
Abstract
Frontotemporal lobar degeneration is the most common cause of dementia of non-Alzheimer's type worldwide. It manifests, clinically, with behavioural changes and language impairment and is pathologically associated with tau- or ubiquitin-positive inclusions detected in neurons and glial cells of the frontal and temporal lobes in the brain. Genetic variations in the microtubule-associated protein tau and progranulin genes explain almost 50% of familial cases, whilst variations in TAR DNA-binding protein, charged multivescicular body protein 2B, valosin-containing protein and fused in sarcoma genes contribute to <5% of cases. The rapidly developing investigative techniques available to geneticists such as genome-wide association studies, whole-exome sequencing and, soon, whole-genome sequencing promise to contribute to the unravelling of the genetic architecture of this complex disease and, in the future, to the development of more sensitive, accurate and effective diagnostic and treatment measures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Frontototemporal lobar degeneration (FTLD) is second only to Alzheimer’s disease (AD) as a cause of dementia in patients under 65 years of age (Neary et al. 1998; Ratnavalli et al. 2002; Knopman et al. 2004). Within the spectrum of FTLD, familial forms of frontotemporal dementia (FTD) occur in 30–50% of cases, presenting mainly as behavioural variant FTD (bvFTD) and less frequently as semantic dementia (SD) or progressive non-fluent aphasia (PNFA). An autosomal-dominant mode of inheritance can occur in 10–27% of all FTD patients (Seelaar et al. 2010). Up to 50% of familial cases show association with chromosome 17: mutations in the microtubule-associated protein tau (MAPT) and progranulin (PGRN) are considered as the main genetic cause of FTD (Seelaar et al. 2010). Variations in TAR DNA-binding protein (TDP-43) on chromosome 1, charged multivescicular body protein 2B (CHMP2B) on chromosome 3, valosin-containing protein (VCP) and the intraflagellar transport 74 (IFT74; Momeni et al. 2006c) on chromosome 9, fused in sarcoma (FUS) on chromosome 16 and transmembrane protein 106 B (TMEM106B) on chromosome 7 (identified recently in FTLD-TDP; Van Deerlin et al. 2010) all together contribute to <5% of the cases (Seelaar et al. 2010).
In the following sections, we will (1) focus specifically on the contribution to FTLD exerted by MAPT and PGRN, (2) critically discuss implications of CHMP2B and VCP and (3) analyse the status of the genome-wide association study (GWAS) approach in FTD. The types of mutations, their effects and their functional consequences for MAPT, PGRN, CHMP2B and VCP are summarized in Table 1.
Genetics of FTD
Microtubule-Associated Protein Tau
Gene and Protein
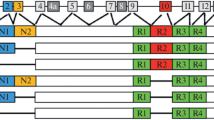
MAPT is located on chromosome 17q21.1 and encodes the microtubule-associated protein tau with varying length due to alternative splicing of exons 2, 3 and 10, leading to 6 different isoforms found in adult human brain (Goedert et al. 1989; Fig. 1a). The N-terminus identifies the so-called projection domain, whilst the C-terminus holds the microtubule-binding domain (MT-binding domain) which is composed of three or four repeats (3R or 4R) depending on the alternative splicing of exon 10 (Fig. 1a; van Swieten and Spillantini 2007). In normal adult brain, the ratio of 3R/4R is approximately equal to 1 (Rovelet-Lecrux et al. 2009). Tau’s primary function is to bind and stabilize the microtubules (MTs) which contributes to maintaining cell shape and trafficking of molecules in the axon (Hutton et al. 1998; Jho et al. 2010); further, tau seems to have a role in neuronal polarity and signal transduction (Caffrey and Wade-Martins 2007). Tau is one of the major representatives of neuronal proteins associated with MTs; it not only regulates MTs’ structure but also, via the N-terminus projection domain, mediates their interaction with other elements of the cytoskeleton and with components of the neural plasma membrane (Jho et al. 2010). Lastly, it has been suggested that this domain might also be involved in regulating the space between MTs (Caffrey and Wade-Martins 2007). A more detailed and comprehensive analysis of tau protein and its functions is discussed in Morris et al. (2011).

a The MAPT gene consists of 13 exons, of which three (exons 2, 3 and 10) are alternatively spliced and three are rarely transcribed (exons 4a, 6 and 8). The six isoforms of MAPT are here represented showing the three and four repeats (3R and 4R) that compose the MT-binding domain. Most or all of the reported variants are shown with amino acid change and position. b Two major haplotypes, H1 and H2, are the result of an inversion of ∼900 kb on the long arm of chromosome 17. This genomic region encompasses the genes CRHR1, IMP5, MAPT and NSF 1–13. Genotypes on each haplotype are inherited as a block and are in LD. The MAPT haplotype can undergo rearrangements and has been linked to several disease phenotypes ranging from FTLD, behavioural and motor issues to mental retardation, facial dysmorphism and possible autism
Mutations and Clinicopathological Aspects
To date, 72 variants (44 of which seem to exert pathogenic effects) lead to deletions, missense, silent and splice site mutations in MAPT (van Swieten and Spillantini 2007; http://www.molgen.ua.ac.be/FTDMutations, accessed May 2011). Mutations in MAPT can be divided into three different categories: (1) exonic point mutations in exons 1, 9–13 (Hutton et al. 1998; Poorkaj et al. 1998), (2) intronic mutations altering the splicing of exon 10 (Hutton et al. 1998; Spillantini et al. 1998; Malkani et al. 2006) and (3) structural mutations (see “Structural Changes on Chromosome 17: A Novel Rare FTLD Mutation?” below; Rovelet-Lecrux et al. 2009, 2010). Exonic variants lead mainly to missense mutations, of which the vast majority is located at the level of the four microtubule-binding domains (Fig. 1a); these cause a decrease in the ability of tau in binding MTs (Lee et al. 2001; Goedert and Jakes 2005). Missense mutations such as R5L, K257T, I260V, P301L, P301S, Q336R, V337M and R406W seem involved in promoting tau’s self-aggregation in the form of abnormal inclusions within neurons and glial cells (Lee et al. 2001; Goedert and Jakes 2005; Goedert 2005). Furthermore, a minority of missense mutations (N279K, N296H, S305N), together with deletions (ΔK280, ΔN296) and intronic mutations (intron 9 g(−10)t and intron 10 +3, +11, +12, +13, +14, +16) affect exon 10 splicing, causing changes in the ratio between 3R/4R and between the different isoforms (Lee et al. 2001; Goedert and Jakes 2005; Goedert 2005; http://www.molgen.ua.ac.be/FTDMutations, accessed May 2011). For a more extensive review on tau mutations and their effects, see Goedert (2005).
Abnormal tau protein identifies tauopathies, neurological disorders characterized by a common pathology defined by tau aggregates. Tau inclusions localize, topographically, to neurons and/or glial cells, mainly, in the frontal and temporal cortex, hippocampus and subcortical nuclei, and, more rarely, in the midbrain, brain stem, cerebellum and spinal cord (Seelaar et al. 2010). To date, tau lesions have been reported in AD (Goedert et al. 1996), Pick’s disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD) and multiple system tauopathy with dementia (MSTD; Spillantini et al. 1997; Goedert et al. 2006; Lee et al. 2001; Goedert 2004, 2005; Mackenzie et al. 2010; Table 2). Notably, the major component of insoluble tangles in FTLD-tau, PSP, CBD and AGD is 4R tau (Caffrey and Wade-Martins 2007). Tau pathology is overall associated with mutations in MAPT (but can also be observed in apparent absence of MAPT abnormalities) and results either from harmful loss of function (due to mutations that affect the MT-binding domain) or from toxic gain of function (increased self-aggregation properties; Kurz and Perneczky 2009). Tau hyperphosphorylation seems to be mediated by the activity of glycogen synthase kinases (GSKs), and it was suggested that increased expression of GSKs is associated with tau hyperphosphorylation, leading to tau pathology (Kurz and Perneczky 2009).
