
Abstract
Frontotemporal dementia (FTD) is a progressive brain disease characterized by atrophy of the frontal and anterior temporal lobes. The prevalence has been estimated between 10 and 30 per 100,000, and patients have severe changes in personality and behavior. The disease has a strong genetic component, and in up to 40 % of cases, a positive family history has been observed. To date, seven disease genes have been identified, of which MAPT, GRN, and C9orf72 are most frequently mutated. In contrast to familial FTD, far less is known about sporadic FTD. GWAS reported TMEM106B as an important risk factor for FTD, and recently, new loci have been associated with the disease. In this chapter, we summarize the current insights into the genetics of FTD based on neuropathological and functional data.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Frontotemporal dementia (FTD) is a devastating presenile dementia characterized by atrophy of the frontal and anterior temporal lobes, and the prevalence, in the age group between 45 and 65 years, has been estimated between 10 and 30 per 100,000 [1]. It is a heterogeneous group of disorders and associated with genes that typically cause a clinical picture of amyotrophic lateral sclerosis (ALS), the most common type of motor neuron disease (MND) [2].
Clinically, there is progressive deterioration of either behavior or language, which translates into two FTD subtypes: the behavioral variant of frontotemporal dementia (bvFTD) and primary progressive aphasia (PPA). In bvFTD, patients have severe changes in behavior and personality, as indicated by early disinhibition, apathy, loss of sympathy, perseverative and stereotypic behavior, and hyperorality [3]. PPA can be further subdivided into semantic variant PPA (svPPA) and nonfluent variant PPA (nfvPPA), based on specific speech and language features. A third recognized subtype for patients not fitting either category is logopenic variant primary progressive aphasia (lvPPA) [4, 5]. The bvFTD accounts for more than 50 % of the FTD patients, and PNFA is the second most prevalent presentation of FTD, accounting for 25 %. The svPPA presents in 20–25 % of the FTD patients, and lvPPA is not considered part of the FTD group of disorders [6, 7]. Other conditions closely related to FTD are MND, progressive supranuclear palsy (PSP) syndromes, corticobasal syndrome (CBS), FTD with parkinsonism (FTDP), and argyrophilic grain disease (AGD) [8].
At the neuropathological level, the different subtypes of FTD present with diverse patterns of frontal and anterior temporal lobe atrophy. The term “frontotemporal lobar degeneration” (FTLD) is often used for these pathological conditions that present with FTD. FTLD is a proteinopathy characterized by abnormal, ubiquitinated protein inclusions in the cytoplasm or nuclei of neuronal and glial cells. Neuronal loss and astrocytosis are seen in cortices of atrophied frontal and temporal lobes, and neuropathological subcategories are based on the major constituent of inclusions [9]. Initially, two pathological categories were described: FTLD-tau including Pick’s disease, in which neurons and glial cells contained inclusions of hyperphosphorylated tau protein, and FTLD-Ubiquitin or FTLD-U with unknown inclusions. In 80–95 % of the FTLD-U group, inclusions were later found to be mainly composed of TDP-43 (FTLD-TDP), and a considerable number of TDP-43-negative FTLD-U cases had inclusion of FUS (FTLD-FUS) [10, 11]. In a small number of FTLD-U patients, the inclusion protein remains unknown (FTLD-UPS) [12].
In up to 40 % of FTD cases, a positive family history is observed and inheritance varies among the different clinical subtypes [13–15]. Family history is most prominent in bvFTD, especially when MND is present. Research on these families has led to the discovery of multiple disease-causing genes. Mutations in microtubule-associated protein tau (MAPT) [16–18], granulin (GRN) [19, 20], and chromosome 9 open reading frame 72 (C9orf72) [21, 22] together explain the majority of familial FTD cases [1, 23]. Mutations in valosin-containing protein (VCP) and charged multivesicular body protein 2B (CHMP2B) are rare, each explaining less than 1 % of the familial FTD [1]. Mutations in common ALS genes TAR DNA-binding protein (TARDBP) and fused in sarcoma (FUS) are a very rare cause of familial FTD [24–27]. Association between affected gene and associated neuropathology is observed, and the association between disease gene and clinical phenotype is limited (Table 5.1) [1]. In contrast to familial forms of FTD, far less is known about the genetics of sporadic forms of the disease. To date, two genome-wide association studies (GWAS) have been published in which TMEM106B at chromosome 7p21 has been reported as a risk factor for FTLD-TDP [28] and novel genetic risk loci and pathways have been associated with FTD [29].
MAPT
In the late 1990s, linkage at chromosome 17q21 was found for multiple families in FTD and parkinsonism linked to chromosome 17 (FTDP-17). Three research groups found mutations in and around MAPT exon 10 and proved that dysfunction of the protein tau is sufficient to cause neurodegeneration and dementia [16–18]. Accumulation of tau occurs primarily as insoluble fibrils in neuronal cell bodies (neurofibrillary tangles (NFTs)) and processes (neuropil threads or dystrophic neurites), but can also accumulate in astrocytes and microglia. Tau deposits are characteristic for a large number of neurodegenerative diseases known as the “tauopathies,” which include Alzheimer’s disease (AD), Pick’s disease (PiD), PSP, corticobasal degeneration (CBD), and AGD [30]. In AD, all six brain isoforms of tau are found in inclusions [31], and in FTDP-17, inclusions are heterogeneous, with different mutations associated with a different isoform composition. For PSP, CBD, and AGD, inclusions consist mainly of 4-repeat tau [32–34], and PiD has predominantly 3-repeat tau deposits [35]. In populations of European descent, MAPT is characterized by two haplotypes, H1 and H2, which result from a 900-kb inversion polymorphism. Inheritance of the H1 haplotype is a risk factor for PSP, CBD, and idiopathic Parkinson’s disease. The H2 haplotype is associated with increased expression of exon 3, suggesting that inclusion of this exon might be protective [36–39].
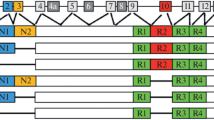
Tau proteins are microtubule-associated proteins that are abundant in the central nervous system (CNS) and mainly concentrated in axons [40]. The MAPT gene is located on chromosome 17q21 and consists of 16 exons, as depicted in Fig. 5.1. In adult human brain, alternative mRNA splicing of exons 2, 3, and 10 produces six tau isoforms ranging from 352 to 441 amino acids. Three of these isoforms contain 4 microtubule-binding repeats (4R), whereas the other three contain only 3 microtubule-binding repeats (3R). The extra repeat domain is encoded by exon 10. The presence or absence of a 29 amino acid (exon 2) or a 58 amino acid (exons 2 and 3) insert in the N-terminal half of the protein divides the isoforms further in 0N, 1N, and 2N (Fig. 5.1). The ratio of 4R and 3R tau isoforms in the cerebral cortex of healthy adults is equal, while in fetal brain, only 3R0N is expressed. Most adult rodents express isoforms with four repeats [41]. Tau is enriched in neurons and has an important function in promoting microtubule assembly and stability [42–44]. The C-terminal repeat domains bind microtubules directly, and 4R tau promotes microtubule assembly 3-fold stronger than 3R tau [45, 46]. The N-terminal inserts are part of the projection domain, establishing the interaction of tau and microtubules with the plasma membrane [47, 48]. In addition to its role in microtubule assembly, tau mediates transport of vesicles and organelles along the microtubules by regulating attachment and detachment of motor proteins [49, 50]. The motility of motor proteins dynein and kinesin is differentially regulated and dependent on tau concentration and isoform, pointing to a balanced spatiotemporal regulation of axonal transport [51]. Literature also suggests a physiological role for tau in dendrites, which might be an important function mediating synaptic impairment early in the disease process [52, 53].

Human brain MAPT isoforms. MAPT and the six isoforms found in adult human brain. Exons 1, 4, 5, 7, 9, and 11–13 are constitutively spliced (blue), and exons 0, which is part of the promoter, and 14 are noncoding (white). Exons 6 and 8 are not transcribed in human brain (orange), and the unusually long exon 4a (gray) is expressed only in the peripheral nervous system in higher-molecular-weight proteins termed “big tau.” Alternative splicing of exons 2 (green), 3 (purple), and 10 (red) gives rise to the six isoforms (352–441 amino acids), and the repeats of tau (R1–R4) are indicated
Phosphorylation is the major posttranslational modification of tau protein, and in diseased brain, tau is abnormally hyperphosphorylated, generating paired helical filaments (PHFs) and neurofibrillary tangles [54–56]. The degree of tau phosphorylation influences its interaction with microtubules. Abnormal hyperphosphorylated tau sequesters normal tau, leading to misfolding, co-aggregation, and inhibition of microtubule assembly [57]. Microtubule-associated proteins 1 and 2 are also sequestered from pre-assembled microtubules, leading to their disruption [58–61]. There are 80 serine/threonine and 5 tyrosine potential phosphorylation sites, and normal brain tau contains 2–3 mol of phosphate per mole of tau protein. Mass spectrometry and antibody staining have identified more than 40 serine/threonine hyperphosphorylation sites of tau in AD [62, 63]. The phosphorylation of tau itself is developmentally regulated, and fetal tau is more phosphorylated than adult brain tau [64]. Several protein kinases are known to phosphorylate tau, and hyperphosphorylation probably involves coordinated action of several of them [65, 66]. When tau hyperphosphorylation induces dissociation from microtubules, it is redistributed from axonal to somatodendritic compartments. The increased pool of hyperphosphorylated tau is thought to promote assembly into fibrillar aggregates. In addition to phosphorylation, tau undergoes other posttranslation modifications, such as acetylation, glycation, glycosylation with O-linked N-acetylglucosamine, nitration, ubiquitination, sumoylation, prolyl isomerization, and truncation, all of which affect tau phosphorylation and/or aggregation [30].
