
Abstract
Dehydroepiandrosterone (DHEA) is the most abundant circulating steroid hormone in humans, produced by the adrenals, the gonads and the brain. DHEA was previously shown to bind to the nerve growth factor receptor, tropomyosin-related kinase A (TrkA), and to thereby exert neuroprotective effects. Here we show that DHEA reduces microglia-mediated inflammation in an acute lipopolysaccharide-induced neuro-inflammation model in mice and in cultured microglia in vitro. DHEA regulates microglial inflammatory responses through phosphorylation of TrkA and subsequent activation of a pathway involving Akt1/Akt2 and cAMP response element-binding protein. The latter induces the expression of the histone 3 lysine 27 (H3K27) demethylase Jumonji d3 (Jmjd3), which thereby controls the expression of inflammation-related genes and microglial polarization. Together, our data indicate that DHEA-activated TrkA signaling is a potent regulator of microglia-mediated inflammation in a Jmjd3-dependent manner, thereby providing the platform for potential future therapeutic interventions in neuro-inflammatory pathologies.
Similar content being viewed by others


Introduction
Dehydroepiandrosterone (DHEA, 3β-hydroxy-5-androstene-17-one) and its conjugate ester, DHEA sulfate (DHEA-S), are the most abundant circulating steroid hormones in humans.1, 2, 3, 4 DHEA is produced by the adrenals, the gonads1 and the central nervous system.3, 5, 6, 7 In the brain, DHEA is synthesized by neurons and astrocytes,8, 9 which is why DHEA is also considered a neurosteroid.
DHEA can bind to many different receptors and activate several pathways,2, 10 including GABA-A, NMDA and sigma 1 receptors,2, 11 G protein-coupled receptors12, 13, 14 or nuclear receptors, such as the peroxisome proliferator-activated receptor alpha (PPARα), the pregnane X receptor, the estrogen receptors α and β (ERα and ERβ, respectively) or androgen receptors.2, 10, 15 Furthermore, DHEA was found to bind and activate the nerve growth factor (NGF) high-affinity receptor, tropomyosin-related kinase A (TrkA),16 other Trk receptors, such as TrkB and TrkC,17 as well as the pan-neurotrophin receptor p75NTR.16
Circulating DHEA can cross the blood–brain barrier (BBB) and reach the brain due to its lipophilic nature.18 In contrast, DHEA-S, which is a hydrophilic molecule, does not readily cross the BBB.3, 19 In humans, the concentration of DHEA in the brain is 4–6.5 times higher than in the plasma.20 In rodents, DHEA(S) serum concentrations are very low, as rodent adrenals lack the P450c17 enzyme that converts pregnenolone to DHEA.3, 21 However, in rodents, like in humans, DHEA is synthesized in the brain.3, 22 This fact, together with DHEA's propensity to cross the BBB, suggests that rodents represent an appropriate animal model for studying the function of DHEA in the central nervous system.3
In humans, serum concentration of DHEA(S) peak during the third decade of life and decline with advancing age.1 The age-related decline of circulating DHEA(S) concentration has been associated with development of neurological, neuropsychiatric and neurodegenerative diseases, such as depression and Alzheimer’s disease.3, 23, 24, 25, 26, 27 Administration of DHEA to animal models of multiple sclerosis or Parkinson’s Disease had beneficial effects.28, 29, 30, 31, 32, 33 This could be attributed to its neuroprotective functions3, 12, 13, 16, 32, 34, 35, 36, 37 as well as to its anti-inflammatory properties,3, 28, 29, 38, 39, 40, 41, 42, 43, 44, 45 including modulatory effects on microglial function.29, 38, 39
Microglial cells are resident immune cells of the central nervous system.46, 47 In their inactive state, they are highly ramified cells, constantly surveying their environment.47 When activated by pathogens or an injury they acquire an ameboid phenotype, produce nitric oxide and reactive oxygen species, and express pro-inflammatory cytokines, chemokines and MHCII.47, 48, 49, 50, 51 Normally, the acute phase of inflammation is followed by a resolution phase, during which microglia return to a quiescent state.49, 51 If resolution is perturbed, chronic inflammation may lead to neurodegeneration.49, 51 Chronic neuro-inflammation is a feature of many neurodegenerative diseases, such as Alzheimer’s disease and multiple sclerosis.49, 51
Given the beneficial role of DHEA in animal models of neurodegenerative disease and the crucial role of microglia-related inflammation in such pathologies, we sought to study if DHEA modulates microglial function in acute neuro-inflammation and to understand the underlying molecular mechanisms. Our findings show that DHEA reduces the pro-inflammatory responses of microglia in the context of acute lipopolysaccharide (LPS) -induced brain inflammation. This effect is at least in part mediated by the TrkA receptor, which, upon interaction with DHEA, triggers phosphorylation of Akt1 and Akt2 and subsequent activation of cAMP response element-binding protein (CREB). CREB in turn induces gene expression of the histone 3 lysine 27 (H3K27) demethylase Jumonji d3 (Jmjd3), which controls pro-inflammatory gene expression and thus regulates microglia-mediated inflammation. This study provides novel insights into the mechanisms regulating microglia-driven innate immune responses.
Materials and methods
Mice
Eight-week old male C57BL/6 mice (Janvier Labs, Le Genest-Saint-Isle, France) were used. An estimate of at least 3–5 mice per group was used in experiments. No blinding or randomization was performed. Mice were injected on 2 consecutive days intraperitoneally (i.p.) with freshly prepared DHEA (80 mg kg−1) in a 500 μl solution containing phosphate-buffered saline (PBS) with 4.5% ethanol and 1% bovine serum albumin (BSA) or with the same solution alone as control. One hour after the second dose of DHEA or control solution, mice were injected i.p. with LPS (5 mg kg−1) (Ultrapure LPS, Escherichia coli 0111:B4, Invivogen, San Diego, CA, USA). After 2 or 4 h of LPS treatment, mice were euthanized and organs were retrieved for further analysis. Animal experiments were approved by the Landesdirektion, Dresden, Germany.
Microglial cell sorting
Isolated brains were grinded on a 100 μm pore-sized cell strainer in PBS supplemented with 0.5% fetal bovine serum (FBS, Life Technologies, Paisley, UK) on ice. Cells were centrifuged at 1200 r.p.m. for 7 min at 4 °C and diluted in PBS containing 0.5% FBS. Two hundred μl of anti-myelin magnetic beads (MACS Miltenyi Biotec, Bergisch Gladbach, Germany) were added to each sample and cells were incubated for 15 min at 4 °C. Then PBS with 0.5% BSA was added to each tube, samples were centrifuged at 300 g for 10 min at 4 °C, the cell pellet was diluted in PBS with 0.5% BSA and passed through a LS column (Miltenyi Biotec).
Cells were stained with 1:100 anti-CD45-PE (553081, BD, Heidelberg, Germany), 1:100 anti-Ly6G-APC (560599, BD) and 1:100 anti-CD11b-PerCP (101230, Biolegend, Fell, Germany) and sorted with a BDFACS ARIA apparatus (BD) using the BD FACSDIVA v8.0.1 software (BD). Cells were collected in lysis buffer supplemented with beta-mercaptoethanol (NucleoSpin RNA kit, Macherey-Nagel, Duren, Germany).