The mean onset age in FTD cases caused by MAPT mutations is around 55 years, and the duration of the disease is on average 9 years (ranging between 5 and 20 years; Seelaar et al. 2010). Genetic defects in MAPT are mainly associated with clinical bvFTD, to a lesser extent with SD and, rarely, with PNFA (more associated with PGRN mutations; Mesulam et al. 2007; Rohrer et al. 2009; Seelaar et al. 2010).
Structural Changes on Chromosome 17: A Novel Rare FTLD Mutation?
Recently, motor skills impairment together with behavioural problems and poor social interactions were associated with microduplication on chromosome 17q21.31 (Grisart et al. 2009). The authors suggested that overexpression of MAPT in neurons could contribute to the observed behavioural problems and that duplication of the corticotropin-releasing hormone receptor 1 gene (CRHR1), located 59 Kb centromeric from MAPT, could explain (1) the impaired motor skills (due to overexpression of CRHR1 in the cerebellum) and (2) the social impairment (CRHR1 being involved also in stress responses; Grisart et al. 2009). The occurrence of phenotypical features such as behavioural and social impairment in the presence of structural changes at the MAPT locus suggested that rearrangements at this locus might also be associated with FTLD. Screening of a genetic region encompassing PGRN and MAPT failed to indentify abnormal copy number variations (CNVs) in 39 FTLD patients (negative for mutations in those two genes; Skoglund et al. 2009). Also, no abnormal CNVs for MAPT were found in 70 FTLD patients without MAPT mutations (12 of which pathologically proven FTLD-TAU; Lladó et al. 2007). Even though these studies were not able to establish a link between FTLD and structural changes in loci encompassing MAPT and PGRN, further FTD cohorts were investigated, leading to different results (Rovelet-Lecrux et al. 2009, 2010). In the first study, the authors identified a heterozygous 17.3-Kb deletion responsible for the removal of exons 6–9 of MAPT in one FTD patient (Rovelet-Lecrux et al. 2009). Functional assessment suggested that this deletion caused the loss of the first MT-binding domain and, consequently, a decrease in the binding abilities of tau to the MTs plus a toxic gain of function such as the sequestration of another MT-associated protein, namely, MAPB1 (also involved in MT stabilization; Rovelet-Lecrux et al. 2009). The same group reported a 439-Kb duplication in the region encompassing CRHR1, intramembrane protease 5 (IMP5), MAPT and saitohin (STH) in a patient affected by behavioural disorder and amnestic syndrome (Rovelet-Lecrux et al. 2010). As sequencing did not reveal pathogenic variants neither in MAPT nor PGRN (Rovelet-Lecrux et al. 2010), it was suggested that the duplication could possibly explain the disease phenotype. Even though we should be cautious at drawing conclusions, these findings represent the first evidence of a possible link between rearrangements at the MAPT locus and the FTLD phenotype.
MAPT Haplotype and Neurological Disorders
MAPT is located in a region of ∼900 kb which has undergone an inversion (in the European Caucasian population) ∼1.9–2.7 Mya, resulting into the two haplotypes, H1 (direct orientation) and H2 (inverted orientation) (Zody et al. 2008; Vandrovcova et al. 2010; Fig. 1b). These haplotypes do not recombine for ∼1.5 Mb (Pittman et al. 2004) and influence differently cortical genes expression (Myers et al. 2007a). This chromosomal region is known for the high tendency of rearrangements, especially due to the presence of low-copy repeats (LCRs) at the two extremes of the inversion block (Vandrovcova et al. 2010). In general, the H1 haplotype has been associated with PSP, CBD, AD, Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS; Zody et al. 2008; Vandrovcova et al. 2010), whilst the H2 haplotype has been associated with the 17q21.31 microdeletion syndrome (Koolen et al. 2006; Shaw-Smith et al. 2006; Kirchhoff et al. 2007). More specifically, the H1 haplotype is a strong risk factor for PSP and CBD, and association was refined to the sub-haplotype H1c through single nucleotide polymorphism (SNP) rs242557 (located in the MAPT promoter region; Pittman et al. 2005); also, increased risk for late-onset AD was associated with sub-haplotype H1c in a region between rs242557 and rs 2471738 (the latter located in intron 9 of MAPT; Myers et al. 2007b), although this was not replicated (Mukherjee et al. 2007); neither GWAS in AD showed a clear association with the MAPT region (Naj et al. 2011; Hollingworth et al. 2011; Harold et al. 2009; Lambert et al. 2009). In the case of PD, the association seems due to the H1 haplotype, although there is some lack of consistency among studies (Vandrovcova et al. 2010). Nevertheless, recent GWAS and meta-analysis studies supported H1 haplotype involvement in PD (Simon-Sanchez et al. 2009; Nalls et al. 2011). In the case of FTD, different studies could not identify a clear risk effect of MAPT haplotype; in fact, correlation was rather pertaining to the clinical phenotype (Vandrovcova et al. 2010). For example, the H1/H1 genotype was linked to the parkinsonism phenotype in FTDP-17 (Baba et al. 2005), the H1/H2 genotype to the dementia phenotype (Baba et al. 2005), and the H2 haplotype or H2 haplotype and H2/H2 genotype correlated to onset age modulation (Borroni et al. 2005; Laws et al. 2007) and overrepresentation in familial FTD, respectively (Ghidoni et al. 2006). Further analysis and discussion of the MAPT haplotype can be found in (Vandrovcova et al. 2010).
It is probably noteworthy to consider that the MAPT haplotype seems to have a rather subtle effect on disease phenotypes. In fact, this region being characterized by high linkage disequilibrium (LD), it seems (and has resulted) hard to identify functional variants causing the associations. This suggests that, possibly, the effect exerted by the variability within and the overall involvement of MAPT haplotype in neurological disorders might act at the level of gene expression with a range of cis- and trans-acting elements such as non-coding regions and/or regulatory elements such as LCRs or transcription factors or non-coding microRNAs (miRNAs), being actors of a more complex and disease-associated expression mechanism.
Progranulin
Gene and Protein
Progranulin gene (PGRN) is located on chromosome 17q21.32 and codes for a 593-amino-acid-long protein, which is the precursor of granulins (GRNs). GRNs are ∼6-kDa peptides that result from posttranslational processing of mature GRN which results from the cleavage of the secretory signal peptide at PGRN’s N-terminus (Fig. 2; Gijselinck et al. 2008). PGRN is ubiquitously expressed in different tissues, especially in mitotic active tissues such as epithelial or hematopoietic tissues. In the central nervous system (CNS), PGRN is found in the cerebral cortex, cerebellum and hippocampus (Daniel et al. 2000), and it is upregulated in activated microglial cells (Baker et al. 2006). PGRN is involved in different biological processes such as cell cycle progression, cell growth regulation, wound healing and inflammation (van Swieten and Heutink 2008). Furthermore, PGRN activates several kinase-dependant signalling cascades (controlling cell cycle and motility), stimulates the induction of vascular endothelial growth factor and seems to have a role in brain development (Mackenzie and Rademakers 2007). Finally, PGRN has also been associated with tumorigenesis (He and Bateman 2003); interestingly, it acts as an anti-inflammatory agent, whilst GRNs have a pro-inflammatory effect (He and Bateman 2003). Recently, the notion that PGRN is involved in inflammatory processes was confirmed and supported by the findings of Tang et al. (2011) who provided evidence that PGRN acts as antagonist of the tumour necrosis factor α (TNFα) in binding the TNF receptor. Specifically, PGRN was able to reverse inflammatory arthritis in mice, and it was suggested having potential therapeutic effects in TNFα-mediated pathologies (Tang et al. 2011).