Pathological MAPT Mutations
Up to 44 different clinical MAPT mutations in 134 FTD families have been reported in exons 1, 9, 10, 11, 12, and 13, intron 9, and the first 19 nucleotides of intron 10 (detailed information on each MAPT mutation is available at www.molgen.ua.ac.be/ftdmutations) [67, 68]. Except for mutations P301L in exon 10 and E10 + 16 in intron 10, identified in 32 families and 27 families, respectively, MAPT mutations are rare and seen in single families. Mutations are mainly clustered in exons 9–12, encoding the four microtubule-binding domains of tau, and can be divided into two functional groups. The first group has a primary effect at the protein level and includes missense and deletion mutations in the coding region of the gene. These mutations reduce the ability of tau to interact with microtubules, and some mutations promote filament assembly [68, 69]. The second group affects alternative splicing of tau pre-mRNA. This includes intronic and exonic mutations that alter alternative mRNA splicing of MAPT exon 10. The intronic mutations are clustered at the exon 10/intron 10 junction that is predicted to form a stem-loop structure protecting the 5′-splice site [70, 71]. Most of these mutations increase exon 10 inclusion and raise the normal 4R/3R isoform ratio from 1 to 2–3 [16]. However, a few mutations such as Δ(delta)K280 enhance exon 10 exclusion and result in increased expression of 3R tau [72]. In FTDP-17, the early clinical symptoms vary with the type of mutation found, and there is considerable phenotypic variability [73].
Tau Animal Models
After the identification of pathogenic mutations in MAPT in FTDP-17, several groups reported on the formation of NFTs and neuronal death in mouse models expressing human mutant tau, reviewed by Götz et al. [74]. To study the link between NFTs and brain dysfunction, transgenic mice were created expressing human P301L mutant tau that could be suppressed by doxycycline. This model (rTg4510) develops progressive age-related NFTs, neuronal loss, and behavioral impairment [75]. After switching off transgenic tau expression, memory function recovers and the number of neurons stabilizes, while NFTs continue to accumulate. Additional studies in rTg4510 showed that neuron loss can occur independently of NFT pathology and not all regions with NFT pathology undergo neuron loss, together implying that NFTs alone are not sufficient to cause cognitive decline or neuronal death [76, 77]. In vivo imaging shows that tangle-bearing neurons are long-lived and soluble tau species may be the critical components underlying neurodegeneration [78]. In transgenic mice expressing mutant repeat domains of human tau, similar observations were made [79]. After turning off expression of pro-aggregant Δ(delta)K280 mutant repeat domains of tau, memory and long-term potentiation recovers, while neuronal loss and aggregates persist. When anti-aggregant mutations were added, neuronal death was prevented, but tau aggregates are still formed [80].
There is accumulating evidence for intercellular transfer of tau aggregates [81–83]. In one of the first studies, brain homogenates from mice expressing human mutant P301S tau protein (with silver-positive tau inclusions) were injected into mice expressing human wild-type tau. This induced filament formation of wild-type human tau and spreading of pathology over time to anatomically connected brain regions [81]. Additional work showed that synthetic tau fibrils assembled from human mutant tau protein promoted the formation of tau inclusions in presymptomatic mice transgenic for human mutant P301S tau protein. In these mice, spreading of tau pathology was also initiated [84]. This has been further supported by experiments in a mouse model expressing human tau P301L restricted to layer II of the entorhinal cortex, recreating an early stage of NFT pathology [85]. Recently, similar results were obtained after injection of brain homogenates from cases of human tauopathies (including AD, PiD, AGD, PSP, and CBD) [86]. One other study demonstrated the induction and propagation of pathology when tau oligomers from AD brain were injected into wild-type mice, suggesting that they might play a critical role in initiation and spreading of tau pathology [87]. In vitro studies also showed induction and propagation of tau misfolding, and involvement of the endocytic pathway has been suggested [83, 88–91]. These studies show that tau fibrils spread from cell to cell by converting soluble tau into aggregates, and this mechanism might be responsible for disease propagation.
Besides the mouse, other organisms have been used to study FTD, such as the fruit fly Drosophila melanogaster, the nematode Caenorhabditis elegans, and the zebrafish Danio rerio. When wild-type and FTDP-17 mutant forms of human tau (R406W) were expressed in Drosophila, flies exhibited adult-onset progressive neurodegeneration, early death, enhanced toxicity of mutant tau, and abnormal tau accumulation without NFT formation [92]. Expression of wild-type and mutant human tau (P301L and V337M) in the C. elegans led to progressive uncoordinated locomotion, accumulation of insoluble tau species, neurodegeneration, and loss of neurons [93]. Expression of mutant tau caused an earlier and more severe phenotype compared to wild-type tau. Stable expression of human tau with the P301L mutation in the zebrafish also recapitulates key pathological features of tauopathies, including tau hyperphosphorylation, tangle formation, behavioral disturbances, and neuronal cell death [94].
GRN
In a large number of families linked to chromosome 17q21, mutations in MAPT could not be identified. In 2006, systematic analyses of the 17q21 region led to the discovery of mutations in GRN as a cause of FTLD-U [19, 20]. All GRN-associated FTLD patients present with TDP-43-positive inclusions in the affected cortical regions and basal ganglia (FTLD-TDP), often accompanied by irregular and short dystrophic neurites [95, 96]. The inclusions are found in neurons and glial cells and can be cytoplasmic as well as intranuclear. NFTs, neuritic plaques, and Lewy bodies have occasionally been observed in GRN brains [97–100].
In humans, the GRN gene is located upstream to the MAPT gene and comprises 12 coding exons (Fig. 5.2) [101]. The gene encodes progranulin, a 593-amino-acid-long protein containing a secretory signal sequence and seven and a half repeats of a unique 10–12 cysteine-containing motif. This motif is highly conserved in evolution and a reminiscent of the epithelial growth factor [102–105]. Progranulin is expressed in several tissues of the periphery as well as in neurons and microglia in the central nervous system, and little or no expression has been detected in adult astrocytes and oligodendrocytes [102, 106–108]. A GWAS implicated sortilin (SORT1) as an important regulator of GRN levels in human plasma, and cellular studies showed that SORT1 is a major neuronal receptor for GRN [109, 110]. SORT1 facilitates extracellular GRN clearance via endocytosis and delivery to the lysosomes. When GRN interaction with SORT1 is impaired, it is possible to restore extracellular GRN levels in FTD patient-derived lymphocytes and induced pluripotent stem cells (iPSC) differentiated to neurons [111].

GRN gene and protein structure. Schematic representation of human GRN gene structure (top) and progranulin protein structure (bottom). The GRN gene spans approximately 8 kb genomic sequence on chromosome 17q21. The gene structure indicates noncoding exon 1 (white) and the coding exons 2–13 (blue). The full-length progranulin protein presents a signal peptide (SP) and seven and a half tandem repeats of a conserved 10–12 cysteine-containing motif (granulins A to G and paragranulin P). Once the signal peptide is cleaved off, the mature protein gets secreted; it can be further processed by cleavage in the linker regions between the granulin domains. The lettered boxes in progranulin refer to individual granulin domains (~6 kDa)
GRN plays a key role in several biological processes including cell growth, wound repair, inflammation, and neuron development [112]. Studies in different cellular models show that GRN expression enhances neuronal survival and promotes neurite outgrowth and branching, while siRNA-mediated GRN knockdown results in reduced neurite arborization and dendritic protrusions [113–117]. The exact mechanism through which GRN exerts its neurotrophic function is not completely understood; however, compelling evidence suggests the involvement of the phosphatidylinositol 3-kinase/Akt signaling pathway and glycogen synthase kinase-3 beta regulation [118–120]. GRN-deficient cells show increased caspase activation, decrease in cellular survival, and vulnerability to a number of cellular stressors, including oxygen and glucose deprivation, oxidative stress, and sublethal doses of N-methyl-D-aspartic acid, kinase inhibitors, and proteasomal inhibitors [118, 121–123].
It has been hypothesized that GRN performs a neuroprotective role in the CNS by stimulating production of anti-inflammatory Th2 cytokines and supporting neuroprotective inflammation by binding to TNF receptors [124, 125]. The anti-inflammatory effects of GRN are evident in Grn−/− mice that show an exacerbated inflammatory reaction [123]. Moreover, increased GRN expression has been observed in microglia following a variety of acute and chronic insults in several neurodegenerative disorders including Creutzfeldt-Jakob disease [126], ALS [127, 128] and FTLD [99]. It also attracts and activates microglia, enhancing endocytosis of extracellular peptides such as amyloid β (beta) 1–42 [129]. Interestingly, microglia of patients with GRN mutations are able to upregulate GRN despite reduced levels in blood, cerebrospinal fluid, and neurons in unaffected brain regions [99] suggesting GRN plays a central role in the inflammatory response.