Cell culture
Primary microglial cells were isolated from brains of C57BL/6 mice. The brains were transferred to an enzymatic solution, consisting of 0.25 mg ml−1 papain (Sigma-Aldrich, Munich, Germany), 1.2 U ml−1 dispase II (Roche, Mannheim, Germany), 1 mm cysteine (Sigma-Aldrich) and 0.5 mm EDTA (Sigma-Aldrich) diluted in DMEM (Invitrogen, Darmstadt, Germany), minced and incubated at room temperature for 30 min under continuous stirring. The enzymatic reaction was quenched by the addition of PBS with 20% FBS. After centrifugation at 1200 r.p.m. for 10 min at room temperature, cells were re-suspended in 0.5 mg ml−1 DNase I (ThermoFisher Scient, Waltham, MA, USA) in PBS and incubated for 5 min at room temperature. Cells were gently dissociated and passed through a 70 μm pore-sized cell strainer. Isolated cells were put in culture in DMEM/F12 with Glutamax, 10% FBS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin (all from Invitrogen) and 5 ng ml−1 of murine recombinant granulocyte and macrophage colony-stimulating factor (Peprotech, Rocky Hill, NJ, USA) in poly-l-lysine (Sigma-Aldrich) -coated flasks and maintained in culture at 37 °C and 5% CO2. Four days thereafter, medium was changed every 2–3 days. When a continuous feeder layer had formed (14–17 days after isolation), loosely attached microglial cells were collected by gentle shaking of the flasks and centrifugation of the supernatants. Before treatments, microglial cells were cultured for 3 days in medium without granulocyte and macrophage colony-stimulating factor.
The murine brain-derived microglia cell line BV2 (ICLC, Genova, Italy) was cultured in RPMI medium (Invitrogen) supplemented with 10% FBS at 37 °C and 5% CO2.
Cell treatments
Microglial cells were treated with 10−7 m DHEA (Sigma-Aldrich) or control solution (containing same dilution of ethanol), and DHEA-7-(O-carboxymethyl oxime)-BSA (DHEA-BSA) (Steraloids, Newport, RI, USA) or control solution (with same BSA concentration) with or without LPS (100 ng ml−1). All treatments were done in phenol red-free RPMI 1% FBS and 1% Pen/Strep. FBS and DHEA-BSA stock solutions were stripped with charcoal prior to use, as described,35, 52 in order to remove endogenous steroids or non-conjugated DHEA, respectively. In some experiments cells were treated with 100 ng ml−1 NGF (2.5 S) (Millipore, Darmstadt, Germany). In other experiments cells were pre-treated for 30 min with 1 μm TrkA inhibitor (inhibitor of the kinase activity of TrkA) (Calbiochem—Merck Millipore), 1 μm CREB/CBP interaction inhibitor (Merck Millipore), 1 μm Wortmannin (R&D Systems, Wiesbaden-Nordenstadt Germany), 10 μm UO126 (Merck Millipore) or 10 μm U-73122 (Merck Millipore). The number of independent in vitro experiments was estimated based on our experience with these methods. No randomization was performed.
Cell transfection and generation of stable clones
BV2 cells (50 000 cells per well in a 24-well plate) were transfected with siRNA targeting TrkA (10 nm), Jmjd3 (30 nm) or control non-targeting siRNA (10 or 30 nm). Primary microglia cells were transfected with siRNA against Akt1 (30 nm) or siRNA against Akt2 (30 nm) or control siRNA (30 nm). All transfections were performed using Lipofectamine RNAimax (Invitrogen, Darmstadt, Germany) and the reverse transfection method according to manufacturer’s instructions. All siRNAs were from Dharmacon.
For generation of stable clones, BV2 cells seeded in six-well plates (1 × 106 cells per well) were transfected with 4 μg DNA empty pCMV plasmid or pCMV-KCREB plasmid (both kindly provided by A Mauviel, Curie Institute, Paris, France) and 8 μl Lipofectamine LTX Reagent (Invitrogen) according to manufacturer‘s instructions. pCMV-KCREB over-expresses CREB containing mutations in its DNA-binding domain. Two days after transfection, 375 μg ml−1 Geneticin (G418) (Life Technologies) was added to the medium to select transfected clones. Stably transfected clones were maintained by culturing in G418-suplemented medium.
RNA extraction and gene expression analysis
RNA was isolated from frozen brain tissues with the Trizol Reagent (Invitrogen). Total RNA was isolated from cell cultures using the NucleoSpin RNA isolation kit (Macherey-Nagel). Genomic DNA contamination was eliminated by DNase I treatment. One microgram of RNA was reverse transcribed with the iScript cDNA Synthesis kit (Bio-Rad, Munich, Germany). cDNA was analyzed by qPCR using the SsoFast Eva Green Supermix (Bio-Rad), a CFX384 real-time System C1000 Thermal Cycler (Bio-Rad) and the Bio-Rad CFX Manager 3.1 software. The relative amount of the different mRNAs was quantified with the ΔΔCt method while 18 S RNA was used for normalization.53 Primers used are described in Supplementary Table 1.
Western blot
Whole-cell extracts were collected in lysis buffer (10 mm Tris pH 7.4, 1% SDS, 1 mm sodium orthovanadate, 1:400 benzonase (Sigma), supplemented with Mini Protease Inhibitor and Phosphatase Inhibitor Cocktail (Roche)) and denatured by heating at 95 °C for 3 min. Thirty to sixty microgram of proteins were subjected to SDS–polyacrylamide gel electrophoresis, and transferred onto Hybond nitrocellulose membranes (GE Healthcare Europe, Freiburg, Germany). Membranes were blocked for 1 h in 5% non-fat milk diluted in TBS-T buffer (0.15 m NaCl, 2.7 mm KCl, 24.8 mm Tris-base, 0.1% Tween-20), and immunoblotted with anti-phosphoTrkA (Tyr490) (Abcam, Cambridge, MA, USA, ab1445, 1:500), TrkA (Abcam, ab76291, 1:1000), anti-iNos (Abcam, ab15323 1:1000), anti-Jmjd3 (Abcam, ab154126, 1:1000), anti-arginase 1 (Abcam ab60176, 1:1000), anti-phosphoCREB (Cell Signaling Technology, Danvers, MA, USA, #9196, 1:1000), anti-phosphoAkt (Ser473) (Cell Signaling, #4060, 1:1000), anti-phosphoAkt1 (Ser473) (Abcam, ab81283, 1:1000), anti-phosphoAkt2 (Ser474) (Cell Signaling, #8599, 1:1000) and anti-Actin (1:1000, Abcam, ab3280) overnight in 5% BSA or 5% non-fat milk in TBS-T buffer at 4 °C and then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (R&D Systems; Santacruz Biotechnologies, Heidelberg, Germany; and Jackson Immunoresearch, Hamburg, Germany). Band intensity of western blots was quantified with the open source Fiji software (http://fiji.sc/).
Cytokine measurement
Brain lysates were prepared in RIPA Buffer (50 mm Tris-HCl, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, protease inhibitors) by mechanical homogenization. Supernatants were collected from 80-90% confluent cell cultures. Cytokines were measured in brain lysates and cell supernatants using the Meso Scale Discovery (MSD) Technology following manufacturer’s instructions.