The PGRN gene consists of 12 coding exons and one non-coding exon (exon 0). PGRN protein is a precursor of granulins. Most or all of the reported variants are shown with amino acid change and position
Mutations and Clinicopathological Aspects
Mutations in PGRN associated with FTLD were reported for the first time in 2006 (Baker et al. 2006; Cruts et al. 2006). Those studies showed that null mutations in PGRN result in a decrease or absence of the mutated transcript (haploinsufficiency; Gijselinck et al. 2008). Currently, 148 variants have been reported, of which 69 seem to exert pathogenic effects (Fig. 2; http://www.molgen.ua.ac.be/FTDMutations, accessed May 2011); these variants lead mainly to nonsense and frameshift mutations (Mackenzie and Rademakers 2007), but some also to missense, silent and intronic mutations (Gijselinck et al. 2008). Nonsense, frameshift and intronic (IVS1 +5G>C) mutations cause haploinsufficiency due either to mRNA nonsense-mediated decay or to nuclear degradation of transcripts retaining the first intron of PGRN (Gijselinck et al. 2008). Even if nonsense and frameshift mutations represent the main cause of PGRN haploinsufficiency, some missense mutations were reported as contributing to a decrease in secreted PGRN (van der Zee et al. 2007). In the case of A9D (Mukherjee et al. 2006, 2008), an in vitro model showed that even though there was no gross difference in the mRNA level for mutation carriers compared with normal controls, the amino acid change negatively affected the secretion of the mutant protein, suggesting a functional haploinsufficiency due to cytoplasmic missorting (Gijselinck et al. 2008; Shankaran et al. 2008). Other missense mutations, such as C105R, I124T, G168S, P248L, R432C, W541 and R556C, seem more likely to affect PGRN functionality (van der Zee et al. 2007; Gijselinck et al. 2008). Moreover, some of these (I124T, P248L, S258N, A324T and R432C) seem to affect the structure and stability of PGRN (Gijselinck et al. 2008). In the cases of P248L and R432C, translated products are inefficiently transported within the secretory pathway, resulting in a reduction of PGRN secretion (Shankaran et al. 2008). In addition to the aforementioned small in/dels (or frameshift mutations; Gijselinck et al. 2008), larger deletions (54–69 Kb comprising PGRN; Gijselinck et al. 2008) or nearly a complete deletion of the gene with removal of exons 1–11 (Rovelet-lecrux et al. 2008) can lead to loss of functionality in PGRN. This latter rearrangement, due to a non-homologous recombination, was seen in an FTD patient (proband) and in a sibling with PD, suggesting that recombination on the long arm of chromosome 17 can affect not only the MAPT haplotype but also the region containing PGRN, which is 1.7 Mb centromeric from MAPT (Rovelet-lecrux et al. 2008). Investigation of such rearrangements and CNVs in this chromosomal region should become routine in research, as also suggested in van Swieten and Heutink (2008).
The overall frequency of PGRN mutations in FTLD is 15–24%. Mutations in PGRN have also been associated with different types of neurological disorders, such as AD, PD, PSP (rarely), corticobasal syndrome (CBS), mild parkinsonism and, very rarely, with FTD-MND (frontotemporal dementia with motor neuron disease; Josephs et al. 2007; Le Ber et al. 2007; Benussi et al. 2008, 2009; Gijselinck et al. 2008; Pickering-Brown et al. 2008). The mean onset age is around 60 years (ranging from 35 to 89 years) and the average duration of the disorder is 8 years (with a wide range of 3–22 years; Seelaar et al. 2010).
Clinically, individuals carrying mutations in PGRN show mainly apathy and social withdrawal among their behavioural disturbances; also, language impairment as well as hallucinations and delusions can be part of the clinical spectrum (Seelaar et al. 2010). PGRN mutations have been associated with asymmetrical frontal, temporal and inferior parietal lobe atrophy (Rohrer et al. 2010) and, at the molecular level, mainly with tau-negative and ubiquitin-positive inclusions (FTLD-U), refined in the nomenclature FTLD-TDP (TDP-43 being the main protein associated with ubiquitin inclusions). Nonetheless, it has been reported that the effects of PGRN mutations can go beyond FTLD-U pathology, extending, in some cases, to even tau or α-synuclein pathology (Gijselinck et al. 2008).
In an attempt to explain the link between PGRN mutations and TDP-43 pathobiology, a study in HeLa (derived from cervical cancer) and H4 (neuroglioma) cells suggested that PGRN haploinsuffciency could influence the pathological cleavage of TDP-43 via caspase-3 activity (Zhang et al. 2007); in contrast, in another report, accumulation of TDP-43 fragments in HeLa cells seemed independent from PGRN knockdown (Shankaran et al. 2008). In a further study that investigated on one side siRNA-mediated knockdown of PGRN in cell cultures (HeLa cell line) and on the other PGRN knockdown in a mouse model, accumulation of TDP-43 C-terminus phosphorylated fragments could not be detected (Dormann et al. 2009). Mouse primary neuronal cultures showed that PGRN depletion activated caspase-3 activity, leading to neuronal death through enhanced vulnerability, but not to TDP-43 cleavage (Guo et al. 2010) and, further, that PGRN depletion was associated with TDP-43 abnormal cytoplasmic cellular localization (Guo et al. 2010). Other data supported the fact that PGRN deficiency in cultured murine neuronal cells increases caspase activity, leading to apoptotic cell death and neurons’ vulnerability (Kleinberger et al. 2010). In the same study, a decreased solubility of full-length TDP-43 was reported, and it was suggested that the formation of C-terminus truncated and phosphorylated TDP-43 fragments could depend on either caspase or non-caspase-mediated mechanisms (Kleinberger et al. 2010). Given that the contribution of PGRN haploinsufficiency to the cleavage of TDP-43 needs to be further investigated, these reports seem contradictory, suggesting that PGRN haploinsufficiency might lead to the accumulation of C-terminus truncated TDP-43 fragments through different pathways, one of which could be caspase-mediated, or that accumulation of cleaved TDP-43 fragments is independent from PGRN expression. Keeping in mind that (1) differences in results might also be due to the differences in experimental designs and used cell lines (neuronal vs. non-neuronal) and (2) TDP-43 can accumulate after impairment of the Ub–proteasome system (bringing also VCP variability into the picture—for VCP, see section below; Gitcho et al. 2009), further investigations and functional studies are needed to shed light on the pathological mechanisms correlated with PGRN deficiency. Not least, a recent study showed that secreted PGRN binds to sortilin (SORT1), a neuronal cell surface binding site which mediates PGRN’s endocytosis and lysosomal localization (Hu et al. 2010). It was shown that sortilin (1) (if ablated) can modulate PGRN protein level in brain and plasma (Hu et al. 2010), (2) might act as a modulator of PGRN levels (Carrasquillo et al. 2010) and (3) could, to a certain extent, regulate PGRN’s biology and activity. Thus far, not only it is noteworthy to consider that genetic variability in SORT1 might affect, generally, sortilin functionality and, specifically, its interaction with PGRN, but also that it is out of doubt that the effect of sortilin-mediated PGRN endocytosis and its role in FTLD-TDP pathophysiology needs to be consistently further investigated.
In conclusion, keeping in mind that PGRN is involved in many different biological mechanisms, it is reasonable to expect that several modifiers [TMEM106B, currently under investigation (see “GWAS of FTLD-TDP Cases” further below), and “SORT1” (Carrasquillo et al. 2010)] regulate PGRN, leading, together with PGRN variability, not only to the wide range of age of onset but also to the highly heterogeneous clinical and pathological presentation (Cruts and Van Broeckhoven 2008).
PGRN Expression/Plasma Levels: A Useful Diagnostic Tool?
Given the clinical and pathological heterogeneity of FTLD cases associated with PGRN mutations, it seems difficult to draw conclusions about the role of mutated PGRN in the process of neurodegeneration. Wild-type PGRN acts in several different biological processes; therefore, it is involved in different pathways, interacts with different substrates and is likely to be modulated by several factors. PGRN nonsense, frameshift and, in some cases, intronic and missense mutation carriers show a reduced level of PGRN protein (≥50%), a condition considered to contribute to the disease. Recently, several attempts have been made to link the genetic variability of PGRN to PGRN expression/plasma levels, aiming to understand whether PGRN expression/plasma levels could be used to identify mutation carriers and, eventually, as a prognostic tool.