Pathological GRN Mutations
Confirmed pathogenic mutations in GRN cases have been so far identified only in FTLD-TDP, and GRN mutation screening in patients with ALS [130, 131], Parkinson’s disease [132], and non-FTD cases with TDP-43 pathology [133] yields no positive results. To date, 69 pathogenic mutations have been observed in patients with FTLD-TDP, mostly being loss-of-function mutations suggesting that GRN haploinsufficiency is the major pathogenic mechanism (detailed information on each GRN mutation is available at www.molgen.ua.ac.be/ftdmutations) [67]. GRN deficiency occurs through frame-shift, splice site, and nonsense mutations, introducing a premature termination codon with activation of nonsense-mediated mRNA decay and reduction of progranulin protein levels [19, 20, 134]. Loss of mRNA translation and consequent protein haploinsufficiency is also observed when mutations are present in the Kozak sequence of the GRN gene preventing translation [19, 20, 135, 136] or result in genomic deletion of one copy of the gene [137, 138].
The GRN R493X and T272fs mutations are among the most frequent mutations found in GRN patients [139, 140]. Patients carrying these mutations do not show significant phenotypic differences with other GRN mutation carriers. Interestingly, the occurrence of a founder effect has been suggested for both mutations [139, 141]. In addition, missense and silent mutations have been observed scattered along the GRN gene. For a large number of them, it is still debated if they play an active role in the disease pathogenesis. Some mutations are predicted to affect protein function in silico, they have been found in patients only, or experimental studies demonstrated a potential pathogenic effect causing reduced protein expression by cellular mechanisms other than GRN mRNA reduction [142, 143]. Mutations A9D, P248L, and R432C have been reported to affect protein secretion and stability thereby reducing the amount of available GRN. The A9D mutation [144] occurs within the hydrophobic core of the signal peptide sequence of GRN impairing its secretion. The mutated protein fails to undergo N-glycosylation, suggesting its missorting into the cytosol [135, 143]. The P248L and R432C mutations might contribute to disease phenotype by adversely affecting the neurotrophic properties of GRN [114].
GRN mutations pathologically confirmed in FTD with TDP-43 pathology are so far all heterozygote. The only exception is a single report of two siblings clinically diagnosed with adult-onset neuronal ceroid lipofuscinosis (NCL), a lysosomal storage disorder with severe accumulation of lipofuscin, and progressive neurodegeneration [145]. Interestingly, Grn−/− mice also show an increase of the aging pigment lipofuscin accompanied by tissue vacuolization in the hippocampus. Although we are still awaiting for the neuropathological assessment, the inclusion of GRN as genetic cause of NCL [146] has important consequences for the role of GRN in lysosome biology.
GRN Animal Models
To date, five independent Grn−/− mouse models have been established and extensively characterized (reviewed in [112, 147]). Although in humans GRN loss alone is sufficient to cause neurodegeneration, none of the mouse models fully recapitulate the human disease. Their most consistent behavioral phenotype is reduction in social interactions already observed at young age. Neuropathologically, Grn−/− mice all display pronounced progressive microgliosis and astrocytosis as well as ubiquitin accumulation in the cortex, hippocampus, thalamus, and brain stem. Accumulation of ubiquitinated proteins suggests disturbance of the proteasomal and/or autophagy-lysosomal pathways, which is supported by accumulation of the autophagy-related receptor p62 and increased expression of lysosomal protease cathepsin D [148]. Neuronal loss is in general not remarkable, and it is present only in selected areas; however, Grn−/− mice show impaired neuronal function with reduced synaptic connectivity and impaired synaptic plasticity. These findings support in part the results from GRN knockdown or knockout studies in primary hippocampal neuronal and slice cultures [149, 150]. Interestingly, altered synaptic vesicle numbers are also found in FTD patients carrying GRN mutations [149]. Grn−/− mice do not show TDP-43 fragmentation, cytoplasmic mislocalization, abnormal phosphorylation, and accumulation in aggregates typical of FTLD-TDP associated with GRN mutations [148, 151]. In the insoluble fraction of Grn−/− mice brain lysates, only full-length TDP-43 has been detected with a significant increase from 12 months forward. One study did show the appearance of phosphorylated TDP-43 in the cytosol of neurons in the hippocampal and thalamic areas [123, 152], and studies in mouse cortical neurons and GRN patient iPSC differentiated to neurons demonstrated increased translocation of TDP-43 from the nucleus to the cytoplasm [118, 121].
C9orf72
In 2011, two independent consortia identified the pathological expansion of a noncoding GGGGCC hexanucleotide repeat in C9orf72 as a cause of familial FTD and ALS, explaining several linkages and GWAS performed since 2006 linking chromosome 9p to these disorders [21, 22, 153, 154]. The findings where confirmed by other studies in a wide series of populations [155, 156], and repeat expansions for C9orf72 have since then also been confirmed in families affected with AD, CBS, and ataxia syndromes [157, 158].
The C9orf72 gene is transcribed in three major transcripts and located on the short arm of chromosome 9. The location of the repeat is between noncoding exons 1a and 1b, which is the first intron following noncoding exon 1 of transcript variants 1 and 3 and the upstream regulatory region of transcript variant 2 (Fig. 5.3). In the general population, the median length of the hexanucleotide repeat sequence is two repeats (range 0 to ±20). The C9orf72 transcript variants 1 and 3 encode for a 481 amino acid protein, whereas transcript variant 2 encodes for a shorter 222 amino acid protein. These protein isoforms share the first 221 N-terminal amino acids but differ in their C-terminal region. C9orf72 is detected throughout the CNS, with the highest expression level observed within the cerebellum [21, 22, 154, 159, 160]. Currently, there are no specific antibodies for C9orf72, and therefore, the exact subcellular localization is not very well established. In neuronal cell lines transfected with C9orf72, the protein is detected both in the nucleus and cytoplasm and in the medium [161], and additional studies have suggested C9orf72 is also present in the membrane fraction of cells after subcellular fractionation experiments [162, 163]. Further experiments are needed to conclusively determine where the protein is localized.

Human C9orf72 gene structure. The structure of the C9orf72 gene and its major transcripts. Transcript variant 1 (NM_145005.5) in green, transcript variant 2 (NM_018325.3) in orange, and transcript variant 3 (NM_001256054.1) in purple according to the NCBI RefSeq Database. The arrow (red) marks the hexanucleotide repeat location. (GGGGCC)n indicates repetition of this sequence n times, with a median length of two repeats in the general population (range 0 to ±20). Colored blocks indicate coding exons and white blocks indicate noncoding exons
C9orf72 is highly conserved in evolution, and bioinformatics approaches showed that the protein is a novel homologue of differentially expressed in normal and neoplastic (DENN) proteins [164, 165]. DENN proteins are guanine exchange factors (GEF) that activate Rab GTPases, and these findings suggest a role for C9orf72 in Rab GTPase-dependent membrane trafficking. Support comes from a study in which several Rab proteins involved in endocytosis and autophagy were found to co-localize or co-immunoprecipitate with C9orf72 [161]. Knockdown of C9orf72 in neuronal cell lines also led to defects in autophagic processing as well as endocytosis. Furthermore, human neurons differentiated from iPSC with the C9orf72 repeat expansion were more sensitive to autophagy-inhibiting drugs [166]. Functional studies are required in order to conclusively demonstrate that C9orf72 encodes a GEF and Rab proteins might be activated.
Effect of the Pathogenic C9orf72 Mutation
The frequency of C9orf72 repeat expansions ranges from 12 to 25 % in familial and 6–7 % in sporadic FTD patients and 10–50 % in familial and 5–7 % in sporadic ALS cases. This makes C9orf72 repeat expansions the most common cause of the FTD/ALS complex of diseases [155, 167, 168]. The causal effect of the C9orf72 mutation is not yet completely understood, and currently, three mechanisms have been implicated for the disease.
There is accumulating evidence that the expanded repeat is associated with reduced expression of C9orf72 transcripts, and studies in human postmortem brain tissue, patient-derived iPSC, and lymphoblasts suggest that a loss of function might be relevant for the disease pathogenesis [21, 154, 159, 160, 162, 166, 169, 170]. Epigenetic changes could be responsible for the decrease in C9orf72 expression levels observed in patients, and this mechanism has been described previously for Friedreich’s ataxia and fragile X syndrome [171, 172]. This hypothesis is supported by two studies describing hypermethylation of CpG islands and abnormal histone binding associated with the repeat expansion [159, 173].