Statistical analysis
All values are expressed as the mean±s.e.m. Comparison between groups was made with Mann–Whitney U-test; P⩽0.05 was the significance level.
Results
DHEA reduces LPS-induced microglial inflammation
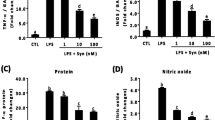
Firstly, we tested whether DHEA regulates neuro-inflammation in an acute LPS-induced brain inflammation model. Notably, DHEA systemically administrated to mice can be detected by liquid chromatography-tandem mass spectrometry in brain regions, such as the cortex, the hippocampus, the hypothalamus and the cerebellum (data not shown). Four hours after systemic LPS administration in C57BL/6 mice, we found a strong induction of pro-inflammatory gene expression such as Tnf, Il6, Il12/23p40 and Mcp1 (Figures 1a and d), as well as enhanced protein concentrations of cytokines, such as Il1beta and Cxcl1 (Figures 1e and f) in whole-brain extracts. A similar inflammatory response was observed upon LPS administration in microglia isolated by fluorescence-activated cell sorting from the brain (Figures 1g and h). Intraperitoneal administration of DHEA before LPS significantly reduced LPS-induced gene expression of pro-inflammatory cytokines and of the chemokine Mcp1 as well as protein concentrations of Il1beta and Cxcl1 in whole-brain extracts (Figures 1a–f). In addition, DHEA pre-treatment decreased LPS-induced Il6 and Mcp1 expression in sorted microglia (Figures 1g and h). The anti-inflammatory effect of DHEA was reproduced in vitro; DHEA blocked the LPS-induced upregulation of iNos, as well as of pro-inflammatory cytokines and chemokines in cultured murine primary microglia (Figures 1i and j) and in the BV2 microglial cell line (Figure 1k).

DHEA inhibits microglia-driven inflammation. (a–d) DHEA (80 mg kg−1) or ethanol-containing control solution (Eth.) was administered i.p. to male C57BL/6 mice twice, at 24 h and 1 h before LPS treatment. Mice were killed 4 h after LPS treatment. Real-time PCR was performed in whole-brain extracts for the indicated genes using 18 S RNA for normalization. Normalized gene expression of brains from mice treated only with ethanol-containing control solution (Eth.) was set as 1. Data are expressed as mean±s.e.m. (n=3;5;5). (e and f). Il1beta and Cxcl1 protein levels were measured in brain lysates from the same mice as in (a–d). Data are expressed as mean±s.e.m. (n=3;5;5). (g and h). Microglial cells were sorted by FACS as CD11b+CD45interLy6G− cells from brains of mice treated as described in (a–f); gene expression was analyzed by qPCR. Normalized gene expression in samples from mice treated with control solution (Eth.) was set as 1; mean±s.e.m. (n=3;5;5). (i and j). Cultured primary murine microglial cells were treated with DHEA (10−7 m) or control solution (containing ethanol at same dilution (Eth.)) twice, at 24 h and 1 h before LPS (100 ng ml−1) incubation (for 4 h). Gene expression was analyzed by qPCR; normalized gene expression of cells treated with LPS and control solution (Eth.) was set in each experiment as 1; mean±s.e.m. (n=3 independent experiments). (k) BV2 microglial cell line was pre-treated for 24 h with DHEA (10−7 M) or ethanol at same dilution (Eth.), as control, then they were treated with a second dose of DHEA (10−7 M) or ethanol-containing control solution for 1 h, followed by stimulation or not by LPS (100 ng ml−1) for 4 h. The normalized gene expression of samples treated with LPS and control solution (Eth.) was set as 1 in each experiment; mean±s.e.m. (n=4 independent experiments). *P⩽0.05 (MWU test). DHEA, dehydroepiandrosterone; FACS, fluorescence-activated cell sorting; LPS, lipopolysaccharide; MWU, Mann–Whitney U-test; qPCR, quantitative PCR.
We also assessed whether DHEA administration had systemic anti-inflammatory properties, by examining livers and spleens that derived from the same mice as the analyzed brain lysates. LPS-induced enhanced expression of inflammatory genes, such as Tnf or iNos in these organs was not affected by DHEA pre-treatment (data not shown). Hence, at least in this specific experimental setting, the anti-inflammatory effect of DHEA was observed specifically in the brain, suggesting that DHEA may preferentially target brain immune cells, such as microglia.
DHEA-treated microglia acquire an M2-like phenotype
Similar to macrophages, microglial cells also acquire different polarization phenotypes, the two extremes of which are the M1 (pro-inflammatory) and the M2 (anti-inflammatory) phenotype.44 Interestingly, along with reduced inflammation, whole-brain extracts from mice treated with DHEA and LPS displayed increased gene expression of M2 polarization markers, such as arginase 1, Ym1 (chitinase-like protein 3), Fizz1 and Klf4 (Kruppel like factor 4) (Figures 2a and d), as well as increased protein concentrations of Il10 (Figure 2e), as compared to brain extracts from mice treated with LPS and control solution. Furthermore, DHEA enhanced gene expression of M2 polarization markers (Figure 2f), secretion of Il10 (Figure 2g) and protein expression of arginase 1 in BV2 microglial cells (Figure 2h).

DHEA induces microglial M2 polarization. (a–d) DHEA (80 mg kg−1) or ethanol-containing control solution (Eth.) was administrated i.p. to male C57BL/6 mice twice, at 24 h and 1 h, before LPS treatment for another 4 h. Real-time PCR was performed in whole-brain extracts for the indicated genes using 18 S for normalization. Normalized gene expression of brains from mice treated only with ethanol-containing control solution (Eth.) was set as 1. Data are expressed as mean±s.e.m. (n=3;5;5). (e). Il10 protein concentration was determined in brain lysates of mice treated as in (a–d). Data are expressed as mean±s.e.m. (n=3;5;5). (f) BV2 cells were pre-treated for 24 h with DHEA (10−7 m) or ethanol at same dilution (Eth.) as control, then they were treated with a second dose of DHEA (10−7 m) or the ethanol-containing control solution for 1 h and finally they were stimulated or not with LPS (100 ng ml−1) for 4 h. The normalized gene expression of samples treated with control solution (Eth.) in the absence of LPS was set as 1 in each experiment; mean±s.e.m. (n=4 independent experiments). (g) BV2 cells were treated as described in (f) and cell culture supernatants were analyzed for Il10 protein levels. Il10 levels of samples treated with control solution (Eth.) and LPS were set as 1 in each experiment; mean±s.e.m. (n=5 independent experiments). (h) BV2 cells were treated with DHEA (10−7 m) or ethanol-containing control solution (Eth.) for 24 h and cell lysates were analyzed by western blot for Arginase 1. Actin was used as a loading control. Three independent experiments were quantified. The intensity of Arg1 and Actin bands was quantified; in each experiment the ratio Arg1/Actin was set as 1 for samples treated with control solution (Eth.); mean±s.e.m. (n=5). *P⩽0.05 (MWU test). DHEA, dehydroepiandrosterone; LPS, lipopolysaccharide; MWU, Mann–Whitney U-test.