In a study investigating patients with different neurodegenerative dementias (FTLD, AD, CBS, PSP and ALS), a correlation between mutations in PGRN and PGRN expression levels was established, showing a decrease of PGRN expression levels in both an FTLD patient and an unaffected normal control (Coppola et al. 2008). Sequencing of these individuals resulted in the isolation of a point mutation (IVS7 −1G>C) and a microdeletion (c.675_676delCA), respectively (Coppola et al. 2008). Also, three other cases (one FTD, one CBS and one normal control) showed decreased PGRN expression levels, but no variants/mutations were identified (Coppola et al. 2008). Finally, as well as in a previous study on ALS cases (Malaspina et al. 2001), a correlation between greater PGRN mRNA levels and disease status, in this case AD, was reported (Coppola et al. 2008). Another study showed that PGRN protein was strongly reduced (3.93 times) in both the plasma and cerebrospinal fluid (CSF) of affected and unaffected individuals carrying PGRN L271LfsX10 and Q341X mutations (Ghidoni et al. 2008). In this case, the dosage of plasma PGRN was suggested as being a useful tool for discriminating carriers of PGRN mutations and, eventually, for developing and monitoring treatment options (Ghidoni et al. 2008). Another study, in alignment with findings by Ghidoni et al., showed that PGRN plasma levels were considerably reduced in PGRN mutation carriers (symptomatic and asymptomatic) when compared with no mutation carriers or normal controls (Finch et al. 2009). Specifically, reduced PGRN plasma levels suggested and contributed to the identification of a loss-of-function deletion in an AD patient (Finch et al. 2009). This result is of interest because it shows that screening of PGRN plasma levels could predict the presence of loss-of-function mutations in PGRN; at the same time, it slightly contradicts findings in Coppola et al., as above, where PGRN mRNA levels were found to be increased in AD patients. In another study, the null mutation IVS1 +5G>C, the null mutation M1, frameshift mutations, several predicted pathogenic missense mutations and some benign missense polymorphisms were all investigated (Sleegers et al. 2009): serum PGRN levels were reduced in both affected and unaffected null mutation carriers. Serum PGRN levels for predicted pathogenic missense mutations (C139R and R564C) were lower than in controls, but higher than in null mutation carriers; the levels for carriers of benign missense polymorphisms were not significantly different from controls (Sleegers et al. 2009). Finally, in a recent study, PGRN mutations in patients with FTD and CBS were associated with abnormally low PGRN levels (Schofield et al. 2010), whilst elevated levels of PGRN were detected in patients with PNFA (Schofield et al. 2010).
All these studies taken together show that the evaluation of PGRN expression/plasma levels can help in discriminating PGRN mutation carriers (both symptomatic and asymptomatic) from no mutation carriers or normal controls, in most cases. As such, this method (1) seems an inexpensive, useful tool for primary screening of individuals affected by neurological disorders or asymptomatic individuals of a family with neurological disorders history to predict PGRN loss-of-function mutations and (2) may hold promise for the future use of PGRN plasma/serum levels as a biomarker in neurodegenerative diseases. Moreover, it is of relevance that investigation of PGRN expression/plasma levels can, possibly, detect microdeletions or larger out-of-frame deletions that might be missed by sequencing, gene dosage or microsatellite length polymorphisms analysis.
However, there are other points that might be taken into account:
-
1.
In each case, independently from the plasma-level measurements, the subject’s DNA needs to be screened for PGRN mutations.
-
2.
If a subject carries a PGRN mutation in a pre-symptomatic status, then the plasma level of PGRN could predict the progression from being asymptomatic to symptomatic for that subject whilst ageing and reaching onset age.
-
3.
We need to establish whether the PGRN expression/plasma levels are changed in carriers of mutations in other candidate genes such as MAPT; otherwise, PGRN is not a general biomarker of neurodegeneration, but only reflecting the PGRN mutations that have been or are to be found in the DNA of the patients.
-
4.
If the PGRN expression/plasma levels are only changed in PGRN mutation carriers and each mutation exerts its effect differently, then the usefulness of measuring PGRN plasma levels will be only limited to monitoring the effect of future pharmaceutical intervention measures that target specific PGRN abnormalities.
-
5.
In the best established and mostly replicated biomarkers of neurodegeneration, namely, Aβ and tau in the CSF of Alzheimer’s patients, these proteins are measured because they are the universal protein markers of the pathology of AD (regardless of carrying mutations in MAPT or APP or PSEN1 and PSEN2). In patients with PGRN mutations, there is a direct correlation between the mutation and abnormal expression/plasma levels of PGRN. This knowledge, however, does not transcend the information we can obtain by screening patients’ DNA.
-
6.
We need to keep in mind that PGRN expression and plasma levels could be modulated by other factors (independently from PGRN mutations) as the cases under investigation of TMEM106B (Finch et al. 2011; Cruchaga et al. 2011) and SORT1 (Hu et al. 2010; Carrasquillo et al. 2010). Of course, genetic variability in TMEM106B, SORT1 and PGRN is likely to increase the level of complexity of the pathogenic mechanisms.
Not least, it seems that there is a certain heterogeneity of the PGRN expression/plasma levels in different neurological disorders such as AD, FTD, CBS, ALS and PNFA, suggesting that the analysis of PGRN expression/plasma levels needs to be carefully correlated with the corresponding syndrome; for example, the elevated levels of PGRN in patients with PNFA or the inconsistency of the data found in AD among different studies (Coppola et al. 2008; Finch et al. 2009) need to be critically considered.
Charged Multivescicular Body Protein 2B
Gene and Protein
The charged multivesicular body protein 2B gene (CHMP2B) is located on chromosome 3p11.2 and codes for a 213-amino-acid-long protein, characterized by a coiled coil domain at the N-terminus and a microtubule-interacting transport (MIT) and microtubule-interacting region (MIR) at the C-terminus (Fig. 3). CHMP2B is part of the endosomal sorting complex required for transport III complex and is involved in sorting and trafficking surface receptors or proteins into intraluminal vesicules for lysosomal degradation and binding the Vps4 protein responsible for the dissociation of ESCRT components (Williams and Urbé 2007; Urwin et al. 2009). Impairment of this machinery could lead to the disruption of endosomal trafficking, lack of trophic support for the cell, aberrant cellular signalling and impairment of autophagy (Urwin et al. 2009). CHMP2B is expressed in all major regions of the brain, including, frontal and temporal lobes, cerebellum and hippocampus (Skibinski et al. 2005).

The CHMP2B gene consists of six coding exons. The CHMP2B protein presents two functional domains: the coiled coil domain and the microtubule-interacting transport (MIT) and MIR. Most or all of the reported variants are shown with amino acid change and position
Mutations and Clinicopathological Aspects
After the report of a familial case of FTD linked to chromosome 3 (FTD3; Brown et al. 1995; Gydesen et al. 2002), Skibinski et al. (2005) reported a variant in CHMP2B leading to the two aberrant transcripts CHMP2Bintron5 and CHMP2BΔ10. In these last 5 years, different studies tried to elucidate the relevance of CHMP2B in FTD, identifying, to date, 11 variants (of which four seem to exert a pathogenic effect; Fig. 3; http://www.molgen.ua.ac.be/FTDMutations, accessed May 2011). Several C-terminus truncation mutations and missense mutation (most of the latter ones found in the C-terminus region of the protein) were reported mainly in FTD cases (Skibinski et al. 2005; Cannon et al. 2006; Momeni et al. 2006a; Rizzu et al. 2006; van der Zee et al. 2008; Ferrari et al. 2010; Ghanim et al. 2010), but some also in CBS (van der Zee et al. 2008).
Clinically, FTD3 patients have a mean onset age of 57 years and present with personality change (mainly social behavioural disinhibition), hyperorality, dyscalculia, a range of speech disturbances and dystonic postures (Urwin et al. 2009). Symmetrical frontal and temporal cortical atrophy is observed with TDP-43-negative and ubiquitin- and/or p62-positive neuronal inclusions (either in the dentate gyrus or sparse in the frontal or other cortical areas; Urwin et al. 2009). The protein p62 is part of the ubiquitin–proteosome system (UPS); therefore, FTD3 is now also named FTLD-UPS (Mackenzie et al. 2009).