A second mechanism associated with repeat expansion diseases is RNA toxicity, caused by sequestration of normal transcripts and RNA-binding proteins by RNA molecules including the expanded repeat [174]. The presence of the expanded repeat in pre-mRNA transcripts could prevent normal splicing toward a mature mRNA, and the resulting repeat-containing RNA species could then aggregate into RNA foci and have a toxic effect. In support of this hypothesis, repeat-containing RNA aggregates have been reported in brains of patients and in patient-derived iPSC [21, 162, 163, 175, 176]. In vitro experiments using GGGGCC-repeat RNA oligonucleotides showed that they fold into stable RNA G-quadruplex structures, which suggests that the inclusion of the expanded repeat into the C9orf72 transcripts in vivo could interfere with the normal processing of the pre-mRNA for transcripts 1 and 3 [177, 178]. This is supported by in vitro transcription assays that show the presence of abortive transcripts caused by G-quadruplex assembly, and these findings were confirmed in patient-derived B lymphocytes [179]. There is also evidence for the sequestration of RNA-binding proteins (RBPs) by RNA molecules including the transcribed repeat expansion, which could cause a depletion of RBPs available for normal RNA metabolism. RNA-binding assays, using r(GGGGCC)10 repeat sequences, identified Pur α as a repeat binding protein. Loss of Pur α reduced cell viability of Neuro-2a cells and overexpression of Pur α in Drosophila, and Neuro-2a cells mitigated the repeat-mediated neurotoxicity [180]. The interaction of Pur α is supported by co-localization studies, and also binding of several heterogeneous nuclear ribonucleoprotein (hnRNP) family members to the hexanucleotide repeat has been reported [163, 181].
A third hypothesis is that repeat-associated non-ATG-initiated (RAN) translation takes place within the expanded repeat sequence, resulting in the production and aggregation of dipeptide repeat proteins. In support of this hypothesis, aggregates positively stained with antibodies raised against putative hexanucleotide repeat RAN-translated peptides have been detected in human postmortem brain and cultured patient-derived iPSC [162, 170, 182]. The presence of stable RNA G-quadruplex structures could induce RAN translation similar to what has been reported for expanded CAG repeats in spinocerebellar ataxia 8 and myotonic dystrophy type 1 [183]. Sequencing results of DNAse 1-treated pre-mRNA from postmortem tissue matched the C9orf72 genomic sequence and therefore support this hypothesis [182]. Interestingly, a C9orf72 antisense transcript containing intron 1 sequences including repeat sequences was detected raising the possibility that the dipeptide repeat proteins might be translated from this transcript [184]. Foci containing this antisense RNA transcript are observed in the same brain regions as sense foci, but they are present in fewer cells and the average number of foci per cell appears higher [175]. Recently, a study in U2OS cancer cells and human astrocytes showed that synthetic dipeptides containing 20 repeats of sense glycine-proline (GR) or antisense proline-arginine (PR) associate with nucleoli and affect cell morphology and viability. Exposure of cultured cells to these GR and PR translation products also led to altered pre-mRNA splicing and changes in ribosomal RNA biogenesis [185].
Animal Models for C9orf72
One of the first animal models created for C9orf72 was a zebrafish model in which the translation of the zC9orf72 orthologue was blocked using antisense morpholino oligonucleotides (AMO). This model system provided evidence that a decrease in C9orf72 expression level could be important for the disease process [169]. The knockdown resulted in disrupted branching and shortening of motor neuron axons when compared to noninjected or mismatch AMO-injected fish; this phenotype could be rescued by injecting the mRNA for the human C9orf72 long transcript. In addition, knockdown of C9orf72 in zebrafish resulted in motor deficits associated with axonopathy, such as deficits in touch-evoked escape response and reduced mobility. Partial knockdown of endogenous C9orf72 to 30–40 % of normal levels in a mouse model by antisense oligonucleotide administration was well tolerated for 18 weeks, which does not support that a loss of function is the main mechanism of the mutation [176]. Studies in patient fibroblasts and motor neurons derived from patient iPSC also did not confirm this hypothesis [163, 176]. A Drosophila model expressing the expanded hexanucleotide repeat (rGGGGCC) provides evidence that RNA foci or aberrantly processed pre-mRNAs are toxic. Neuronal toxicity in Drosophila and Neuro-2a cells was observed, and the expression of rGGGGCC repeats caused progressive neurodegeneration in Drosophila eye and a reduction in locomotor activity [180]. A more recent study differentiated between repeat RNA and DPR protein toxicity and showed that the major toxic species in their Drosophila model system are the DPR proteins; however, additional contribution of RNA toxicity is not ruled out [186].
VCP and CHMP2B
Mutations in two other genes, VCP and CHMP2B, lead to FTLD-TDP and account for a minority of familial cases [1]. Mutations in VCP were identified in 2004 by linkage analysis studies in families with inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD) [187]. VCP mutations are also a cause of familial ALS [188]. The protein is ubiquitously expressed and a highly conserved member of the AAA(+)-ATPase superfamily. The protein is associated with a wide variety of cellular activities, including cell cycle control and membrane fusion. It is also a multi-ubiquitin chain-targeting factor and is required in the degradation of many ubiquitin-proteasome pathway substrates [189]. IBMPFD mutations cluster in the N-terminal CDC48 domain, mediating substrate recognition and cofactor binding. Studies in patient tissue, transgenic mice, and cell models suggest that VCP is a key regulator at the intersection of autophagy and the ubiquitin-proteasome system [190, 191]. Mouse models expressing mutant VCP recapitulate aspects of the disease, including age-dependent degeneration in muscle, brain, and bone, and progressive widespread TDP-43 pathology [192, 193].
Mutations in CHMP2B were found in 2005 via linkage analysis in a large Danish FTD family [194], and mutations are also a cause of familial ALS [195]. The ubiquitinated neuronal cytoplasmic inclusions do not stain for tau, TDP-43, or FUS, and the classification is FTLD-UPS. CHMP2B is a component of the endosomal sorting complex required for transport III (ESCRT-III) and expressed in neurons of all major brain regions. The ESCRT-III complex is required for function of the multivesicular body (MVB), an endosomal structure that fuses with the lysosome to degrade endocytosed proteins. Mutations affect the C-terminal region of the protein due to aberrant splicing, and mutant CHMP2B disrupts the fusion of endosomes with lysosomes in cell culture models [196]. Mice expressing C-terminally truncated and mutant CHMP2B develop axonal swellings and have reduced survival. The mice also develop ubiquitinated protein inclusions negative for TDP-43 and FUS, mimicking the inclusions found in patients with CHMP2B mutations [197].
TARDBP and FUS
Mutations in TARDBP and FUS are found, although these are very rare in FTD and more common in ALS [24–27]. FTD associated with mutations in GRN, VCP, or C9orf72 is consistently characterized by the presence of TDP-43 pathology, and a considerable number of tau-/TDP-negative FTLD cases have inclusion of FUS, suggesting that dysregulation of both proteins might be important in the pathogenesis of FTD [10, 95].
TDP-43 is a 414 amino acid protein with two RNA recognition motifs and a carboxy-terminal glycine-rich domain, encoded by the TARDBP gene on chromosome 1. TDP-43 is a member of the hnRNP family of proteins and regulates RNA processing in a variety of ways. It is involved in mRNA transport, stability and turnover, splicing, and translation. The protein is highly conserved, is predominantly nuclear, and can shuttle between the nucleus and the cytoplasm [198–200]. Essentially, all ALS- and FTD-associated mutations are missense changes in the C-terminal glycine-rich region, involved in protein-protein interactions. In FTLD-TDP brains, TDP-43 is redistributed to the cytoplasm, hyperphosphorylated, ubiquitinated, and cleaved into the C-terminal fragments (CTFs) [95]. Experimental studies do not conclusively demonstrate whether TDP-43-mediated neurodegeneration results from a gain or loss of function of the protein. There is evidence for both: gains of functions mediated by aggregation and abnormal cytoplasmic function or loss of functions mediated by nuclear depletion, CTFs, and RNA dysregulation [201, 202].
FUS was first reported in 2009 to be the cause of ~3 % familial ALS cases and subsequently found to be the marker for the pathology of many remaining tau-/TDP-negative FTLD cases [10, 203, 204]. The FUS gene encodes a 526 amino acid protein and forms a gene family together with EWSR1 and TAF15 (FET). Increased insolubility of all FET proteins has been found in FTLD-FUS, but not in ALS-FUS [205]. It is highly conserved, ubiquitously expressed, and mainly localized to the nucleus. The protein has a high degree of functional homology with TDP-43 and is involved in multiple steps of RNA processing [206]. Most mutations are missense mutations affecting the C-terminus, disrupting the binding of FUS to Transportin. This nuclear import receptor shuttles proteins from the cytoplasm to the nucleus leading to an accumulation of mutant FUS in the cytoplasm [207]. Also for FUS, it is still unclear whether the cytosolic deposition causes loss of essential nuclear functions, a gain of toxic functions in the cytosol, or both.
Susceptibility Genes and Risk Loci
Compared to Mendelian FTD genes, little is known about susceptibility genes contributing to the risk of developing the disease. In 2010, Van Deerlin and colleagues identified TMEM106B in a genome-wide association study as a risk factor for FTLD-TDP [28]. Three SNPs encompassing the TMEM106B gene were found significant in a series of 515 FTLD-TDP patients, and variants specifically increased the risk in GRN mutation carriers. The initial GWAS finding has been successfully replicated in several FTD cohorts, strongly supporting a key role for TMEM106B in FTLD-TDP pathogenesis [208–210]. TMEMB106B is a 274 amino acid transmembrane type 2 protein with a highly glycosylated luminal domain [211]. It is cytoplasmically expressed in neurons, glia, and endothelial cells/pericytes of human brain samples from normal individuals, while in neurons of FTLD-TDP cases, TMEM106B appears to extend beyond the cell body into neuronal processes. In multiple immortalized cell lines and primary cortical neurons, TMEM106B is localized to late endosomes or lysosomes, and increased levels affect endolysosomal and progranulin pathways [212–214]. A recent GWAS points to novel associations, and the immune system processes, lysosomal, and autophagy pathways are potentially involved in FTD. The new loci need to be replicated in future studies to determine their possible association with the disease [29].