Jmjd3 contributes to the anti-inflammatory effects of DHEA in microglia
Jmjd3 is a H3K27 demethylase that has a key role in regulation of macrophage54, 55, 56, 57, 58, 59 and microglial60 inflammatory responses. Jmjd3 expression is induced in macrophages by inflammatory stimuli, such as LPS56 and acts to finetune the LPS-induced transcriptional responses in macrophages by binding to the transcription start sites of genes encoding inflammatory cytokines and chemokines, proteins for microbial recognition, adhesion molecules, immune receptors or transcription factors, negatively regulating their expression.56
LPS treatment for 2 h significantly induced Jmjd3 gene expression in whole-brain extracts (Figure 3a). The induction of Jmjd3 mRNA was also obvious in microglia sorted from brains of LPS-treated mice (Figure 3b), as well as in cultured primary microglia treated ex vivo with LPS (Supplementary Figure 1). Interestingly, pre-treatment of mice with DHEA significantly increased Jmjd3 expression in whole-brain extracts and in sorted microglia, as compared to LPS treatment with vehicle (Figures 3a and b). In BV2 cells DHEA also induced Jmjd3 expression (Figure 3c). We next examined whether Jmjd3 is required for the anti-inflammatory effect of DHEA. siRNA-mediated Jmjd3 knockdown (Figure 3c and Supplementary Figure 2) abrogated the inhibitory effect of DHEA on LPS-induced expression of Tnf (Figure 3d) and iNos (not shown), hence, suggesting that Jmjd3 may contribute to the anti-inflammatory effect of DHEA in microglia.

Jmjd3 mediates the anti-inflammatory effect of DHEA. (a and b) Mice were injected i.p. with DHEA (80 mg kg−1) or ethanol-containing control solution (Eth.) 24 h and 1 h before LPS treatment for another 2 h. Whole brains (a) and FACS sorted microglia (b) were analyzed for Jmjd3 gene expression by qPCR using 18 S RNA for normalization. Normalized Jmjd3 expression of samples deriving from mice treated only with control solution (Eth.) was set as 1; mean±s.e.m.; n=4. (c) BV2 cells were transfected with siRNA targeting Jmjd3 or with control siRNA. Twenty-four hours after transfection they were treated with DHEA (10−7m) or control solution (ethanol at same concentration (Eth.)) and on the next day they were treated with a second dose of DHEA (10−7 m) or control solution together with LPS (100 ng ml−1) for 6 h. Proteins were analyzed by western blot for Jmjd3 and actin. The western blot shown here is one out of two experiments. (d) BV2 cells were transfected with siRNA targeting Jmdj3 or control siRNA and on the next day they were treated with DHEA (10−7M) or ethanol-containing control solution (Eth.). After further 24 h, they were treated with a second dose of DHEA (10−7 m) or control solution for 1 h and finally they were stimulated or not by LPS (100 ng ml−1) for 4 h. Normalized Tnf expression of samples treated with siCtrl, ethanol-containing control solution (Eth.) and LPS was set in each experiment as 1; mean±s.e.m. (n=3 independent experiments). *P⩽0.05; ns (MWU test). DHEA, dehydroepiandrosterone; FACS, fluorescence-activated cell sorting; LPS, lipopolysaccharide; MWU, Mann–Whitney U-test; ns, non-significant; qPCR, quantitative PCR.
TrkA mediates the anti-inflammatory effects of DHEA
We continued to address the signaling mechanisms involved in the regulation of Jmjd3 gene expression by DHEA. First, to assess, whether DHEA mediates its effects through membrane or intracellular receptors, we treated BV2 cells with a membrane-impermeable DHEA-BSA conjugate12 or BSA as a control in the absence or presence of LPS. DHEA-BSA was charcoal-stripped before use in order to remove any free steroid molecules. DHEA-BSA blocked LPS-induced expression of Il6, iNos, Tnf and Il12/23p40 (Supplementary Figure 3A), whereas it increased the gene expression of arginase 1, Fizz1 and Ym1 (Supplementary Figure 3B). These data suggest that membrane-initiated DHEA signals are sufficient to suppress LPS responses and induce M2 polarization in microglia. Thus, the anti-inflammatory effect of DHEA on microglia must be mediated by membrane-localized receptors.
We have previously shown that DHEA binds to the NGF receptor TrkA.16 Microglial cells express TrkA, as also macrophages do (Supplementary Figure 4). Previous reports have also demonstrated expression of TrkA in microglial cells and macrophages.61, 62, 63, 64 Furthermore, TrkA protein expression does not significantly change after LPS treatment in BV2 cells (Supplementary Figure 5). DHEA induced TrkA phosphorylation in microglial cells (Figure 4a) and co-treatment of BV2 cells with DHEA and a TrkA inhibitor abolished the positive effect of DHEA on Jmjd3 levels under both baseline conditions and upon LPS stimulation (Figures 4b and c). Furthermore, knockdown of TrkA by siRNA transfection (Supplementary Figure 6) as well as treatment with TrkA inhibitor reversed the inhibitory effect of DHEA on LPS-induced iNos mRNA and iNos protein expression (Figures 4d and e). In accordance with these findings, the prototype ligand of TrkA, NGF, reduced the expression of iNos in LPS-activated microglia; the inhibitory effect of NGF on iNos protein levels was reversed by TrkA inhibition (Supplementary Figure 7). These data point to an anti-inflammatory role of signaling induced by TrkA ligation and activation in microglia and that TrkA mediates the effects of DHEA on microglia polarization.

TrkA mediates the anti-inflammatory effect of DHEA in microglia. (a) BV2 cells were treated with DHEA (10−7 m) for 10 or 20 min or with ethanol-containing control solution (Eth.) for 20 min. Proteins were analyzed by western blot for phospho-TrkA and Actin. (left) A representative out of four blots. (right) The intensity of the bands of phospho-TrkA and Actin was quantified; the ratio phospho-TrkA/Actin was calculated for each sample and in each experiment it was set as 1 for control-treated samples (Eth. for 20 min); mean±s.e.m. (n=4 independent experiments). (b and c) BV2 cells were pre-treated with TrkA inhibitor (1 μm) or control dimethyl sulfoxide (DMSO) solution for 30 min and then they were treated with DHEA (10−7 m) or ethanol-containing control solution (Eth.) for 24 h in the absence (b) or presence (c) of 100 ng ml−1 LPS. Western blots were performed to measure the levels of Jmjd3 and Actin. The intensity of the bands of Jmjd3 and Actin was quantified and the ratio Jmdj3/Actin was set in each experiment as 1 for samples treated with DMSO and control solution (Eth.) (b), or for samples treated with DMSO, control solution (Eth.) and LPS (c). Data are mean±s.e.m. (n=3 independent experiments). (d) BV2 cells were transfected with siRNA against TrkA or control siRNA, on the next day they were pre-treated with DHEA (10−7 m) or ethanol-containing control solution (Eth.) for 24 h and thereafter, they were treated with a second dose of DHEA (10−7 m) or control solution (Eth.). One hour later they were treated or not with LPS (100 ng ml−1) for 4 h. Normalized gene expression in samples treated with siCtrl, control solution (Eth.) and LPS was set as 1; mean±s.e.m. (n=3 independent experiments). (e) BV2 cells were pre-treated with TrkA inhibitor (1 μm) or control DMSO solution and with DHEA (10−7m) or ethanol-containing control solution (Eth.) for 24 h. On the next day they were treated a second time with DHEA or control solution (Eth.) and with LPS (100 ng ml−1) for 6 h. iNos and Actin protein levels were analyzed by western blot. Here one representative out of three blots is shown. (right) The intensity of the bands of iNos and Actin was quantified and the ratio iNos/Actin was set in each experiment as 1 for samples treated with DMSO, control solution (Eth.) and LPS (n=3 independent experiments). *P⩽0.05; ns (MWU test). DHEA, dehydroepiandrosterone; LPS, lipopolysaccharide; MWU, Mann–Whitney U-test; ns, not significant.