Comment on CHMP2B Mutations
Variability in CHMP2B has been associated with a small minority of FTD cases (FTD3), even though findings over the past 5–6 years have been unequivocal.
C-terminus Truncation Mutations
CHMP2Bintron5 (Skibinski et al. 2005), CHMP2BΔ10 (Skibinski et al. 2005), Q165X (van der Zee et al. 2008) and R186X (Momeni et al. 2006a) cause, a priori, the loss of the Vsp4-binding domain in the C-terminus of CHMP2B protein (Fig. 3). Specifically, CHMP2Bintron5 and Q165X have been investigated in functional studies, revealing that aberrant cytoplasmic phenotype and impairment of late endosomal trafficking can contribute to neurodegenerative processes in FTD (Skibinski et al. 2005; van der Zee et al. 2008; Urwin et al. 2009, 2010). Nevertheless, no functional studies are available to assess the phenotype associated with R186X. R186X was found in two asymptomatic siblings of a familial case of FTD with an apparent autosomal-dominant mode of inheritance (and not in neurologically normal controls; Momeni et al. 2006a); it is not clear whether this mutation is a non-pathogenic CHMP2B C-terminus truncating mutation or a pathogenic mutation with incomplete penetrance or a pathogenic mutation causing variable age of onset. If the asymptomatic state would persist in the carriers, one could argue that, possibly, not all reported C-terminus truncation mutations are pathogenic (Momeni et al. 2006a; Lee et al. 2007). Moreover, functional analysis of CHMP2BΔ10 showed that its transcription rate is significantly reduced (10% of the wild type; Urwin et al. 2010) and that it seems to not have real implication in neurodegeneration (Ahmad et al. 2009). It is noteworthy that screening of all the open reading frames in the region linked to FTD3 in one affected member who carried the original CHMP2B mutation reported in (Skibinski et al. 2005) did not reveal any other pathogenic variant (Momeni et al. 2006b).
Missense Mutations
Most of them locate to the C-terminus of the protein, suggesting that this might be, to a certain extent, a polymorphic region. Mutation I29V was found in a neurologically normal control (Cannon et al. 2006; Rizzu et al. 2006), and mutations D148Y and N143S were neither associated with pathogenicity nor with aberrant endosomal phenotype (Skibinski et al. 2005; Lee et al. 2007; van der Zee et al. 2008). The other reported mutations are: S187N, identified in an FTD case but also in neurologically normal controls (Ferrari et al. 2010; Ghanim et al. 2010); Q206H (Parkinson et al. 2006), for which no functional studies are available; and S194L, for which in silico analysis predicted possible pathogenic effects (Ghanim et al. 2010).
In conclusion, considering that (1) the two mutations in the Danish (Skibinski et al. 2005) and Belgian (van der Zee et al. 2008) families most likely cause the disease and (2) mutations causing C-terminus truncation have, possibly, a detrimental effect on the normal function of the protein (Skibinski et al. 2005; van der Zee et al. 2008; Urwin et al. 2010), but also (3) some reports have led to conflicting results, further genetic screening and functional studies are recommended in order to shed light on the pathogenic role of CHMP2B variants in FTD.
Valosin-Containing Protein
Gene and Protein
The valosin-containing protein (VCP) gene is located on chromosome 9p13.3 and encodes an 806-amino-acid-long protein (monomer). The VCP hexamer is a member of the AAA-ATPase (ATPases associated with various cellular activities) superfamily and consists of six monomers forming a ring around a central pore with two AAA + protein domains (D1 and D2 domains; Fig. 4; Weihl et al. 2009). VCP is involved in various cellular processes like cell cycle regulation, post-mitotic Golgi reassembly, suppression of apoptosis, DNA damage response and protein degradation through the UPS and the endoplasmic reticulum-associated protein degradation pathways (Kondo et al. 1997; Rabouille et al. 1998; Rabinovich et al. 2002; Wang et al. 2004; Forman et al. 2006). Fundamentally, VCP is involved in protein homeostasis, maintaining the proper balance between protein synthesis and protein degradation (Balch et al. 2008). VCP is expressed in most mammalian tissues (Sugita and Sudhof 2000).

The VCP gene consists of 17 coding exons. The VCP protein presents three functional domains: the cofactor binding domain encoded by exons 1, 2, 3, 4 and 5 and two ATPase domains (D1 and D2) that form a hexameric complex. The D1 and D2 domains are encoded by exons 6, 7, 8, 9, 10 and 12, 13, 14, respectively. D1 and D2 are linked by the two L1 and L2 domains. Most or all of the reported variants are shown with amino acid change and position
Mutations and Clinicopathological Aspects
Mutations in VCP cause inclusion body myopathy (IBM) associated with Paget’s disease of the bone (PDB) and frontotemporal dementia (FTD) or IBMPFD (Weihl et al. 2009). IBMPFD is autosomal-dominantly inherited (Ju and Weihl 2010). To date, more than 18 variants (of which 17 seem to exert pathogenic effects) have been reported in VCP (Fig. 4; http://www.molgen.ua.ac.be/FTDMutations, accessed May 2011). R155C was the first pathogenic missense mutation reported in FTD (Watts et al. 2004). Missense mutations associated with IBMPFD have been identified in different domains: the N-terminus domain (I27V, R93C, R95C/G, P137L, R155H/P/C/S/L, G157R and R159H/C); the linker L1 connecting the N-terminus and D1 domains (R191Q and L198W); the D1 domain (A232E, T262A, N387H); and the L2 linker (A439S; Ju and Weihl 2010). Initial functional studies reported that the R155H mutation did not affect VCP’s ability to hexamerize or hydrolyze ATP (Weihl et al. 2006), whilst further in vitro studies showed that A232E and R155P were associated with an increase in basal ATPase activity (Halawani et al. 2009). A very recent study highlighted that K524A mutant and the triple mutants R93C-R155C-K524A block protein degradation, rendering cells highly susceptible to ER stress-induced cell death (Poksay et al. 2011). To date, functional studies yielded conflicting results, but it seems that it is acceptable to consider VCP’s mutations mainly affecting autophagic structures and their maturation contributing, in this fashion, to IBMPFD pathogenesis (Ju and Weihl 2010). For a detailed and comprehensive analysis of all VCP mutations and their effects, please refer to Ju and Weihl (2010).
IBMPFD presents with three predominant phenotypic features: first, patients develop disabling weakness with a mean onset of 45 years (90% of cases); second, patients have osteolytic lesions consistent with PDB (51% of cases); and third, prominent language and behaviour dysfunction can be detected, with a mean onset of 54 years (32% of cases; Weihl et al. 2009). IBMPFD CNS pathology is tau-negative and ubiquitin-positive, consistent with a FTLD with ubiquitin-positive inclusions (FTLD-U; Forman et al. 2006); specifically, TDP-43 and VCP are associated with the ubiquitin inclusions which are intranuclear, selectively distinguishing it from other FTLD-U subtypes and placing it in the category of FTLD-U subtype 4 (Forman et al. 2006; Neumann et al. 2007). Moreover, VCP is also present in the inclusions of multiple diseases including ALS, PD and Huntington’s disease (HD; Weihl et al. 2009).