In 2011, UBQLN2 was added to the list of ALS-FTD genes [215]. The gene encodes the ubiquitin-like protein ubiquilin 2, a member of the ubiquitin family, which regulates the degradation of ubiquitinated proteins. In ~20 % of UBQLN2 mutation carriers, progressive dementia similar to FTD was identified, but all patients eventually developed motor symptoms. P62, encoded for by the SQSTM1 gene, is another protein at the intersection of ALS and FTD. It was found following the observation of involvement of P62 in ALS and has been subsequently reported in ALS and in FTD [216–218]. Inclusion positive for p62 can be found in C9orf72 expansion mutation patients, both with and without TDP-43 pathology [219].
Another risk factor for FTLD-TDP comes from a variant located in the 3′UTR of GRN in a binding site for the microRNA mir-659 [220]. In a series of pathologically confirmed FTLD-TDP patients without GRN mutations, Rademakers and colleagues observed that homozygous T-allele carriers had a 3.2-fold increase risk to develop FTLD-TDP as compared to homozygote C-allele carriers. They hypothesized that this variant reduces GRN protein expression through mir-659-dependent translational inhibition. It is still unclear whether GRN levels play a direct role in general FTLD population as two other studies failed to replicate the original findings [221, 222]. However, the risk allele is overrepresented in cases with hippocampal sclerosis [223, 224] and was found associated with a lower GRN serum level in two independent studies [225, 226], suggesting that decreased GRN expression may be a risk factor for FTLD-TDP and other dementias.
Conclusion
In recent years, remarkable progress has been made regarding the understanding of the genetic causes and neuropathological features of FTD. The recent discovery of the C9orf72 repeat expansions as the most common cause of the FTD/ALS complex of diseases showed that there is still a lot to be discovered. We know, for example, very little about susceptibility genes contributing to the risk of developing FTD and not all proteinopathies are fully characterized. The biological significance of the identified FTD genes has been studied extensively in cellular and animal models, as described in this chapter. However, diagnosis of FTD remains challenging, and no cure is available yet. Interestingly, many of the FTD genes seem to be connected at the molecular level. TMEM106B has originally been found as a risk factor for GRN mutation carriers, and several studies now describe TMEM106B as a genetic modifier for FTD with the C9orf72 repeat expansions [227, 228]. Patients with mutations in C9orf72 in combination with mutations in other genes involved in FTD/ALS have also been reported [229, 230]. Furthermore, FTD cases associated with GRN and C9orf72 mutations both present with TDP-43 pathology, and several studies showed that TDP-43 binds the 3′-UTR of GRN and regulates its expression [199, 231]. Understanding the full biology and connecting all identified FTD genes and risk factors with clinical phenotypes are of high importance for developing therapeutic approaches and offering reliable genetic advice to patients.
Abbreviations
- aFTLD-U:
-
Atypical frontotemporal lobar degeneration with ubiquitinated inclusions
- AGD:
-
Argyrophilic grain disease
- BIBD:
-
Basophilic inclusion body disease
- C9orf72 :
-
Chromosome 9 open reading frame 72
- CBD:
-
Corticobasal degeneration
- CHMP2B :
-
Charged multivesicular body protein 2B
- FTD-3:
-
Frontotemporal dementia linked to chromosome 3
- FTLD:
-
Frontotemporal lobar degeneration
- FUS :
-
Fused in sarcoma
- GRN :
-
Granulin
- MAPT :
-
Microtubule-associated protein tau
- MSTD:
-
Multiple system tauopathy with dementia
- NFT-dementia:
-
Neurofibrillary tangle predominant dementia
- ni:
-
No inclusions
- NIFID:
-
Neuronal intermediate filament inclusion disease
- PiD:
-
Pick’s disease
- PSP:
-
Progressive supranuclear palsy
- TARDBP :
-
TAR DNA-binding protein
- TDP:
-
TDP-43
- UPS:
-
Ubiquitin proteasome system
- VCP :
-
Valosin-containing protein
- WMT-GGI:
-
White matter tauopathy with globular glial inclusions
References
Sieben A, Van Langenhove T, Engelborghs S, Martin J-J, Boon P, Cras P, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012;124(3):353–72.
Bennion Callister J, Pickering-Brown SM. Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp Neurol. 2014;262:84–90. PubMed PMID: 24915640.
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–77. PubMed PMID: 21810890. PubMed Central PMCID: 3170532.
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–14. PubMed PMID: 21325651. PubMed Central PMCID: 3059138.
Rascovsky K, Grossman M. Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int Rev Psychiatry. 2013;25(2):145–58. PubMed PMID: 23611345. PubMed Central PMCID: 3906583.
Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62(6):925–30. PubMed PMID: 15956163.
Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122(2):137–53. PubMed PMID: 21614463. PubMed Central PMCID: 3232515.
Riedl L, Mackenzie IR, Forstl H, Kurz A, Diehl-Schmid J. Frontotemporal lobar degeneration: current perspectives. Neuropsychiatr Dis Treat. 2014;10:297–310. PubMed PMID: 24600223. PubMed Central PMCID: 3928059.
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117(1):15–8. PubMed PMID: 19015862. Pubmed Central PMCID: 2710877.
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(Pt 11):2922–31. PubMed PMID: 19674978. Pubmed Central PMCID: 2768659.
Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol. 2008;116(2):147–57. PubMed PMID: 18536926.
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. PubMed PMID: 19924424. Pubmed Central PMCID: 2799633.
Goldman JS, Adamson J, Karydas A, Miller BL, Hutton M. New genes, new dilemmas: FTLD genetics and its implications for families. Am J Alzheimers Dis Other Demen. 2007–2008;22(6):507–15. PubMed PMID: 18166610.
Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73(18):1451–6. PubMed PMID: 19884572. PubMed Central PMCID: 2779007.
Seelaar H, Kamphorst W, Rosso SM, Azmani A, Masdjedi R, de Koning I, et al. Distinct genetic forms of frontotemporal dementia. Neurology. 2008;71(16):1220–6. PubMed PMID: 18703462.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–5. PubMed PMID: 9641683.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815–25. PubMed PMID: 9629852.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95(13):7737–41. PubMed PMID: 9636220. PubMed Central PMCID: 22742.
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–9. PubMed PMID: 16862116.
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–4. PubMed PMID: 16862115.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. PubMed PMID: 21944778. Pubmed Central PMCID: 3202986.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. PubMed PMID: 21944779. Pubmed Central PMCID: 3200438.
Cohn-Hokke PE, Elting MW, Pijnenburg YA, van Swieten JC. Genetics of dementia: update and guidelines for the clinician. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(6):628–43. PubMed PMID: 22815225.
Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65(4):470–3. PubMed PMID: 19350673.
Borroni B, Archetti S, Del Bo R, Papetti A, Buratti E, Bonvicini C, et al. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res. 2010;13(5):509–17. PubMed PMID: 20645878.
Huey ED, Ferrari R, Moreno JH, Jensen C, Morris CM, Potocnik F, et al. FUS and TDP43 genetic variability in FTD and CBS. Neurobiol Aging. 2012;33(5):1016 e9–17. PubMed PMID: 21943958.
Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74(5):366–71. PubMed PMID: 20124201.
Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42(3):234–9. PubMed PMID: 20154673. Pubmed Central PMCID: 2828525.
Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JB, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014;13(7):686–99. PubMed PMID: 24943344.
Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12(6):609–22. PubMed PMID: 23684085.
Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8(1):159–68. PubMed PMID: 1530909.
Flament S, Delacourte A, Verny M, Hauw JJ, Javoy-Agid F. Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol. 1991;81(6):591–6. PubMed PMID: 1831952.
Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH, et al. Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol. 1994;145(6):1496–508. PubMed PMID: 7992852. Pubmed Central PMCID: 1887493.
Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M, et al. Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol. 2002;61(6):547–56. PubMed PMID: 12071638.
Delacourte A, Robitaille Y, Sergeant N, Buee L, Hof PR, Wattez A, et al. Specific pathological Tau protein variants characterize Pick’s disease. J Neuropathol Exp Neurol. 1996;55(2):159–68. PubMed PMID: 8786374.
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8(4):711–5. PubMed PMID: 10072441.
Houlden H, Baker M, Morris HR, MacDonald N, Pickering-Brown S, Adamson J, et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56(12):1702–6. PubMed PMID: 11425937.
Pastor P, Ezquerra M, Munoz E, Marti MJ, Blesa R, Tolosa E, et al. Significant association between the tau gene A0/A0 genotype and Parkinson’s disease. Ann Neurol. 2000;47(2):242–5. PubMed PMID: 10665497.
Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37(2):129–37. PubMed PMID: 15654335.
Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101(4):1371–8. PubMed PMID: 3930508. Pubmed Central PMCID: 2113928.
Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry. 1992;31(43):10626–33. PubMed PMID: 1420178.
Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116(2):207–25. PubMed PMID: 599557.
