et al. 2005; Brown et al. 2010; Giraud et al. 2010; Dang et al. 2011). Generally, hypoxia-related inflammatory signaling occurs in activated astroglia and microglia comparably although at different degrees and time intervals. This together with the sudden infiltration of peripheral immune cells such as T cells and macrophages often makes it impossible to attribute a specific immune-relevant function to one of the two glia cell types. Therefore, hypoxic in vitro models could better provide insight into cell-specific responses but being aware that cell–cell communication between different brain cell types is switched off. We have recently analyzed the immortalized neonatal murine BV-2 microglia-like cell line to study their competence to respond to hypoxia and being regulated by sex steroids (Habib P et al. Hypoxia-induced expression of aquaporin-4, cyclooxygenase 2 and hypoxia inducible factor 1alpha in mouse cortical astroglia is inhibited by sex steroids, in revision). We could show that E2 and P dampen certain aspects of inflammatory signaling and abrogate hypoxia-induced phagocytotic activity. These data confirmed observations published by Baker et al. who have described the anti-inflammatory potential of E2 via ERb signaling in this cell line (Baker et al. 2004). The present study extends these findings to primary rat microglia. It is noteworthy that there exist subtle but meaningful differences between BV-2 cells and primary microglia. Primary rat microglia cells but not murine BV-2 microglia-like cells express GPR30, and Pgrmc1 and mPRA only appear to be up-regulated in BV-2 cells after hypoxia, whereas these receptors are constitutively expressed in primary rat microglia. In common, both microglia cell types similarly respond to sub-lethal oxidative challenges in vitro by expressing subsets of pro-inflammatory and oxidative stress-related factors with the exception of CCL5 and IL10 which were only augmented after hypoxia and down-regulated by sex steroids in BV-2 cells but not in the primary microglia. An intriguing aspect of both studies is that both gonadal steroids reverse the hypoxia-inducible and pro-inflammatory M1 phenotype and strengthen the protective M2 subtype. Our findings clearly demonstrate that the levels of the two M2 subtype markers Trem2 and Arg1 which are reduced after hypoxia are either restored or even boosted after P and E2 application, respectively. Trem2 mediates diverse functions on microglia including inhibition of Toll-like receptor signaling, reduction of phagocytotic activity, and release of pro-inflammatory cytokines suggesting an anti-inflammatory influence (Bajramovic 2011). The Arg1 enzyme competes with the NOS enzymes for the substrate, thus reducing pro-inflammatory NO production, and controls T lymphocyte activation thereby suppressing local inflammation (Bronte et al. 2003). Macrophages and microglia can adopt different phenotypes and polarization which are crucial for shaping inflammatory responses to injury. Using BV-2 cells, the M2 subtype protected the hippocampal neurons against oxygen–glucose deprivation (Girard et al. 2013). Although the phenotype-specific role of microglia in ischemic stroke is still poorly understood, first evidence comes from a recent animal study revealing that the local microglia and invading macrophages assume the alternatively activated and protective M2 phenotype at the initial hypoxic stage (Hu et al. 2012b). As time after stroke onset progresses, these innate immune cells transform into the M1-polarized phenotype. In support for this assumption are data provided by Frieler et al. who have shown that knockdown of mineralocorticoid receptors which are normally exacerbating cell degeneration after stroke reduced the infarct volume by suppressing classical M1 markers and preserving alternative M2 markers (Frieler et al. 2011).
In general, neuroprotection by E2 and P after an acute ischemic event in the brain may be the result of a multi-faceted action of these steroid hormones on different neuropathological processes at the same time and during the course of stroke-related pathophysiology. Besides dampening excitotoxicity and edema formation as well as interfering directly with detrimental intracellular signaling cascades, the fine-tuning of local inflammation appears to have a major impact on stroke outcome. Considering the previous data showing reduced activation of microglia after hypoxia (Dang et al. 2011) and function of microglia-like BV-2 cells (Habib et al. 2013), we propose that E2 and/or P-mediated attenuation of microglia-dependent inflammatory responses in the acute ischemic phase take part in mediating neuroprotection. This might involve the preservation of the M2 and prevention of the M1 microglia phenotype. In addition, local astroglia is implicated in the balance of inflammatory processes, and together with peripheral immune cells, these different glia cell types, macrophages and lymphocytes appear to be a good target for steroid regulation. Further studies have to address the precise roles of these cells and their intercommunication during ischemia-related damaging events.