Akt1 and Akt2 mediate the effect of DHEA on Jmjd3 gene expression
We next assessed the intermediate pathways linking DHEA-mediated TrkA activation to Jmjd3 expression. TrkA activates three different signaling cascades: the Shc-Ras-Raf-MEK-ERK, the Shc-PI3K-Akt and the PLCgamma-PKCdelta pathway.65, 66 We first pre-treated microglial cells with UO126 (a MEK inhibitor), Wortmannin (a PI3K inhibitor) or U-73122 (a PLCgamma inhibitor) to block each of these three pathways and incubated the cells with DHEA and LPS. Wortmannin blocked the effect of DHEA on Jmjd3 gene expression (Figure 5a), while the other two inhibitors had no effect (Figure 5a). These findings suggest that the effect of DHEA on promoting Jmjd3 expression is mediated by the PI3K/Akt pathway, but not by the ERK or the PKCdelta pathways. In accordance, DHEA induced phosphorylation of Akt in a TrkA-dependent manner (Figure 5b).

DHEA induces Jmjd3 gene expression through activation of the Akt1/Akt2 pathway. (a) BV2 cells were pre-treated with UO126 (10 μm), U-73122 (10 μm), Wortmannin (1 μm) or DMSO as control for 30 min, and then with DHEA (10−7 m) or ethanol-containing control solution (Eth.) for 24 h. On the next day, treatments were repeated, and 1 h later, cells were treated with LPS (100 ng ml−1) for 4 h. Jmjd3 expression was analyzed by qPCR and 18 S RNA was used for normalization. Normalized gene expression of samples treated with DMSO, control solution (Eth.) and LPS was set as 1; mean±s.e.m. (n=4 independent experiments). (b) BV2 cells pre-treated for 30 min with TrkA inhibitor (1 μm) or same amount of DMSO were stimulated for 30 min with DHEA (10−7 m) or ethanol-containing control solution (Eth.). Cell lysates were analyzed by western blot for phospho-Akt and Actin levels. (left) A representative blot out of three; (right) the intensity of the bands of phospho-Akt and Actin was quantified and the ratio phospho-Akt/Actin was set as 1 in samples treated with DMSO and control solution (Eth.); mean±s.e.m. (n=3 independent experiments). (c) BV2 cells were pre-treated with Wortmannin (1 μm) or control DMSO solution for 30 min followed by treatment with DHEA (10−7m) or ethanol-containing control solution (Eth.) for 30 min. Phospho-Akt1, phospho-Akt2 and Actin were analyzed by western blot. (d) Primary microglial cells were transfected with siRNAs against Akt1, Akt2 or control siRNA and on the next day they were treated with DHEA (10−7m) or ethanol-containing control solution (Eth.) for 24 h. Thereafter, cells were treated with a second dose of DHEA (10−7m) or control solution (Eth.), and 1 h later cells were stimulated or not with 100 ng ml−1 LPS for 4 h. Expression of iNos was analyzed by qPCR and 18 S RNA was used for normalization. Normalized iNos expression of samples treated with siCtrl, control solution (Eth.) and LPS was set in each experiment as 1; mean±s.e.m. (n=3 independent experiments) *P⩽0.05; ns (MWU test). DHEA, dehydroepiandrosterone; LPS, lipopolysaccharide; MWU, Mann–Whitney U-test; ns, not significant.
The Akt family consists of three isoforms, Akt1, Akt2 and Akt3.67, 68 All isoforms are expressed in macrophages.69, 70 However, Akt1 and Akt2 are the isoforms predominantly expressed and mostly studied in the context of macrophage polarization.71, 72, 73 DHEA induced phosphorylation of both Akt1 and Akt2 to a similar degree. DHEA-induced Akt1 and Akt2 phosphorylation was abolished in the presence of Wortmannin (Figure 5c). We then established siRNA-mediated silencing of Akt1 and Akt2 in primary microglia, which was efficient and specific for each isoform (Supplementary Figure 8). Suppression of either Akt1 or Akt2 abrogated the inhibitory effect of DHEA on iNos expression in LPS-activated primary microglia (Figure 5d). Furthermore, siRNA-mediated silencing of Akt1 and Akt2 reduced the stimulatory effect of DHEA on Jmjd3 expression in LPS-stimulated BV2 cells (not shown). Hence, Akt1 and Akt2 both mediate the effect of DHEA on regulation of microglia activation. As Akt1 and Akt2 have opposing roles in M1 or M2 polarization of macrophages,71, 72, 73 we assessed if siRNA-mediated knockdown of Akt1 and Akt2 in primary microglia also affected their polarization. Silencing of Akt1 or Akt2 did not alter LPS- or IL4-induced microglia polarization in contrast to the role of these Akt isoforms in macrophage polarization (data not shown).
CREB is activated by TrkA signaling and induces Jmjd3 gene expression
CREB is a leucine zipper transcription factor that is activated by DHEA and NGF as well as by Akt signaling.35, 74, 75, 76 It regulates several immune-related genes, such as Il6, Il10 or Tnf.77 We therefore tested, whether CREB is activated by DHEA in microglia. CREB phosphorylation was induced by DHEA and this effect was abolished by Wortmannin (Figure 6a), suggesting that CREB is activated by DHEA through an Akt-dependent signaling cascade. Next, we generated BV2 clones overexpressing CREB carrying mutations in its DNA-binding domain (KCREB mutant clones). Although DHEA blocked LPS-induced iNos expression in control clones, it failed to do so in the mutant clones (Figure 6b). Of note, LPS more efficiently induced iNos expression in the KCREB clones indicating a potential anti-inflammatory transcriptional role of CREB (Figure 6b). Finally, a CREB/CBP interaction inhibitor abrogated the effect of DHEA on Jmjd3 expression (Figure 6c). Hence, our results indicate that CREB mediates the anti-inflammatory effects of DHEA. Therefore, we suggest that CREB is activated by DHEA through a TrkA-Akt1/2-CREB signaling pathway and induces Jmdj3 expression. Jmjd3 in turn negatively regulates the expression of LPS-induced pro-inflammatory genes (Figure 6d).