Hirokawa N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Curr Opin Cell Biol. 1994;6(1):74–81. PubMed PMID: 8167029.
Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3(10):1141–54. PubMed PMID: 1421571. Pubmed Central PMCID: 275678.
Goode BL, Chau M, Denis PE, Feinstein SC. Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenerative disease. J Biol Chem. 2000;275(49):38182–9. PubMed PMID: 10984497.
Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33(32):9511–22. PubMed PMID: 8068626.
Brandt R, Leger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol. 1995;131(5):1327–40. PubMed PMID: 8522593. Pubmed Central PMCID: 2120645.
Pooler AM, Hanger DP. Functional implications of the association of tau with the plasma membrane. Biochem Soc Trans. 2010;38(4):1012–5. PubMed PMID: 20658995.
Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol. 1998;143(3):777–94. PubMed PMID: 9813097. Pubmed Central PMCID: 2148132.
Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci. 1999;112(Pt 14):2355–67. PubMed PMID: 10381391.
Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319(5866):1086–9. PubMed PMID: 18202255. Pubmed Central PMCID: 2866193.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models6. Cell. 2010;142(3):387–97. PubMed PMID: 20655099.
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68(6):1067–81. PubMed PMID: 21172610. PubMed Central PMCID: 3026458.
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261(13):6084–9. PubMed PMID: 3084478.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83(13):4913–7. PubMed PMID: 3088567. Pubmed Central PMCID: 323854.
Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–3. PubMed PMID: 14032480.
Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2(7):783–7. PubMed PMID: 8673924.
Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10(6):1089–99. PubMed PMID: 8318230.
Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet. 1986;2(8504):412–6. PubMed PMID: 2874414.
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91(12):5562–6. PubMed PMID: 8202528. PubMed Central PMCID: 44036.
Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A. 1997;94(1):298–303. PubMed PMID: 8990203. PubMed Central PMCID: 19321.
Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013;33 Suppl 1:S123–39. PubMed PMID: 22710920.
Mercken M, Vandermeeren M, Lubke U, Six J, Boons J, Van de Voorde A, et al. Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol. 1992;84(3):265–72. PubMed PMID: 1384266.
Kanemaru K, Takio K, Miura R, Titani K, Ihara Y. Fetal-type phosphorylation of the tau in paired helical filaments. J Neurochem. 1992;58(5):1667–75. PubMed PMID: 1560225.
Dolan PJ, Johnson GV. The role of tau kinases in Alzheimer’s disease. Curr Opin Drug Discovery Dev. 2010;13(5):595–603. PubMed PMID: 20812151. PubMed Central PMCID: 2941661.
Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15(3):112–9. PubMed PMID: 19246243.
Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33(9):1340–4. PubMed PMID: 22581678. PubMed Central PMCID: 3465795.
Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24(4):277–95. PubMed PMID: 15365985.
Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282(5395):1914–7. PubMed PMID: 9836646.
D’Souza I, Schellenberg GD. Determinants of 4-repeat tau expression. Coordination between enhancing and inhibitory splicing sequences for exon 10 inclusion. J Biol Chem. 2000;275(23):17700–9. PubMed PMID: 10748133.
D’Souza I, Schellenberg GD. tau Exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5′ and 3′ splice sites. J Biol Chem. 2002;277(29):26587–99. PubMed PMID: 12000767.
Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64(2):414–21. PubMed PMID: 9973279. PubMed Central PMCID: 1377751.
D’Souza I, Schellenberg GD. Regulation of tau isoform expression and dementia. Biochim Biophys Acta. 2005;1739(2–3):104–15. PubMed PMID: 15615630.
Gotz J, Deters N, Doldissen A, Bokhari L, Ke Y, Wiesner A, et al. A decade of tau transgenic animal models and beyond. Brain Pathol. 2007;17(1):91–103. PubMed PMID: 17493043.
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–81. PubMed PMID: 16020737. PubMed Central PMCID: 1574647.
Roberson ED. Mouse models of frontotemporal dementia. Ann Neurol. 2012;72(6):837–49. PubMed PMID: 23280835. PubMed Central PMCID: 3539234.
Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, et al. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168(5):1598–607. PubMed PMID: 16651626. PubMed Central PMCID: 1606598.
de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature. 2010;464(7292):1201–4. PubMed PMID: 20357768. PubMed Central PMCID: 3091360.
Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011;31(7):2511–25. PubMed PMID: 21325519.
Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28(3):737–48. PubMed PMID: 18199773.
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–13. PubMed PMID: 19503072. PubMed Central PMCID: 2726961.
Clavaguera F, Hench J, Lavenir I, Schweighauser G, Frank S, Goedert M, et al. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014;127(2):299–301. PubMed PMID: 24362441.
Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845–52. PubMed PMID: 19282288. PubMed Central PMCID: 2676015.
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33(3):1024–37. PubMed PMID: 23325240. PubMed Central PMCID: 3575082.
de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73(4):685–97. PubMed PMID: 22365544. PubMed Central PMCID: 3292759.
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110(23):9535–40. PubMed PMID: 23690619. PubMed Central PMCID: 3677441.
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. PubMed PMID: 23050084. PubMed Central PMCID: 3463004.
Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. 2013;288(3):1856–70. PubMed PMID: 23188818. PubMed Central PMCID: 3548495.
Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286(17):15317–31. PubMed PMID: 21372138. PubMed Central PMCID: 3083182.
Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287(23):19440–51. PubMed PMID: 22461630. PubMed Central PMCID: 3365982.
Santa-Maria I, Varghese M, Ksiezak-Reding H, Dzhun A, Wang J, Pasinetti GM. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J Biol Chem. 2012;287(24):20522–33. PubMed PMID: 22496370. PubMed Central PMCID: 3370237.
Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293(5530):711–4. PubMed PMID: 11408621.
Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003;100(17):9980–5. PubMed PMID: 12872001. PubMed Central PMCID: 187908.
Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S, et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119(5):1382–95. PubMed PMID: 19363289. PubMed Central PMCID: 2673864.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3. PubMed PMID: 17023659.
Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114(1):49–54. PubMed PMID: 17458552.
Behrens MI, Mukherjee O, Tu PH, Liscic RM, Grinberg LT, Carter D, et al. Neuropathologic heterogeneity in HDDD1: a familial frontotemporal lobar degeneration with ubiquitin-positive inclusions and progranulin mutation. Alzheimer Dis Assoc Disord. 2007;21(1):1–7. PubMed PMID: 17334266.
Leverenz JB, Yu CE, Montine TJ, Steinbart E, Bekris LM, Zabetian C, et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130(Pt 5):1360–74. PubMed PMID: 17439980.
Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129(Pt 11):3081–90. PubMed PMID: 17071926.
Spina S, Murrell JR, Huey ED, Wassermann EM, Pietrini P, Baraibar MA, et al. Clinicopathologic features of frontotemporal dementia with progranulin sequence variation. Neurology. 2007;68(11):820–7. PubMed PMID: 17202431.
Bhandari V, Bateman A. Structure and chromosomal location of the human granulin gene. Biochem Biophys Res Commun. 1992;188(1):57–63. PubMed PMID: 1417868.
Bateman A, Bennett HP. Granulins: the structure and function of an emerging family of growth factors. J Endocrinol. 1998;158(2):145–51. PubMed PMID: 9771457.
Cadieux B, Chitramuthu BP, Baranowski D, Bennett HP. The zebrafish progranulin gene family and antisense transcripts. BMC Genomics. 2005;6:156. PubMed PMID: 16277664. PubMed Central PMCID: 1310530.
Kao AW, Eisenhut RJ, Martens LH, Nakamura A, Huang A, Bagley JA, et al. A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc Natl Acad Sci U S A. 2011;108(11):4441–6. PubMed PMID: 21368173. PubMed Central PMCID: 3060230.
Pera EM, Hou S, Strate I, Wessely O, De Robertis EM. Exploration of the extracellular space by a large-scale secretion screen in the early Xenopus embryo. Int J Dev Biol. 2005;49(7):781–96. PubMed PMID: 16172975.
Bhandari V, Giaid A, Bateman A. The complementary deoxyribonucleic acid sequence, tissue distribution, and cellular localization of the rat granulin precursor. Endocrinology. 1993;133(6):2682–9. PubMed PMID: 8243292.
Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48(7):999–1009. PubMed PMID: 10858277.
Petkau TL, Neal SJ, Orban PC, MacDonald JL, Hill AM, Lu G, et al. Progranulin expression in the developing and adult murine brain. J Comp Neurol. 2010;518(19):3931–47. PubMed PMID: 20737593.
Carrasquillo MM, Nicholson AM, Finch N, Gibbs JR, Baker M, Rutherford NJ, et al. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet. 2010;87(6):890–7. PubMed PMID: 21087763. PubMed Central PMCID: 2997361.
Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68(4):654–67. PubMed PMID: 21092856. PubMed Central PMCID: 2990962.
Lee WC, Almeida S, Prudencio M, Caulfield TR, Zhang YJ, Tay WM, et al. Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Hum Mol Genet. 2014;23(6):1467–78. PubMed PMID: 24163244. PubMed Central PMCID: 3929086.
Kleinberger G, Capell A, Haass C, Van Broeckhoven C. Mechanisms of granulin deficiency: lessons from cellular and animal models. Mol Neurobiol. 2013;47(1):337–60. PubMed PMID: 23239020. PubMed Central PMCID: 3538123.