The Genome-Wide Approach and FTD
GWAS as well as whole-exome sequencing (best combined with array methods that help identify in/dels and/or duplications) are the cutting-edge technologies for a better understanding of the genetic underpinnings of complex diseases (Hardy and Singleton 2009; Singleton et al. 2010). Based on the achievements of the last decades, through the accomplishments of (1) the Human Genome Project (International HapMap Consortium 2003; http://www.ornl.gov/sci/techresources/Human_Genome/home.shtml), (2) the International Human HapMap project (International HapMap Consortium 2005; International HapMap Consortium 2007; http://hapmap.ncbi.nlm.nih.gov) and (3) the 1000 genome project (1000 Genomes Project Consortium 2010; http://www.1000genomes.org), GWAS are, today, provided with extremely powerful tools for detecting associations between common variability and disease. By means of evenly distributed known SNPs and based on our ever developing knowledge on LD blocks, GWAS are able to identify loci associated with disease. On the other hand, whole-exome sequencing has a probably wider and finer applicability to the extent of recognizing almost all the categories of genetic risks associated with disease: (1) high-risk rare alleles, (2) moderate-risk low-frequency alleles, (3) low-risk common alleles. The primary outcome of GWAS is the identification of a locus, a genetic region that might be associated with a trait/disease; the association, normally, is further investigated to discover the possible underlying causal variants through fine mapping, dense genotyping and DNA sequencing. The expected outcome of GWAS is not exclusively the identification of one or several coding changes affecting the functions of a protein but also the identification of variants affecting transcription and translation or variants that are in LD with the causal variants (Hardy and Singleton 2009). GWAS have now reached the level of almost standard technique and have been used to investigate the genetic bases of a large variety of different disorders. For a complete list of GWAS accomplished to date, see http://www.genome.gov/26525384, whilst for a complete list of GWAS on neurological disorders, see http://www.alzgene.org/.
GWAS of FTLD-TDP Cases
A GWAS on 515 FTD patients with TDP-43 pathology was published in early 2010 identifying a possible susceptibility locus, which encompasses the gene TMEM106B, on chromosome 7p21 (Van Deerlin et al. 2010). The study identified three associated SNPs (rs1020004, rs6966915 and rs1990622) which relate, seemingly, to an increased expression of TMEM106B, a condition that could be involved in pathogenic mechanisms leading to FTLD-TDP (Van Deerlin et al. 2010). Two of the three GWAS-associated SNPs were investigated in patients with pure ALS and patients with ALS accompanied with cognitive impairment (Vass et al. 2011) showing no significant correlation in pure ALS cases, whilst ALS patients with comparatively poor cognitive performance carried the TMEM106B risk alleles (T allele for rs1990622; Vass et al. 2011). Furthermore, the distribution and severity of TDP-43 pathology seemed independent from TMEM106B genotypes (Vass et al. 2011). Another study on TMEM106B examined (1) the association of the three GWAS-associated SNPs (rs1020004, rs6966915 and rs1990622) in (a) a heterogeneous population of clinical FTLD (with and without PGRN mutations) and (b) pathologically FTLD-TDP confirmed patients without PGRN mutations; (2) the correlation between the TMEM106B risk alleles and PGRN protein levels in the plasma of normal controls; and (3) the correlation between the TMEM106B risk alleles and TMEM106B and PGRN mRNA levels (Finch et al. 2011). In the case of SNP rs1990622, the cohorts with PGRN mutations showed a higher association with major allele T, whilst cases associated with the minor allele C, specifically homozygous C, showed a later disease onset. No clear association was found in the large cohort of clinical FTLD and in the smaller cohort of FTLD-TDP with no PGRN mutations (Finch et al. 2011). Among neurologically normal controls, the minor alleles of the GWAS SNPs showed association with increased PGRN protein levels in plasma, and higher levels of PGRN mRNA were found in the presence of reduced levels of TMEM106B mRNA (Finch et al. 2011). In summary, the results from the original and this study showed that the highest association with the TMEM106B locus was found in FTLD-TDP cases with PGRN mutations. Replication in clinical FTLD cases did not reveal a significant association for this locus either in the original GWAS (Van Deerlin et al. 2010) or in Finch et al. (2011). The attempt to replicate the association for TMEM106B risk alleles failed in a British (Manchester and London) FTLD cohort (Rollinson et al. 2011), whilst association with rs1990622 was reported in a Belgian FTLD cohort (n = 288, but no significant TMEM106B mRNA differences in patients vs. controls were detected; van der Zee et al. 2011). Taking all these together, TMEM106B seems associated with FTLD, especially in pathologically confirmed FTLD-TDP and PGRN mutation carriers. If on one side these results (provided confirmed by future studies) will expand our knowledge about the genetic elements involved in FTLD-TDP and represent a starting point from where researchers can look into possible novel pathogenic pathways, on the other hand, they are confined to a specific FTLD subgroup, suggesting that the association with TMEM106B might be strictly related to a minority of FTD cases associated with a rare disease haplotype, which cannot be extended to the general FTLD population. Not least, it was suggested that TMEM106B could (1) act as a disease modifier by increasing risk in the general population, (2) affect age of onset and (3) modulate secreted levels of PGRN (Cruchaga et al. 2011).
As a note, successful GWAS require stringent inclusion/exclusion criteria and a well-defined disease phenotype. It is also relevant to perform genotyping on large cohort sizes (in the order of, at least, thousands) and to choose the appropriate genotyping platform and the population-based appropriate panel of SNPs. The power of a study is the probability of detecting a hypothesized effect of a certain size and is the function of (1) the effect of size on the causal variant, (2) the allele frequency of the causal variant and (3) the linkage disequilibrium between the tested variant, the causal variant and the sample size. Based on these observations, the aforementioned studies on TMEM106B confirm that sample size plays a fundamental role, in GWAS, in conferring statistical relevance to the results. If on one side the recruitment of pathologically confirmed samples helps tremendously in defining and characterizing a more homogeneous study population, on the other side, it contributes to limiting the size of the cohorts to be screened, conferring a reduced power to the study and, in addition, leading to a more difficult interpretation of the results. For example, in the case of TMEM106B, the association for SNPs rs1020004, rs6966915 and rs1990622 probably only represents a hint of association in the group of cases characterized, strictly, by TDP-43 pathology and, further, by the presence of PGRN mutations.
GWAS on Clinical FTD
With the aim of progressing in the knowledge of the genetics of FTD and discovering common genetic variants that influence disease risk, progression and variation, we are performing an ongoing GWAS on ∼3,500 FTD cases. Clinically well-defined and characterized samples have been collected from a large number of international research groups from 11 different countries (USA, Canada, UK, Belgium, the Netherlands, Denmark, Sweden, Germany, France, Spain and Italy) at the two institutions where this study is being performed: University College of London and the National Institute of Health. The inclusion and exclusion criteria for sample enrolment were based on the Neary criteria (Neary et al. 1998). Samples from patients with clinical features like behavioural variant; language dysfunction (semantic dementia, progressive non-fluent aphasia); and FTD-MND were included in the study. Most of the collected samples are of clinical extraction having been diagnosed by means of standard physical examination and psychological testing integrated, in most of the cases, with neuroimaging data, whilst a small minority has also a defined pathology. The collected cohort comprises sporadic cases of FTD and just the index patients from familial cases. Even though we are aware of the fact that collecting mainly clinical FTD samples contributes to a certain heterogeneity among samples, we have been careful in excluding patients with logopenic or phonological variant (LPA) which is associated, predominantly, with AD pathology (Seelaar et al. 2010) to rule out a possible overlap with AD that might contribute to misleading interpretation of the results. Genotyping has been performed using Illumina Infinium HD platform with the 660W-Quad BeadChips. In terms of analysis, we foresee that polymorphisms with real effects will not necessarily have the lowest p values in the study, but may be hidden amongst many other polymorphisms that have similar or greater levels of association. The results of this project will be collected in a central database (dbGAP) and made available to the scientific community. The db will contain age at onset, sex and centre codes together with the whole-genome data set: we expect to finalize data collection and to proceed with uploading in fall 2011.
One of the major difficulties underlying complex disease is the presumed and likely heterogeneous nature of the disorder. If on one side the probable high heterogeneity among samples will need to be critically managed, especially during analysis, among the strengths of this consortium and this project are the previous work by the applicants in their patient series to permit further analysis on subsets of the samples and the extremely large size of samples screened and analysed that will confer a considerable power to the study. At the end of this project, we expect to identify loci relevant to disease onset and progression and hope to pursue a better understanding of the genetic basis of the different subtypes of FTD.