CREB is involved in the transcriptional induction of Jmjd3 expression by DHEA. (a) BV2 cells were pre-treated with Wortmannin (1 μm) or same amount of DMSO for 30 min, then they were incubated for 60 min with DHEA (10−7 m) or ethanol-containing control solution (Eth.) and phospho(Ser133)-CREB levels were measured by western blot. (left) A representative out of three western blots; (right) the ratio phospho-CREB/Actin was calculated for each sample and it was set as 1 in samples treated with DMSO and control solution (Eth.); mean±s.e.m. (n=3 independent experiments). (b) BV2 clones expressing CREB with a mutated DNA-binding domain (KCREB) or control clones (transfected with a control plasmid, pcDNA) were pre-treated with DHEA (10−7 m) or ethanol-containing control solution (Eth.) for 24 h, then they received a second dose of DHEA or control solution (Eth.) in the absence or presence of 100 ng ml−1 LPS for 6 h. iNos expression was analyzed by western blot using Actin as loading control. The intensity of iNos and Actin bands was quantified for each sample and in each experiment the ratio iNos/Actin was set as 1 for control pcDNA clones treated with control solution (Eth.) and LPS; mean±s.e.m. (n=3 independent experiments). (c) BV2 cells were pre-treated with DMSO or CREB/CBP inhibitor (1 μm) for 30 min and were then incubated with ethanol-containing control solution (Eth.) or DHEA (10−7 m) for 24 h in the presence of LPS (100 ng ml−1). Jmjd3 and Actin protein levels were analyzed by western blot. (left) A representative out of four blots; (right) the intensity of the bands of Jmjd3 and Actin was measured and the ratio Jmjd3/Actin was set in each experiment as 1 for samples treated with DMSO, control solution (Eth.) and LPS; mean±s.e.m. (n=4 independent experiments). *P⩽0.05; ns (MWU test). (d) Schematic summary: in microglia, DHEA activates TrkA and triggers the signaling cascade PI3K-Akt1/Akt2-CREB that leads to transcriptional induction of Jmdj3, which in turn downregulates the expression of inflammatory genes. CREB, cAMP response element-binding protein; DHEA, dehydroepiandrosterone; LPS, lipopolysaccharide; MWU, Mann–Whitney U-test; ns, not significant.
Discussion
DHEA is an adrenal steroid hormone, which is present in relatively high concentrations in the brain,3, 5, 6, 8, 9 due to its peripheral production and its BBB-penetrable nature, as well as its abundant production by neurons and astrocytes.3, 5, 6, 8, 9 In the present work, we have identified a novel mechanism, by which DHEA reduces endotoxin-induced pro-inflammatory microglia activation.
DHEA is a precursor of androgen and estrogen. However, its effect in microglia described here is, at least in part, independent of its conversion to other steroid hormones, as the membrane-impermeable conjugate DHEA-BSA had a similar anti-inflammatory effect in microglia. These data suggested that the effect of DHEA is mediated through receptors/binding sites localized on the cell membrane. Moreover, DHEA induced within few minutes TrkA phosphorylation, whereas TrkA inactivation by a highly selective TrkA inhibitor abrogated the anti-inflammatory effect of DHEA. This receptor was recently identified as an important receptor for the DHEA-mediated neuroprotective effects.16, 36 In accordance with these findings, NGF, the classical TrkA ligand, can also downregulate inflammatory responses of microglial cells and monocytes/macrophages. Moreover, the anti-inflammatory effect of NGF in microglia was blocked by TrkA inhibition61, 62, 78 (Supplementary Figure 7).
TrkA phosphorylation by DHEA was rapidly followed by Akt phosphorylation. The latter effect was blunted by a TrkA inhibitor, which diminishes ligand-induced TrkA phosphorylation and targets the kinase activity of the receptor.79 This finding suggests that phosphorylation of TrkA is required for downstream activation of the Akt/CREB pathway, leading to enhanced Jmjd3 expression.
It was previously shown that Akt1 and Akt2 deletion impact on macrophage polarization in an opposite fashion; Akt1 deletion induces M1, while Akt2 deletion favors M2 polarization.71, 72, 73 However, in microglia, siRNA-mediated knockdown of Akt1 or Akt2 did not affect their polarization. Further in vivo studies are needed to address the role of Akt1 and Akt2 in microglia polarization, for instance by engaging Akt1- and Akt2-deficient mice. Notably, both Akt1 and Akt2 mediated the effect of DHEA on the CREB/Jmjd3 pathway, suggesting that both Akt1 and Akt2 are involved in signaling initiated upon TrkA activation in microglia. That the effect of DHEA on microglia polarization is mediated by both Akt1 and Akt2 should be verified in vivo with Akt1- and Akt2-deficient mice in the future.
Jmjd3 has been implicated in the regulation of inflammatory responses in macrophages and microglia by binding to the promoter of inflammation-related genes.55, 56, 58, 59, 60 Inflammatory stimuli, such as LPS, induce Jmjd3 gene expression in macrophages.54, 55, 56 We here found that Jmjd3 is upregulated by LPS in microglia as well. Pre-treatment of microglia with DHEA further enhanced the LPS-induced expression of Jmjd3 and reduced in parallel LPS-triggered inflammation. Of note, a several-hour pre-treatment with DHEA was necessary for DHEA to exert its anti-inflammatory actions; simultaneous application of DHEA and LPS did not inhibit the pro-inflammatory effects of LPS (data not shown). This observation stands in accordance with the fact that Jmjd3 gene expression and translation are involved in the anti-inflammatory effects of DHEA.
The signaling pathways mediating the induction of Jmdj3 expression by LPS have not been elucidated so far. Here we show that TrkA activation by DHEA induced Jmjd3 expression through activation of the transcription factor CREB. TrkA signaling did not induce Jmdj3 expression in microglial cells treated with a CREB/CBP interaction inhibitor. Together, these findings support that DHEA-induced Jmjd3 expression depends on CREB activation.
In conclusion, we suggest that DHEA activates through its interaction with TrkA the Akt1/Akt2-CREB pathway leading to enhanced Jmjd3 expression, thereby acting as an endogenous regulator of neuro-inflammatory microglial responses. Thus, in addition to the previously described neuroprotective effects of DHEA, such as support of cell growth and survival of neuronal cells,12, 13, 16, 35, 36, 37 DHEA may also suppress neuro-inflammation. Therefore, application of small lipophilic ligands of TrkA, like DHEA, which can penetrate the BBB and selectively activate TrkA signaling in microglia, could be envisioned as a means to control innate immune responses in the central nervous system, which are relevant in the pathogenesis of a variety of neuro-inflammatory, neurodegenerative and neuropsychiatric disorders.
References
Barrou Z, Charru P, Lidy C . Dehydroepiandrosterone (DHEA) and aging. Arch Gerontol Geriat 1997; 24: 233–241.
Webb SJ, Geoghegan TE, Prough RA . The biological actions of dehydroepiandrosterone involves multiple receptors. Drug Metab Rev 2006; 38: 89–116.
Maninger N, Wolkowitz OM, Reus VI, Epel ES, Mellon SH . Neurobiological and neuropsychiatric effects of dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS). Front Neuroendocrinol 2009; 30: 65–91.
Alesci S, Koch CA, Bornstein SR, Pacak K . Adrenal androgens regulation and adrenopause. Endocr Regul 2001; 35: 95–100.
Compagnone NA, Mellon SH . Neurosteroids: biosynthesis and function of these novel neuromodulators. Front Neuroendocrinol 2000; 21: 1–56.
Friess E, Schiffelholz T, Steckler T, Steiger A . Dehydroepiandrosterone - a neurosteroid. Eur J Clin Invest 2000; 30: 46–50.
Koch C, Bornstein S, Pacak K, Chrousos G . How does metyrapone decrease seizures? Neurosurg Rev 1998; 21: 302–303.