Chitramuthu BP, Baranowski DC, Kay DG, Bateman A, Bennett HP. Progranulin modulates zebrafish motoneuron development in vivo and rescues truncation defects associated with knockdown of Survival motor neuron 1. Mol Neurodegener. 2010;5:41. PubMed PMID: 20946666. PubMed Central PMCID: 2974670.
Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen-West K, et al. Progranulin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener. 2012;7:33. PubMed PMID: 22781549. PubMed Central PMCID: 3508877.
Laird AS, Van Hoecke A, De Muynck L, Timmers M, Van den Bosch L, Van Damme P, et al. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One. 2010;5(10):e13368. PubMed PMID: 20967127. PubMed Central PMCID: 2954192.
Ryan CL, Baranowski DC, Chitramuthu BP, Malik S, Li Z, Cao M, et al. Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci. 2009;10:130. PubMed PMID: 19860916. PubMed Central PMCID: 2779192.
Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181(1):37–41. PubMed PMID: 18378771. PubMed Central PMCID: 2287280.
Almeida S, Zhang Z, Coppola G, Mao W, Futai K, Karydas A, et al. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2012;2(4):789–98. PubMed PMID: 23063362. PubMed Central PMCID: 3532907.
Gao X, Joselin AP, Wang L, Kar A, Ray P, Bateman A, et al. Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3beta. Protein Cell. 2010;1(6):552–62. PubMed PMID: 21204008.
Nedachi T, Kawai T, Matsuwaki T, Yamanouchi K, Nishihara M. Progranulin enhances neural progenitor cell proliferation through glycogen synthase kinase 3beta phosphorylation. Neuroscience. 2011;185:106–15. PubMed PMID: 21540081.
Guo A, Tapia L, Bamji SX, Cynader MS, Jia W. Progranulin deficiency leads to enhanced cell vulnerability and TDP-43 translocation in primary neuronal cultures. Brain Res. 2010;1366:1–8. PubMed PMID: 20888804.
Kleinberger G, Wils H, Ponsaerts P, Joris G, Timmermans J-P, Van Broeckhoven C, et al. Increased caspase activation and decreased TDP-43 solubility in progranulin knockout cortical cultures. J Neurochem. 2010;115(3):735–47.
Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207(1):117–28. PubMed PMID: 20026663. PubMed Central PMCID: 2812536.
De Muynck L, Van Damme P. Cellular effects of progranulin in health and disease. J Mol Neurosci. 2011;45(3):549–60. PubMed PMID: 21611805.
Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. 2011;332(6028):478–84. PubMed PMID: 21393509. PubMed Central PMCID: 3104397.
Baker CA, Manuelidis L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A. 2003;100(2):675–9. PubMed PMID: 12525699. PubMed Central PMCID: 141055.
Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem. 2001;77(1):132–45. PubMed PMID: 11279269.
Philips T, De Muynck L, Thu HN, Weynants B, Vanacker P, Dhondt J, et al. Microglial upregulation of progranulin as a marker of motor neuron degeneration. J Neuropathol Exp Neurol. 2010;69(12):1191–200. PubMed PMID: 21107132.
Pickford F, Marcus J, Camargo LM, Xiao Q, Graham D, Mo JR, et al. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am J Pathol. 2011;178(1):284–95. PubMed PMID: 21224065. PubMed Central PMCID: 3070582.
Del Bo R, Corti S, Santoro D, Ghione I, Fenoglio C, Ghezzi S, et al. No major progranulin genetic variability contribution to disease etiopathogenesis in an ALS Italian cohort. Neurobiol Aging. 2011;32(6):1157–8. PubMed PMID: 19632744. PubMed Central PMCID: 3511779.
Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M, Momeni P, et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry. 2007;78(7):754–6. PubMed PMID: 17371905. PubMed Central PMCID: 2117704.
Nuytemans K, Pals P, Sleegers K, Engelborghs S, Corsmit E, Peeters K, et al. Progranulin variability has no major role in Parkinson disease genetic etiology. Neurology. 2008;71(15):1147–51. PubMed PMID: 18838661.
Yu CE, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E, et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. 2010;67(2):161–70. PubMed PMID: 20142524. PubMed Central PMCID: 2901991.
Behm-Ansmant I, Kashima I, Rehwinkel J, Sauliere J, Wittkopp N, Izaurralde E. mRNA quality control: an ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett. 2007;581(15):2845–53. PubMed PMID: 17531985.
Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15(20):2988–3001. PubMed PMID: 16950801.
Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. 2008;131(Pt 3):732–46. PubMed PMID: 18245784.
Gijselinck I, van der Zee J, Engelborghs S, Goossens D, Peeters K, Mattheijssens M, et al. Progranulin locus deletion in frontotemporal dementia. Hum Mutat. 2008;29(1):53–8. PubMed PMID: 18157829.
Rovelet-Lecrux A, Deramecourt V, Legallic S, Maurage CA, Le Ber I, Brice A, et al. Deletion of the progranulin gene in patients with frontotemporal lobar degeneration or Parkinson disease. Neurobiol Dis. 2008;31(1):41–5. PubMed PMID: 18479928.
Chen-Plotkin AS, Martinez-Lage M, Sleiman PM, Hu W, Greene R, Wood EM, et al. Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol. 2011;68(4):488–97. PubMed PMID: 21482928. PubMed Central PMCID: 3160280.
Benussi L, Ghidoni R, Pegoiani E, Moretti DV, Zanetti O, Binetti G. Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol Dis. 2009;33(3):379–85. PubMed PMID: 19101631.
Borroni B, Bonvicini C, Galimberti D, Tremolizzo L, Papetti A, Archetti S, et al. Founder effect and estimation of the age of the Progranulin Thr272fs mutation in 14 Italian pedigrees with frontotemporal lobar degeneration. Neurobiol Aging. 2011;32(3):555 e1–8. PubMed PMID: 20947212.
Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Human Mutat. 2008;29(12):1373–86. PubMed PMID: 18543312.
Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283(3):1744–53. PubMed PMID: 17984093.
Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60(3):314–22. PubMed PMID: 16983685. PubMed Central PMCID: 2803024.
Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90(6):1102–7. PubMed PMID: 22608501. PubMed Central PMCID: 3370276.
Kollmann K, Uusi-Rauva K, Scifo E, Tyynela J, Jalanko A, Braulke T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim Biophys Acta. 2013;1832(11):1866–81. PubMed PMID: 23402926.
Petkau TL, Leavitt BR. Progranulin in neurodegenerative disease. Trends Neurosci. 2014;37(7):388–98. PubMed PMID: 24800652.
Wils H, Kleinberger G, Pereson S, Janssens J, Capell A, Van Dam D, et al. Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J Pathol. 2012;228(1):67–76. PubMed PMID: 22733568.
Tapia L, Milnerwood A, Guo A, Mills F, Yoshida E, Vasuta C, et al. Progranulin deficiency decreases gross neural connectivity but enhances transmission at individual synapses. J Neurosci. 2011;31(31):11126–32. PubMed PMID: 21813674.
Petkau TL, Neal SJ, Milnerwood A, Mew A, Hill AM, Orban P, et al. Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis. 2012;45(2):711–22. PubMed PMID: 22062772.
Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. 2010;177(1):311–24. PubMed PMID: 20522652. PubMed Central PMCID: 2893674.
Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, et al. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010;24(12):4639–47. PubMed PMID: 20667979. PubMed Central PMCID: 2992364.
Englund E, Gustafson L, Passant U, Majounie E, Renton AE, Traynor BJ, et al. Familial Lund frontotemporal dementia caused by C9ORF72 hexanucleotide expansion. Neurobiol Aging. 2012;33(8):1850 e13–6. PubMed PMID: 22483864.
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11(1):54–65. PubMed PMID: 22154785.
Cruts M, Gijselinck I, Van Langenhove T, van der Zee J, Van Broeckhoven C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36(8):450–59. PubMed PMID: 23746459.
Heutink P, Jansen IE, Lynes EM. C9orf72; abnormal RNA expression is the key. Exp Neurol. 2014;262:102–10. PubMed PMID: 24873727.
Lindquist SG, Duno M, Batbayli M, Puschmann A, Braendgaard H, Mardosiene S, et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet. 2013;83(3):279–83. PubMed PMID: 22650353.
Majounie E, Abramzon Y, Renton AE, Perry R, Bassett SS, Pletnikova O, et al. Repeat expansion in C9ORF72 in Alzheimer’s disease. N Eng J Med. 2012;366(3):283–4. PubMed PMID: 22216764. PubMed Central PMCID: 3513272.
Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126(6):895–905. PubMed PMID: 24166615. PubMed Central PMCID: 3830740.
Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35(7):1779.e5–1779.e13. PubMed PMID: 24559645.
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, et al. C9ORF72, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23(13):3579–595. PubMed PMID: 24549040.
Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–28. PubMed PMID: 24139042.
Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. PubMed PMID: 24154603.
Zhang D, Iyer LM, He F, Aravind L. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet. 2012;3:283. PubMed PMID: 23248642. PubMed Central PMCID: 3521125.
Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29(4):499–503. PubMed PMID: 23329412. PubMed Central PMCID: 3570213.
Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126(3):385–99. PubMed PMID: 23836290. PubMed Central PMCID: 3753484.
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–30. PubMed PMID: 22406228. PubMed Central PMCID: 3322422.
van der Zee J, Gijselinck I, Dillen L, Van Langenhove T, Theuns J, Engelborghs S, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34(2):363–73. PubMed PMID: 23111906. PubMed Central PMCID: 3638346.
Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74(2):180–87. PubMed PMID: 23720273.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–8. PubMed PMID: 23393093.
Greene E, Mahishi L, Entezam A, Kumari D, Usdin K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 2007;35(10):3383–90. PubMed PMID: 17478498. PubMed Central PMCID: 1904289.
Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1(6):397–400. PubMed PMID: 1301913.
Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet. 2013;92(6):981–9. PubMed PMID: 23731538. PubMed Central PMCID: 3675239.
Wojciechowska M, Krzyzosiak WJ. Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet. 2011;20(19):3811–21. PubMed PMID: 21729883. PubMed Central PMCID: 3168290.
Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126(6):845–57. PubMed PMID: 24170096.
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530–9. PubMed PMID: 24170860.
Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. 2012;2:1016. PubMed PMID: 23264878. PubMed Central PMCID: 3527825.
Zamiri B, Reddy K, Macgregor Jr RB, Pearson CE. TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J Biol Chem. 2014;289(8):4653–9. PubMed PMID: 24371143.
Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200. PubMed PMID: 24598541.
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110(19):7778–83. PubMed PMID: 23553836. PubMed Central PMCID: 3651485.
Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125(3):413–23. PubMed PMID: 23381195.
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–46. PubMed PMID: 23415312. Pubmed Central PMCID: 3593233.
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108(1):260–5. PubMed PMID: 21173221. PubMed Central PMCID: 3017129.
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126(6):881–93. PubMed PMID: 24132570.
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, et al. Poly-dipeptides encoded by the C9ORF72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345(6201):1139–145. PubMed PMID: 25081482.
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–194. PubMed PMID: 25103406.
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–81. PubMed PMID: 15034582.
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–64. PubMed PMID: 21145000. PubMed Central PMCID: 3032425.
Dai RM, Li CC. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol. 2001;3(8):740–4. PubMed PMID: 11483959.
Ju JS, Weihl CC. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum Mol Genet. 2010;19(R1):R38–45. PubMed PMID: 20410287. PubMed Central PMCID: 2875057.
Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul Disord: NMD. 2009;19(5):308–15. PubMed PMID: 19380227. PubMed Central PMCID: 2859037.
Badadani M, Nalbandian A, Watts GD, Vesa J, Kitazawa M, Su H, et al. VCP associated inclusion body myopathy and paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS One. 2010;5(10):pii:e13183.
Custer SK, Neumann M, Lu H, Wright AC, Taylor JP. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet. 2010;19(9):1741–55. PubMed PMID: 20147319.
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–8. PubMed PMID: 16041373.
Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One. 2010;5(3):e9872. PubMed PMID: 20352044. PubMed Central PMCID: 2844426.
Urwin H, Authier A, Nielsen JE, Metcalf D, Powell C, Froud K, et al. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet. 2010;19(11):2228–38. PubMed PMID: 20223751. PubMed Central PMCID: 2865375.
Ghazi-Noori S, Froud KE, Mizielinska S, Powell C, Smidak M, et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain. 2012;135(Pt 3):819–32. PubMed PMID: 22366797.
Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20(7):1774–84. PubMed PMID: 11285240. PubMed Central PMCID: 145463.
Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle F, Buratti E, et al. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem. 2012;287(19):15635–47.
Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–96. PubMed PMID: 7745706. PubMed Central PMCID: 189073.
Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. 2011;21(6):904–19. PubMed PMID: 21813273. PubMed Central PMCID: 3228892.
Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13(1):38–50. PubMed PMID: 22127299. PubMed Central PMCID: 3285250.
Kwiatkowski Jr TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–8. PubMed PMID: 19251627.
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–11. PubMed PMID: 19251628.
Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134(Pt 9):2595–609. PubMed PMID: 21856723. PubMed Central PMCID: 3170539.
Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):R46–64. PubMed PMID: 20400460. PubMed Central PMCID: 3167692.
Dormann D, Haass C. Fused in sarcoma (FUS): an oncogene goes awry in neurodegeneration. Mol Cell Neurosci. 2013;56:475–86. PubMed PMID: 23557964.
van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134(Pt 3):808–15. PubMed PMID: 21354975. PubMed Central PMCID: 3044834.
Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76(5):467–74. PubMed PMID: 21178100. PubMed Central PMCID: 3034409.
Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. 2011;68(5):581–6. PubMed PMID: 21220649. PubMed Central PMCID: 3090529.
Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, et al. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem. 2012;287(23):19355–65. PubMed PMID: 22511793. PubMed Central PMCID: 3365973.
Brady OA, Zheng Y, Murphy K, Huang M, Hu F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet. 2013;22(4):685–95. PubMed PMID: 23136129. PubMed Central PMCID: 3554197.
Busch JI, Martinez-Lage M, Ashbridge E, Grossman M, Van Deerlin VM, Hu F, et al. Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol Commun. 2013;1(1):36. PubMed PMID: 24252750. PubMed Central PMCID: 3893524.
Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci. 2012;32(33):11213–27. PubMed PMID: 22895706. PubMed Central PMCID: 3446826.
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset alS and ALS/dementia. Nature. 2011;477(7363):211–5. PubMed PMID: 21857683. PubMed Central PMCID: 3169705.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–6. PubMed PMID: 22084127.
Le Ber I, Camuzat A, Guerreiro R, Bouya-Ahmed K, Bras J, Nicolas G, et al. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 2013;70(11):1403–10. PubMed PMID: 24042580.
Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79(15):1556–62. PubMed PMID: 22972638. PubMed Central PMCID: 3655323.
Liu Y, Yu JT, Sun FR, Ou JR, Qu SB, Tan L. The clinical and pathological phenotypes of frontotemporal dementia with C9ORF72 mutations. J Neurol Sci. 2013;335(1–2):26–35. PubMed PMID: 24090760.
Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17(23):3631–42. PubMed PMID: 18723524. PubMed Central PMCID: 2581433.
Simon-Sanchez J, Seelaar H, Bochdanovits Z, Deeg DJ, van Swieten JC, Heutink P. Variation at GRN 3′-UTR rs5848 is not associated with a risk of frontotemporal lobar degeneration in Dutch population. PLoS One. 2009;4(10):e7494. PubMed PMID: 19847305. PubMed Central PMCID: 2761542.
Rollinson S, Rohrer JD, van der Zee J, Sleegers K, Mead S, Engelborghs S, et al. No association of PGRN 3′UTR rs5848 in frontotemporal lobar degeneration. Neurobiol Aging. 2011;32(4):754–5. PubMed PMID: 19446372.
Dickson DW, Baker M, Rademakers R. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis. 2010;7(1–3):170–4. PubMed PMID: 20197700. PubMed Central PMCID: 2859236.
Pao WC, Dickson DW, Crook JE, Finch NA, Rademakers R, Graff-Radford NR. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25(4):364–8. PubMed PMID: 21346515. PubMed Central PMCID: 3107353.
Hsiung GY, Fok A, Feldman HH, Rademakers R, Mackenzie IR. rs5848 polymorphism and serum progranulin level. J Neurol Sci. 2011;300(1–2):28–32. PubMed PMID: 21047645. PubMed Central PMCID: 3085023.
Kamalainen A, Viswanathan J, Natunen T, Helisalmi S, Kauppinen T, Pikkarainen M, et al. GRN variant rs5848 reduces plasma and brain levels of granulin in Alzheimer’s disease patients. J Alzheimers Dis. 2013;33(1):23–7. PubMed PMID: 22890097.
van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, et al. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 2014;127(3):397–406. PubMed PMID: 24385136.
Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014;127(3):407–18. PubMed PMID: 24442578.
van Blitterswijk M, Baker MC, DeJesus-Hernandez M, Ghidoni R, Benussi L, Finger E, et al. C9ORF72 repeat expansions in cases with previously identified pathogenic mutations. Neurology. 2013;81(15):1332–41. PubMed PMID: 24027057. PubMed Central PMCID: 3806926.
Lashley T, Rohrer JD, Mahoney C, Gordon E, Beck J, Mead S, et al. A pathogenic progranulin mutation and C9orf72 repeat expansion in a family with frontotemporal dementia. Neuropath Appl Neurobiol. 2013;40(4):502–13. PubMed PMID: 24286341.
Polymenidou M, Lagier-Tourenne C, Hutt K, Huelga S, Moran J, Liang T, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–68.
Disclosure
Peter Heutink is a co-applicant on a patent application related to MAPT (PCT/US1999/009529) and C9orf72 (PCT/GB2012/052140) and is co-owner of Synaptologics BV.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Heetveld, S., Rizzu, P., Heutink, P. (2015). Genetics of Frontotemporal Dementia. In: Schneider, S., Brás, J. (eds) Movement Disorder Genetics. Springer, Cham. https://doi.org/10.1007/978-3-319-17223-1_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-17223-1_5
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17222-4
Online ISBN: 978-3-319-17223-1
eBook Packages: MedicineMedicine (R0)