References
Arevalo MA, Santos-Galindo M, Bellini MJ, Azcoitia I, Garcia-Segura LM (2010) Actions of estrogens on glial cells: implications for neuroprotection. Biochim Biophys Acta 1800:1106–1112
Arnold S, Beyer C (2009) Neuroprotection by estrogen in the brain: the mitochondrial compartment as presumed therapeutic target. J Neurochem 110:1–11
Bajramovic JJ (2011) Regulation of innate immune responses in the central nervous system. CNS & Neurolog Disord – Drug Targ 10:4–24
Baker AE, Brautigam VM, Watters JJ (2004) Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor beta. Endocrinology 145:5021–5032
Bali N, Arimoto JM, Morgan TE, Finch CE (2013) Progesterone antagonism of neurite outgrowth depends on microglial activation via Pgrmc1/S2R. Endocrinology 154:2468–2480
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98
Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P (2003) L-arginine metabolism in myeloid cells controls T-lymphocyte function. Trends Immunol 24:302–306
Brown CM, Mulcahey TA, Filipek NC, Wise PM (2010) Production of proinflammatory cytokines and chemokines during neuroinflammation: novel roles for estrogen receptors alpha and beta. Endocrinology 151:4916–4925
Cimarosti H, ÓShea RD, Jones NM, Horn AP, Simao F, Zamin LL, Nassif M, Frozza R, Netto CA, Beart PM, Salbego C (2006) The effects of estradiol on estrogen receptor and glutamate transporter expression in organotypic hippocampal cultures exposed to oxygen-glucose deprivation. Neurochem Res 31:483–490
Dang J, Mitkari B, Kipp M, Beyer C (2011) Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain Behav Immun 25:715–726
Dhandapani KM, Brann DW (2007) Role of astrocytes in estrogen-mediated neuroprotection. Exp Gerontol 42:70–75
Freeman RS, Barone MC (2005) Targeting hypoxia-inducible factor (HIF) as a therapeutic strategy for CNS disorders. Curr Drug Targets CNS Neurol Disord 4:85–92
Frieler RA, Meng H, Duan SZ, Berger S, Schütz G, He Y, Xi G, Wang MM, Mortensen RM (2011) Myeloid-specific deletion of the mineralocorticoid receptor reduces infarct volume and alters inflammation during cerebral ischemia. Stroke 42:179–185
Gelinas DS, McLaurin J (2005) PPAR-alpha expression inversely correlates with inflammatory cytokines IL-1beta and TNF-alpha in aging rats. Neurochem Res 30:1369–1375
Gibson CL, Constantin D, Prior MJ, Bath PM, Murphy SP (2005) Progesterone suppresses the inflammatory response and nitric oxide synthase-2 expression following cerebral ischemia. Exp Neurol 193:522–530
Girard C, Liu S, Adams D, Lacroix C, Sineus M, Boucher C, Papadopoulos V, Rupprecht R, Schumacher M, Groyer G (2012) Axonal regeneration and neuroinflammation: roles for the translocator protein 18 kDa. J Neuroendocrinol 24:71–81
Girard S, Brough D, Lopez-Castejon G, Giles J, Rothwell NJ, Allan SM (2013) Microglia and macrophages differentially modulate cell death after brain injury caused by oxygen-glucose deprivation in organotypic brain slices. Glia 61:813–824
Giraud SN, Caron CM, Pham-Dinh D, Kitabgi P, Nicot AB (2010) Estradiol inhibits ongoing autoimmune neuroinflammation and NFkappaB-dependent CCL2 expression in reactive astrocytes. Proc Natl Acad Sci U S A 107:8416–842
Habib P, Dreymüller D, Ludwig A, Beyer C, Dang J (2013) Sex steroid hormone-mediated functional regulation of microglia-like BV-2 cells during hypoxia. J Steroid Biochem Mol Biol 138:195–205
Hu R, Sun H, Zhang Q, Chen J, Wu N, Meng H, Cui G, Hu S, Li F, Lin J, Wan Q, Feng H (2012a) G-protein coupled estrogen receptor 1 mediated estrogenic neuroprotection against spinal cord injury. Crit Care Med 40:3230–3237
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J (2012b) Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43:3063–3070
Johann S, Beyer C (2012) Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J. Steroid Biochem. Mol. Biol. doi:pii: S0960-0760(12)00239-7. 10.1016/ j.jsbmb.2012.11.006. (in press).