Zwain IH, Yen SSC . Neurosteroidogenesis in astrocytes, oligodendrocytes, and neurons of cerebral cortex of rat brain. Endocrinology 1999; 140: 3843–3852.
Gottfried-Blackmore A, Sierra A, Jellinck PH, McEwen BS, Bulloch K . Brain microglia express steroid-converting enzymes in the mouse. J Steroid Biochem 2008; 109: 96–107.
Prough R, Clark BJ, Klinge CM . Novel mechanisms for DHEA action. J Mol Endocrinol 2016; 56: R139–R155.
Monnet FP, Maurice T . The sigma(1) protein as a target for the non-genomic effects of neuro(active)steroids: Molecular, physiological, and behavioral aspects. J Pharmacol Sci 2006; 100: 93–118.
Charalampopoulos I, Alexaki VI, Lazaridis I, Dermitzaki E, Avlonitis N, Tsatsanis C et al. G protein-associated, specific membrane binding sites mediate the neuroprotective effect of dehydroepiandrosterone. FASEB J 2006; 20: 577.
Charalampopoulos I, Alexaki VI, Tsatsanis C, Minas V, Dermitzaki E, Lasaridis I et al. Neurosteroids as endogenous inhibitors of neuronal cell apoptosis in aging. Ann N Y Acad Sci 2006; 1088: 139–152.
Liu DM, Dillon JS . Dehydroepiandrosterone activates endothelial cell nitric-oxide synthase by a specific plasma membrane receptor coupled to G alpha(i2,3). J Biol Chem 2002; 277: 21379–21388.
Chen F, Knecht K, Birzin E, Fisher J, Wilkinson H, Mojena M et al. Direct agonist/antagonist functions of dehydroepiandrosterone. Endocrinology 2005; 146: 4568–4576.
Lazaridis I, Charalampopoulos I, Alexaki VI, Avlonitis N, Pediaditakis I, Efstathopoulos P et al. Neurosteroid dehydroepiandrosterone interacts with nerve growth factor (NGF) receptors, preventing neuronal apoptosis. Plos Biol 2011; 9: e1001051.
Pediaditakis I, Iliopoulos I, Theologidis I, Delivanoglou N, Margioris AN, Charalampopoulos I et al. Dehydroepiandrosterone: an ancestral ligand of neurotrophin receptors. Endocrinology 2015; 156: 16–23.
Starka L, Duskova M, Hill M . Dehydroepiandrosterone: a neuroactive steroid. J Steroid Biochem 2015; 145: 254–260.
Kishimoto Y, Hoshi M . Dehydroepiandrosterone sulphate in rat brain: incorporation from blood and metabolism in vivo. J Neurochem 1972; 19: 2207–2215.
Maggio M, De Vita F, Fisichella A, Colizzi E, Provenzano S, Lauretani F et al. DHEA and cognitive function in the elderly. J Steroid Biochem Mol Biol 2015; 145: 281–292.
van Weerden WM, Bierings HG, van Steenbrugge GJ, de Jong FH, Schroder FH . Adrenal glands of mouse and rat do not synthesize androgens. Life Sci 1992; 50: 857–861.
Corpechot C, Robel P, Axelson M, Sjovall J, Baulieu EE . Characterization and measurement of dehydroepiandrosterone sulfate in rat brain. Proc Natl Acad Sci USA 1981; 78: 4704–4707.
Souza-Teodoro LH, de Oliveira C, Walters K, Carvalho LA . Higher serum dehydroepiandrosterone sulfate protects against the onset of depression in the elderly: findings from the English Longitudinal Study of Aging (ELSA). Psychoneuroendocrinology 2016; 64: 40–46.
Zhu G, Yin Y, Xiao CL, Mao RJ, Shi BH, Jie Y et al. Serum DHEAS levels are associated with the development of depression. Psychiat Res 2015; 229: 447–453.
Hampl R, Bicikova M . Neuroimmunomodulatory steroids in Alzheimer dementia. J Steroid Biochem 2010; 119: 97–104.
Aldred S, Mecocci P . Decreased dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) concentrations in plasma of Alzheimer's disease (AD) patients. Arch Gerontol Geriat 2010; 51: E16–E18.
Wojtal K, Trojnar MK, Czuczwar SJ . Endogenous neuroprotective factors: neurosteroids. Pharmacol Rep 2006; 58: 335–340.
Du CG, Khalil MW, Sriram S . Administration of dehydroepiandrosterone suppresses experimental allergic encephalomyelitis in SJL/J mice. J Immunol 2001; 167: 7094–7101.
Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK . An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell 2011; 145: 584–595.
Litim N, Morissette M, Di Paolo T . Neuroactive gonadal drugs for neuroprotection in male and female models of Parkinson's disease. Neurosci Biobehav Rev 2015; 67: 79–88.
Belanger N, Gregoire L, Bedard P, Di Paolo T . Estradiol and dehydroepiandrosterone potentiate levodopa-induced locomotor activity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine monkeys. Endocrine 2003; 21: 97–101.
D'Astous M, Morissette M, Tanguay B, Callier S, Di Paolo T . Dehydroepiandrosterone (DHEA) such as 17 beta-estradiol prevents MPTP-induced dopamine depletion in mice. Synapse 2003; 47: 10–14.
Belanger N, Gregoire L, Bedard PJ, Di Paolo T . DHEA improves symptomatic treatment of moderately and severely impaired MPTP monkeys. Neurobiol Aging 2006; 27: 1684–1693.
Li H, Klein GM, Sun P, Buchan AM . Dehydroepiandrosterone (DHEA) reduces neuronal injury in a rat model of global cerebral ischemia. Brain Res 2001; 888: 263–266.
Charalampopoulos L, Tsatsanis C, Dermitzaki E, Alexaki VI, Castanas E, Margioris AN et al. Dehydroepiandrosterone and allopregnanolone protect sympathoadrenal medulla cells against apoptosis via antiapoptotic Bcl-2 proteins. Proc Natl Acad Sci USA 2004; 101: 8209–8214.
Gravanis A, Calogeropoulou T, Panoutsakopoulou V, Thermos K, Neophytou C, Charalampopoulos I . Neurosteroids and microneurotrophins signal through NGF receptors to induce prosurvival signaling in neuronal cells. Sci Signal 2012; 5: pt8.
Charalampopoulos I, Remboutsika E, Margioris AN, Gravanis A . Neurosteroids as modulators of neurogenesis and neuronal survival. Trends Endocrin Met 2008; 19: 300–307.
Wang MJ, Huang HM, Chen HL, Jeng KCG . Dehydroepiandrosterone inhibits lipopolysaccharide-induced nitric oxide production in BV-2 microglia. Journal of Neurochemistry 2001; 77: 830–838.
Barger SW, Chavis JA, Drew PD . Dehydroepiandrosterone inhibits microglial nitric oxide production in a stimulus-specific manner. J Neurosci Res 2000; 62: 503–509.
Du CG, Guan QN, Khalil MW, Sriram S . Stimulation of Th2 response by high doses of dehydroepiandrosterone in KLH-primed splenocytes. Exp Biol Med 2001; 226: 1051–1060.
Kipper-Galperin M, Galilly R, Danenberg HD, Brenner T . Dehydroepiandrosterone selectively inhibits production of tumor necrosis factor alpha and interleukin-6 [correction of interlukin-6] in astrocytes. Int J Dev Neurosci 1999; 17: 765–775.