Summary
Frontotemporal lobar degeneration belongs to the extended category of complex diseases. Geneticists are focused on the dissection of its genetic underpinnings trying to identify the genetic makeup that renders some individuals more susceptible to the development of the disease than others. To date we have made enormous progress in that direction through the latest technologies such as GWAS and whole-exome sequencing, and we know we are on the right track to fill the gaps in our knowledge. We are not far from the time (5–10 years?) when whole-genome sequencing will be available to most of the laboratories. This investigative technique will tremendously boost our insight into the genetic architecture of healthy and disease phenotypes. The final goal is to integrate the growing body of knowledge coming from the genetic, pathological and clinical fields to elucidate how genetic variability and gene expression, together with environmental factors, affect and influence biological pathways and phenotype in FTLD cases. We believe that succeeding in this endeavour is just a matter of vision and time: achieving these goals will tremendously increase our understanding of the etiology of FTLDs, fact that will, in turn, help us in developing more sensitive, accurate and effective diagnostic and treatment measures.
References
Ahmad ST, Sweeney ST, Lee JA, Sweeney NT, Gao FB (2009) Genetic screen identifies serpin5 as a regulator of the toll pathway and CHMP2B toxicity associated with frontotemporal dementia. Proc Natl Acad Sci U S A 106:12168–12173
Baba Y, Tsuboi Y, Baker MC, Uitti RJ, Hutton ML, Dickson DW (2005) The effect of tau genotype on clinical features in FTDP-17. Parkinsonism Relat Disord 11:205–208
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C et al (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319:916–919
Benussi L, Binetti G, Sina E, Gigola L, Bettecken T, Meitinger T et al (2008) A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging 29:427–435
Benussi L, Ghidoni R, Pegoiani E, Moretti DV, Zanetti O, Binetti G (2009) Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol Dis 33:379–385
Borroni B, Yancopoulou D, Tsutsui M, Padovani A, Sawcer SJ, Hodges JR (2005) Association between tau H2 haplotype and age at onset in frontotemporal dementia. Arch Neurol 62:1419–1422
Brown J, Ashworth A, Gydesen S, Sorensen A, Rossor M, Hardy J et al (1995) Familial non-specific dementia maps to chromosome 3. Hum Mol Genet 4:1625–1628
Buee L, Delacourte A (1999) Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick's disease. Brain Pathol 9:681–693
Caffrey TM, Wade-Martins R (2007) Functional MAPT haplotypes: bridging the gap between genotype and neuropathology. Neurobiol Dis 27:1–10
Cannon A, Baker M, Boeve B, Josephs K, Knopman D, Petersen R et al (2006) CHMP2B mutations are not a common cause of frontotemporal lobar degeneration. Neurosci Lett 398:83–84
Carrasquillo MM, Nicholson AM, Finch N, Gibbs JR, Baker M, Rutherford NJ (2010) Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet 87:890–897
Coppola G, Karydas A, Rademakers R, Wang Q, Baker M, Hutton M et al (2008) Gene expression study on peripheral blood identifies progranulin mutations. Ann Neurol 64:92–96
Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N et al (2011) Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 68:581–586
Cruts M, Van Broeckhoven C (2008) Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet 24:186–194
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D et al (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924
Daniel R, He Z, Carmichael KP, Halper J, Bateman A (2000) Cellular localization of gene expression for progranulin. J Histochem Cytochem 48:999–1009
Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K et al (2002) Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946
Dickson DW, Rademakers R, Hutton ML (2007) Progressive supranuclear palsy: pathology and genetics. Brain Pathol 17:74–82
Dormann D, Capell A, Carlson AM, Shankaran SS, Rodde R, Neumann M et al (2009) Proteolytic processing of TAR DNA binding protein-43 by caspases produces c-terminal fragments with disease defining properties independent of progranulin. J Neurochem 110:1082–1094
Ferrari R, Kapogiannis D, Huey ED, Grafman J, Hardy J, Momeni P (2010) Novel missense mutation in charged multivesicular body 1021 protein 2B in a patient with frontotemporal dementia. Alzheimer Dis Assoc Disord 24:397–401
Ferrer I, Santpere G, van Leeuwen FW (2008) Argyrophilic grain disease. Brain 131:1416–1432
Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132:583–591
Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M et al (2011) TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76:467–474
Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA et al (2006) Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol 65:571–581
Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
Ghanim M, Guillot-Noel L, Pasquier F, Jornea L, Deramecourt V, Dubois B et al (2010) CHMP2B mutations are rare in French families with frontotemporal lobar degeneration. J Neurol 257:2032–2036
Ghidoni R, Signorini S, Barbiero L, Sina E, Cominelli P, Villa A et al (2006) The H2 MAPT haplotype is associated with familial frontotemporal dementia. Neurobiol Dis 22:357–362
Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G (2008) Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71:1235–1239
Gijselinck I, Van Broeckhoven C, Cruts M (2008) Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mutat 29:1373–1386
Gitcho MA, Strider J, Carter D, Taylor-Reinwald L, Forman MS, Goate AM et al (2009) VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J Biol Chem 284:12384–12398
Goedert M (2004) Tau protein and neurodegeneration. Semin Cell Dev Biol 15:45–49
Goedert M (2005) Tau gene mutations and their effects. Mov Disord 20(Suppl 12):S45–S52
Goedert M, Jakes R (2005) Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta 1739:240–250
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3:519–526
Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA (1996) Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383:550–553
Goedert M, Klug A, Crowther RA (2006) Tau protein, the paired helical filament and Alzheimer’s disease. J Alz Dis 9:195–207
Grisart B, Willatt L, Destrée A, Fryns JP, Rack K, de Ravel T et al (2009) 17q21.31 microduplication patients are characterised by behavioural problems and poor social interaction. J Med Genet 46:524–530
Guo A, Tapia L, Bamji SX, Cynader MS, Jia W (2010) Progranulin deficiency leads to enhanced cell vulnerability and TDP-43 translocation in primary neuronal cultures. Brain Res 1366:1–8
Gydesen S, Brown JM, Brun A, Chakrabarti L, Gade A, Johannsen P et al (2002) Chromosome 3 linked frontotemporal dementia (FTD-3). Neurology 59:1585–1594
Halawani D, LeBlanc AC, Rouiller I, Michnick SW, Servant MJ, Latterich M (2009) Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol Cell Biol 29:4484–4494
Hardy J, Singleton A (2009) Genomewide association studies and human disease. N Engl J Med 360:1759–1768
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41:1088–1093
He Z, Bateman A (2003) Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med 81:600–612
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 43:429–435
Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR (2010) Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68:654–667
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H et al (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705
International HapMap Consortium (2003) The international HapMap project. Nature 426:789–796
International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320
International HapMap Consortium (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861
Jho YS, Zhulina EB, Kim MW, Pincus PA (2010) Monte Carlo simulations of tau proteins: effect of phosphorylation. Biophys J 99:2387–2397
Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS et al (2007) Neuropathologic features of frontotemporal lobar degeneration with ubiquitinpositive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 66:142–151
Ju JS, Weihl CC (2010) Inclusion body myopathy, Paget's disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum Mol Genet 19(R1):R38–R45
Kirchhoff M, Bisgaard AM, Duno M, Hansen FJ, Schwartz M (2007) A 17q21.31 microduplication, reciprocal to the newly described 17q21.31 microdeletion, in a girl with severe psychomotor developmental delay and dysmorphic craniofacial features. Eur J Med Genet 50:256–263
Kleinberger G, Wils H, Ponsaerts P, Joris G, Timmermans JP, Van Broeckhoven C (2010) Increased caspase activation and decreased TDP-43 solubility in progranulin knockout cortical cultures. J Neurochem 115:735–747
Knopman DS, Petersen RC, Edland SD, Cha RH, Rocca WA (2004) The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology 62:506–508
Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P et al (1997) p47 is a cofactor for p97-mediated membrane fusion. Nature 388:75–78
Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R et al (2006) A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 38:999–1001
Kurz A, Perneczky R (2009) Neurobiology of cognitive disorders. Curr Opin Psychiatry 22:546–551
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41:1094–1099
Laws SM, Perneczky R, Drzezga A, Diehl-Schmid J, Ibach B, Bäuml J (2007) Association of the tau haplotype H2 with age at onset and functional alterations of glucose utilization in frontotemporal dementia. Am J Psychiatry 164:1577–1584
Le Ber I, van der Zee J, Hannequin D, Gijselinck I, Campion D, Puel M et al (2007) Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 28:846–855
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159
Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB (2007) ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol 17:1561–1567
Lladó A, Rodríguez-Santiago B, Antonell A, Sánchez-Valle R, Molinuevo JL, Reñé R et al (2007) MAPT gene duplications are not a cause of frontotemporal lobar degeneration. Neurosci Lett 424:61–65
Mackenzie IR, Rademakers R (2007) The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 8:237–248
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J et al (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117:15–18
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J et al (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4
Malaspina A, Kaushik N, de Belleroche J (2001) Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem 77:132–145
Malkani R, D'Souza I, Gwinn-Hardy K, Schellenberg GD, Hardy J, Momeni P (2006) A MAPT mutation in a regulatory element upstream of exon 10 causes frontotemporal dementia. Neurobiol Dis 22:401–403
Mesulam M, Johnson N, Krefft TA, Gass JM, Cannon AD, Adamson JL et al (2007) Progranulin mutations in primary progressive aphasia. The PPA1 and PPA3 families. Arch Neurol 64:43–47
Momeni P, Rogaeva E, Van Deerlin V, Yuan W, Grafman J, Tierney M et al (2006a) Genetic variability in CHMP2B and frontotemporal dementia. Neurodegener Dis 3:129–133
Momeni P, Bell J, Duckworth J, Hutton M, Mann D, Brown SP et al (2006b) Sequence analysis of all identified open reading frames on the frontal temporal dementia haplotype on chromosome 3 fails to identify unique coding variants except in CHMP2B. Neurosci Lett 410:77–79
Momeni P, Schymick J, Jain S, Cookson MR, Cairns NJ, Greggio E et al (2006c) Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurol 6:44
Morris M, Maeda S, Vossel K, Mucke L (2011) The many faces of tau. Neuron 70:410–426
Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S et al (2006) HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol 60:314–322
Mukherjee O, Kauwe JS, Mayo K, Morris JC, Goate AM (2007) Haplotype-based association analysis of the MAPT locus in late onset Alzheimer's disease. BMC Genet 8:3
Mukherjee O, Wang J, Gitcho M, Chakraverty S, Taylor-Reinwald L, Shears S (2008) Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat 29:512–521
Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L et al (2007a) A survey of genetic human cortical gene expression. Nat Genet 39:1494–1499
Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L (2007b) The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis 25:561–570
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 43:436–441
Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M (2011) Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 377:641–649
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S et al (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM et al (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66:177–183
Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM et al (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67:1074–1077
Pickering-Brown SM, Rollinson S, Du Plessis D, Morrison KE, Varma A, Richardson AM et al (2008) Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131:721–1731
Pittman AM, Myers AJ, Duckworth J, Bryden L, Hanson M, Abou-Sleiman P et al (2004) The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum Mol Genet 13:1267–1274
Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L (2005) Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet 42:837–846
Poksay KS, Madden DT, Peter AK, Niazi K, Banwait S, Crippen D et al (2011) Valosin-containing protein gene mutations: cellular phenotypes relevant to neurodegeneration. J Mol Neurosci 44:91–102
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L et al (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825
Rabinovich E, Kerem A, Fröhlich KU, Diamant N, Bar-Nun S (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 22:626–634
Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G (1998) Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell 92:603–610
Ratnavalli E, Brayne C, Dawson K, Hodges JR (2002) The prevalence of frontotemporal dementia. Neurology 58:1615–1621
Rizzu P, van Mil SE, Anar B, Rosso SM, Donker Kaat L, Heutink P et al (2006) CHMP2B mutations are not a cause of dementia in Dutch patients with familial and sporadic frontotemporal dementia. Am J Med Genet B Neuropsychiatr Genet 141B:944–946
Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J et al (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73:1451–1456
Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC et al (2010) Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. NeuroImage 53:1070–1076
Rollinson S, Mead S, Snowden J, Richardson A, Rohrer J, Halliwell N et al (2011) Frontotemporal lobar degeneration genome wide association study replication confirms a risk locus shared with amyotrophic lateral sclerosis. Neurobiol Aging 32:758.e1–7
Rovelet-Lecrux A, Deramecourt V, Legallic S, Maurage CA, Le Ber I, Brice A et al (2008) Deletion of the progranulin gene in patients with frontotemporal lobar degeneration or Parkinson disease. Neurobiol Dis 31:41–45
Rovelet-Lecrux A, Lecourtois M, Thomas-Anterion C, Le Ber I, Brice A, Frebourg T (2009) Partial deletion of the MAPT gene: a novel mechanism of FTDP-17. Hum Mutat 30:E591–E602
Rovelet-Lecrux A, Hannequin D, Guillin O, Legallic S, Jurici S, Wallon D et al (2010) Frontotemporal dementia phenotype associated with MAPT gene duplication. J Alzheimers Dis 21:897–902
Schofield EC, Halliday GM, Kwok J, Loy C, Double KL, Hodges JR (2010) Low serum progranulin predicts the presence of mutations: a prospective study. J Alzheimers Dis 22:981–984
Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC (2011) Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry 82:476–486
Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B et al (2008) Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem 283:1744–1753
Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S et al (2006) Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet 38:1032–1037
Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D et al (2009) Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41:1308–1312
Singleton AB, Hardy J, Traynor BJ, Houlden H (2010) Towards a complete resolution of the genetic architecture of disease. Trends Genet 26:438–442
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H et al (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37:806–808
Skoglund L, Ingvast S, Matsui T, Freeman SH, Frosch MP, Brundin R et al (2009) No evidence of PGRN or MAPT gene dosage alterations in a collection of patients with frontotemporal lobar degeneration. Dement Geriatr Cogn Disord 28:471–475
Sleegers K, Brouwers N, Van Damme P, Engelborghs S, Gijselinck I, van der Zee J et al (2009) Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 65:603–609
Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B (1997) Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A 94:4113–4118
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A 95:7737–7741
Sugita S, Sudhof TC (2000) Specificity of Ca2+ dependent protein interactions mediated by the C2A domains of synaptotagmins. Biochemistry 39:2940–2949
Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY et al (2011) The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332:478–484
Urwin H, Ghazi-Noori S, Collinge J, Isaacs A (2009) The role of CHMP2B in frontotemporal dementia. Biochem Soc Trans 37:208–212
Urwin H, Authier A, Nielsen JE, Metcalf D, Powell C, Froud K et al (2010) Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet 19:2228–2238
Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239
van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I, Camuzat A et al (2007) Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat 28:416
van der Zee J, Urwin H, Engelborghs S, Bruyland M, Vandenberghe R, Dermaut B et al (2008) CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum Mol Genet 17:313–322
van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R et al (2011) TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134:808–815
van Swieten JC, Heutink P (2008) Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol 7:965–974
van Swieten J, Spillantini MG (2007) Hereditary frontotemporal dementia caused by TAU gene mutations. Brain Pathol 17:63–73
Vandrovcova J, Anaya F, Kay V, Lees A, Hardy J, de Silva R (2010) Disentangling the role of the tau gene locus in sporadic tauopathies. Curr Alzheimer Res 7:726–734
Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D et al (2011) Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol 121:373–380
Wang Q, Song C, Li CC (2004) Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol 146:44–57
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36:377–381
Weihl CC, Dalal S, Pestronk A, Hanson PI (2006) Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum Mol Genet 15:189–199
Weihl CC, Pestronk A, Kimonis VE (2009) Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul Disord 19:308–315
Williams RL, Urbé S (2007) The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol 8:355–368
Zhang YJ, Xu Y, Dickey CD, Buratti E, Baralle F, Bailey R et al (2007) Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci 27:10530–10534
Zody MC, Jiang Z, Fung HC, Antonacci F, Hillier LW, Cardone MF et al (2008) Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet 40:1076–1083
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ferrari, R., Hardy, J. & Momeni, P. Frontotemporal Dementia: From Mendelian Genetics Towards Genome Wide Association Studies. J Mol Neurosci 45, 500–515 (2011). https://doi.org/10.1007/s12031-011-9635-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9635-y