Kaur C, Ling EA (2008) Blood brain barrier in hypoxic-ischemic conditions. Curr Neurovasc Res 5:71–81
Kigerl KA, Gendel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009) Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29:13435–13444
Kipp M, Beyer C (2009) Impact of sex steroids on neuroinflammatory processes and experimental multiple sclerosis. Front Neuroendocrinol 30:188–200
Kipp M, Berger K, Clarner T, Dang J, Beyer C (2012) Sex steroids control neuroinflammatory processes in the brain: relevance for acute ischaemia and degenerative demyelination. J Neuroendocrinol 24:62–70
Li L, Lu J, Tay SS, Moochhala SM, He BP (2007) The function of microglia, either neuroprotection or neurotoxicity, is determined by the equilibrium among factors from activated microglia in vitro. Brain Res 1159:8–17
Linnartz B, Neumann H (2013) Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia 61:37–46
Liu A, Margaill I, Zhang S, Labombarda F, Coqueran B, Delespierre B, Liere P, Marchand-Leroux C, ÓMalley BW, Lydon JP, De Nikola AF, Sitruk-Ware R, Mattern C, Plotkine M, Schumacher M, Guennoun R (2012) Progesterone receptors: a key for neuroprotection in experimental stroke. Endocrinology 153:3747–3757
Liu W, Tang Y, Feng J (2011) Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci 89:141–146
Loane DJ, Byrnes KR (2010) Role of microglia in neurotrauma. Neurotherapeutics 7:366–377
Loram LC, Sholar PW, Taylor FR, Wiesler JL, Strand KA, Berkelhammer D, Day HE, Maier SF, Watkins LE (2012) Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrinology 37:1688–1699
Meffre D, Labombarda F, Delespierre B, Chastre A, De Nikola AF, Stein DG, Schumacher M, Guennoun R (2013) Distribution of membrane progesterone receptors alpha in the male mouse and rat brain and its regulation after traumatic brain injury. Neuroscience 231:111–124
Mor G, Nilsen J, Horvath T, Bechmann I, Brown S, Garcia-Segura LM, Naftolin F (1999) Estrogen and microglia: a regulatory system that affects the brain. J Neurobiol 40:484–496
Morale MC, Serra PA, Lépiscopo F, Tirolo C, Caniglia S, Testa N, Gennuso F, Giaquinta G, Rocchitta G, Desole MS, Miele E, Marchetti B (2006) Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience 138:869–878
Narantuya D, Nagai A, Sheikh AM, Masuda J, Kobayashi S, Yamaguchi S, Kim SU (2010) Human microglia transplanted in rat focal ischemic brain induce neuroprotection and behavioral improvement. PLoS One 5:e11746. doi:10.1371
Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K (2006) Microglia provide neuroprotection after ischemia. FASEB J 20:714–716
Ong SG, Hausenloy DJ (2012) Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol Ther 136:69–81
Pawlak J, Brito V, Küppers E, Beyer C (2005) Regulation of glutamate transporters GLAST and GLT-1 expression in astrocytes by estrogen. Mol Brain Res 138:1–7
Ransohoff RM, Brown MA (2012) Innate immunity in the central nervous system. J Clin Invest 122:1164–1171
Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK (2011) An adiol-ERβ-CtBP transexpression pathway negatively regulates microglial-mediated inflammation. Cell 145:584–595
Sayeed I, Stein DG (2009) Progesterone as a neuroprotective factor in traumatic and ischemic brain injury. Prog Brain Res 175:219–237
Sherwin BB (2009) Estrogen therapy: is time of initiation critical for neuroprotection? Nat Rev Endocrinol 5:620–627
Shin JA, Yang SJ, Jeong SI, Park HJ, Choi YH, Park EM (2013) Activation of estrogen receptor ß reduces blood–brain barrier breakdown following ischemic injury. Neuroscience 235:165–173
Sierra A, Gottfried-Blackmore A, Miner TA, McEwen BS, Bulloch K (2008) Steroid hormone receptor expression and function in microglia. Glia 56:659–674
Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ, Cao Y, Bondar G, Lam J, Ao Y, Sandoval F, Suriany S, Sofroniew MV, Voskuhl RR (2011) Neuroprotection mediated through estrogen receptor-alpha in astrocytes. Proc Natl Acad Sci U S A 108:8867–8872
Spence RD, Wisdom AJ, Cao Y, Hill HM, Mongerson CR, Stapornkul B, Itoh N, Sofroniew MV, Voskuhl RR (2013) Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERα signaling on astrocytes but not through ERß signaling on astrocytes and neurons. J Neurosci 33:10924–10933
Stein DG (2008) Progesterone exerts neuroprotective effects after brain injury. Brain Res Rev 57:386–397
Suzuki S, Brown CM, Wise PM (2009) Neuroprotective effects of estrogens following ischemic stroke. Front Neuroendocrinol 30:201–211
Takahashi N, Tonchev AB, Koike K, Murakami K, Yamada K, Yamashima T, Inoue M (2004) Expression of estrogen receptor-beta in the postischemic monkey hippocampus. Neurosci Lett 369:9–13
Ulbrich C, Zendedel A, Habib P, Kipp M, Beyer C, Dang J (2012) Long-term cerebral cortex protection and behavioral stabilization by gonadal steroid hormones after transient focal hypoxia. J Steroid Biochem Mol Biol 131:10–16
Weinstein JR, Koerner IP, Möller T (2010) Microglia in ischemic brain injury. Future Neurol 5:227–246
Wood H (2011) Estrogen receptor ligands suppress inflammatory responses in astrocytes and microglia. Nat Rev Neurosci 355:7
Wu WF, Tan XJ, Dai YB, Krishnan V, Warner M, Gustafsson JA (2013) Targeting estrogen receptor ß in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 110:3543–3548
Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV (2011) Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener 6:11
Author information
Authors and Affiliations
Corresponding author
Additional information
Jon Dang and Cordian Beyer contributed equally to this study.
Rights and permissions
About this article
Cite this article
Habib, P., Slowik, A., Zendedel, A. et al. Regulation of Hypoxia-Induced Inflammatory Responses and M1-M2 Phenotype Switch of Primary Rat Microglia by Sex Steroids. J Mol Neurosci 52, 277–285 (2014). https://doi.org/10.1007/s12031-013-0137-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-013-0137-y