Padgett DA, Loria RM . Endocrine regulation of murine macrophage function: effects of dehydroepiandrosterone, androstenediol, and androstenetriol. J Neuroimmunol 1998; 84: 61–68.
Straub RH, Konecna L, Hrach S, Rothe G, Kreutz M, Scholmerich J et al. Serum dehydroepiandrosterone (DHEA) and DHEA sulfate are negatively correlated with serum interleukin-6 (IL-6), and DHEA inhibits IL-6 secretion from mononuclear cells in man in vitro: possible link between endocrinosenescence and immunosenescence. J Clin Endocrinol Metab 1998; 83: 2012–2017.
Hoyk Z, Parducz A, Garcia-Segura LM . Dehydroepiandrosterone regulates astroglia reaction to denervation of olfactory glomeruli. Glia 2004; 48: 207–216.
Hazeldine J, Arlt W, Lord JM . Dehydroepiandrosterone as a regulator of immune cell function. J Steroid Biochem 2010; 120: 127–136.
Prinz M, Priller J . Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 2014; 15: 300–312.
Crotti A, Ransohoff RM . Microglial physiology and pathophysiology: insights from genome-wide transcriptional profiling. Immunity 2016; 44: 505–515.
Madry C, Attwell D . Receptors, ion channels, and signaling mechanisms underlying microglial dynamics. J Biol Chem 2015; 290: 12443–12450.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH . Mechanisms underlying inflammation in neurodegeneration. Cell 2010; 140: 918–934.
Casano AM, Peri F . Microglia: multitasking specialists of the brain. Dev Cell 2015; 32: 469–477.
Saijo K, Glass CK . Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 2011; 11: 775–787.
Alexaki VI, Charalampopoulos I, Kampa M, Vassalou H, Theodoropoulos P, Stathopoulos EN et al. Estrogen exerts neuroprotective effects via membrane estrogen receptors and rapid Akt/NOS activation. Faseb J 2004; 18: 1594.
Chung KJ, Chatzigeorgiou A, Economopoulou M, Garcia-Martin R, Alexaki VI, Mitroulis I et al. A self-sustained loop of inflammation-driven inhibition of beige adipogenesis in obesity. Nat Immunol 2017; 18: 654–664.
Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009; 114: 3244–3254.
De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G . The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007; 130: 1083–1094.
De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J 2009; 28: 3341–3352.
Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012; 488: 404–408.
Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 2010; 11: 936–944.
Yan Q, Sun L, Zhu Z, Wang L, Li S, Ye RD . Jmjd3-mediated epigenetic regulation of inflammatory cytokine gene expression in serum amyloid A-stimulated macrophages. Cell Signal 2014; 26: 1783–1791.
Tang Y, Li T, Li J, Yang J, Liu H, Zhang XJ et al. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson's disease. Cell Death Differ 2014; 21: 369–380.
De Simone R, Ambrosini E, Carnevale D, Ajmone-Cat MA, Minghetti L . NGF promotes microglial migration through the activation of its high affinity receptor: modulation by TGF-beta. J Neuroimmunol 2007; 190: 53–60.
Zhu D, Yang N, Liu YY, Zheng J, Ji C, Zuo PP . M2 macrophage transplantation ameliorates cognitive dysfunction in amyloid-beta-treated rats through regulation of microglial polarization. J Alzheimers Dis 2016; 52: 483–495.
la Sala A, Corinti S, Federici M, Saragovi HU, Girolomoni G . Ligand activation of nerve growth factor receptor TrkA protects monocytes from apoptosis. J Leukoc Biol 2000; 68: 104–110.
Ley S, Weigert A, Weichand B, Henke N, Mille-Baker B, Janssen RA et al. The role of TRKA signaling in IL-10 production by apoptotic tumor cell-activated macrophages. Oncogene 2013; 32: 631–640.
Chao MV . Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 2003; 4: 299–309.
Marlin MC, Li G . Biogenesis and function of the NGF/TrkA signaling endosome. Int Rev Cell Mol Biol 2015; 314: 239–257.
Franke TF . PI3K/Akt: getting it right matters. Oncogene 2008; 27: 6473–6488.
Yu H, Littlewood T, Bennett M . Akt isoforms in vascular disease. Vascul Pharmacol 2015; 71: 57–64.
Rotllan N, Chamorro-Jorganes A, Araldi E, Wanschel AC, Aryal B, Aranda JF et al. Hematopoietic Akt2 deficiency attenuates the progression of atherosclerosis. Faseb J 2015; 29: 597–610.
Ding L, Biswas S, Morton RE, Smith JD, Hay N, Byzova TV et al. Akt3 deficiency in macrophages promotes foam cell formation and atherosclerosis in mice. Cell Metab 2012; 15: 861–872.
Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 2009; 31: 220–231.
Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci USA 2012; 109: 9517–9522.
Vergadi E, Vaporidi K, Theodorakis EE, Doxaki C, Lagoudaki E, Ieronymaki E et al. Akt2 deficiency protects from acute lung injury via alternative macrophage activation and miR-146a induction in mice. J Immunol 2014; 192: 394–406.
Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD . Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 1999; 286: 2358–2361.
Riccio A, Pierchala BA, Ciarallo CL, Ginty DD . An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science 1997; 277: 1097–1100.
Du K, Montminy M . CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 1998; 273: 32377–32379.
Wen AY, Sakamoto KM, Miller LS . The role of the transcription factor CREB in immune function. J Immunol 2010; 185: 6413–6419.
Prencipe G, Minnone G, Strippoli R, De Pasquale L, Petrini S, Caiello I et al. Nerve growth factor downregulates inflammatory response in human monocytes through TrkA. J Immunol 2014; 192: 3345–3354.
Wood ER, Kuyper L, Petrov KG, Hunter RN 3rd, Harris PA, Lackey K . Discovery and in vitro evaluation of potent TrkA kinase inhibitors: oxindole and aza-oxindoles. Bioorg Med Chem Lett 2004; 14: 953–957.
Acknowledgements
Supported by grants from the Deutsche Forschungsgemeinschaft (AL1686/3-1 and AL1686/2-2 to VIA; CH279/6-2 and SFB655-project B10 to TC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
About this article

Cite this article
Alexaki, V., Fodelianaki, G., Neuwirth, A. et al. DHEA inhibits acute microglia-mediated inflammation through activation of the TrkA-Akt1/2-CREB-Jmjd3 pathway. Mol Psychiatry 23, 1410–1420 (2018). https://doi.org/10.1038/mp.2017.167
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.167
- Springer Nature Limited
This article is cited by
-
Differential effects of AKT1 and AKT2 on sleep–wake activity under basal conditions and in response to LPS challenge in mice
Sleep and Biological Rhythms (2024)
-
JMJD3 ablation in myeloid cells confers renoprotection in mice with DOCA/salt-induced hypertension
Hypertension Research (2023)
-
Current methods for the microglia isolation: Overview and comparative analysis of approaches
Cell and Tissue Research (2023)
-
Dehydroepiandrosterone exacerbates nigericin-induced abnormal autophagy and pyroptosis via GPER activation in LPS-primed macrophages
Cell Death & Disease (2022)
-
Lactylation: a Passing Fad or the Future of Posttranslational Modification
Inflammation (2022)