
Abstract
Mitochondrial dysfunction has been implicated in the pathogenesis of sporadic, idiopathic Parkinson disease. In some cases, mitochondrial DNA primary genetic abnormalities, or more commonly, secondary rearrangements due to polymerase gamma (POLG1) gene mutation, can directly cause parkinsonism. The case of a Parkinson disease patient with some signs or symptoms suggestive of mitochondrial disease (i.e., ptosis, myopathy, neuropathy) is a relatively common event in the neurological practice. Mitochondrial parkinsonisms do not have distinctive features allowing an immediate diagnosis, and a negative family history does not rule out a possible diagnosis of mitochondrial disorder. In this article, we do not revise the mitochondrial hypothesis of sporadic, idiopathic Parkinson disease, extensively discussed elsewhere, but we review POLG1-related parkinsonism and other well-defined forms of “mitochondrial parkinsonisms”, with mtDNA mutations or rearrangements. Lastly, we try to introduce a possible diagnostic approach for patients with parkinsonism and suspected mitochondrial disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson disease (PD) is a common neurodegenerative movement disorder (Dauer and Przedborski 2003). The cardinal features of PD include resting tremor, rigidity, bradykinesia and postural instability. These symptoms are caused by the progressive loss of the dopamine neurons within the substantia nigra pars compacta and the associated deficiency of the neurotransmitter dopamine in the striatum. Degeneration in other areas (i.e., the locus coeruleus, the dorsal raphe nuclei, and others) may contribute to many of the non-motor symptoms of PD, such as depression, dementia, and autonomic dysfunction (Dauer and Przedborski 2003). In addition, the surviving neurons contain cytoplasmic, eosinophilic inclusions called Lewy bodies (Dauer and Przedborski 2003). PD exists as both familial and idiopathic forms. Linkage studies showed 16 PD-related genetic loci (PARK1-16). Mutations in several genes have been linked to the genetic forms of PD (α-synuclein, Parkin, DJ-1, PINK1, and LRRK2; Bueler 2009). Mitochondrial dynamics (fission, fusion, migration) is important for neurotransmission, synaptic maintenance, and neuronal survival. Recent studies have shown that PINK1 and Parkin play crucial roles in the regulation of mitochondrial dynamics and function. Moreover, mutations in DJ-1 and Parkin render animals more susceptible to oxidative stress and mitochondrial toxins implicated in sporadic PD, lending support to the hypothesis that PD may be caused by gene-environmental factor interactions (Bueler 2009). The etiology of sporadic PD remains largely unclear, but accumulating evidence suggests that mitochondrial dysfunction occurs in brain and peripheral tissues of PD patients (Fukae et al. 2007). Since the discovery that the toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, an inhibitor of the mitochondrial complex I, was an environmental factor capable of causing a disease indistinguishable from sporadic PD, a number of studies have revealed that mitochondrial dysfunction plays an important role in the neurodegenerative process leading to PD (Fukae et al. 2007). Especially complex I dysfunction has been implicated in the pathogenesis of PD (Parker et al. 1989).
Accumulation of clonal, somatic mitochondrial DNA (mtDNA) deletions has been observed in the substantia nigra during aging and in PD, suggesting that mtDNA mutations in some instances may predispose to dopamine neuron death by impairing mitochondrial respiration (Bueler 2009). A role of mtDNA has been also suggested by the finding that normal cells transfected with mtDNA from PD patients (cytoplasmic hybrids (cybrids)) showed 25% decreased complex I activity (Gu et al. 1998). In the cybrid technique, culturable cells depleted of endogenous mtDNA are repopulated with mitochondria (with their own mtDNA) from patients (Mancuso et al. 2008a).
The role of mtDNA variants in PD has been extensively studied (Mancuso et al. 2008a). van der Walt and coworkers (van der Walt et al. 2003) genotyped ten single-nucleotide polymorphisms that define the European mtDNA haplogroups in 609 white PD patient and 340 white controls. Mitochondrial haplogroups are specific and unique sets of common mtDNA polymorphisms that have evolved from the same ancestor. These authors observed that haplotype J and K reduced the incidence of PD by 50% (van der Walt et al. 2003). Consistently with this study, it has been subsequently reported that haplotype cluster UKJT was associated with a 22% reduction in population-attributable risk for PD (Pyle et al. 2005). Autere et al. (2004) observed that the supercluster JTIWX increased the risk of both PD and PD with dementia; this cluster was associated with a twofold increase in nonsynonymous substitutions in the mtDNA genes encoding complex I subunits. Another group evaluated the distribution of mtDNA haplogroups in a large cohort of 620 Italian patients with adult-onset idiopathic PD vs. two groups of ethnic-matched controls (Ghezzi et al. 2005). These authors found that haplogroup K was associated with a lower risk; they reported also that the 10398G polymorphism resulted protective against PD (Ghezzi et al. 2005). This last finding (but not the association between haplogroup K and lower risk) was later confirmed in 271 Spanish PD patients vs. 230 healthy controls (Huerta et al. 2005). Overall, despite some negative studies (Latsoudis et al. 2008), mtDNA haplogroups have been associated with a number of different neurodegenerative diseases, but to date, the only disease consistently associated with a different mtDNA haplogroup frequency is PD (Mancuso et al. 2008a). However, recent data failed to demonstrate a bias towards maternal inheritance in familial PD (Simon et al. 2010).
It is now well established that defects in mtDNA replication can lead to mitochondrial dysfunction and disease. DNA polymerase gamma (POLG) is the only DNA polymerase in human mitochondria and is essential for mtDNA replication and repair (Chan and Copeland 2009). In addition to its 5′ to 3′ polymerase activity, POLG has a 3′ to 5′ exonuclease activity important in the repair process. Mitochondria have their own small 16.5-kb circular double-stranded DNA that encodes 22 tRNAs, 2 rRNAs, and 13 polypeptides that are essential for electron transport and oxidative phosphorylation. Nuclear genes encode the other mitochondrial proteins, including the proteins involved in mtDNA replication, such as POLG subunits 1 and 2 (codified by POLG1 and POLG2 nuclear genes; Chan and Copeland 2009). POLG1 mutations lead to uncorrect nucleotide incorporation in mtDNA (Batabyal et al. 2010). Homozygous knock-in mice expressing a proof-reading-deficient version of POLG show an “mtDNA mutator” phenotype with increased amounts of point mutations and deleted mtDNA. This increase in somatic mtDNA mutations is associated with reduced lifespan and premature onset of aging-related phenotypes, thus providing a link between mtDNA mutations and aging in mammals (Trifunovic et al. 2004).
POLG1 has a polyglutamine tract (poly-Q) in the N-terminal, encoded by a CAG sequence in exon 2 (Chan and Copeland 2009). Commonly, the poly-Q tract comprises ten repeats (10Q, frequency >80%) or 11Q (frequency 6–12%); however the composition of poly-Q alleles has been reported to vary from 6Q to 14Q. POLG1 CAG repeat variants have been associated with male infertility, testicular cancer (Chan and Copeland 2009) and, interestingly, with idiopathic sporadic PD (Luoma et al. 2007). However, the role of the CAG trinucleotide repeat in POLG1 is controversial (Chan and Copeland 2009). Very recently, Eerola et al. (2010) sequenced the poly-Q in 641 North American Caucasian PD patients and 292 controls. Caucasian literature controls were also used. Normal allele was defined either as 10/11Q or as 10Q according to the previous literature (Eerola et al. 2010). Variant alleles defined as non-10Q were significantly increased in the PD patients compared to the North American controls as well as compared to the larger set of 897 controls, strengthening the evidence for involvement of POLG1 CAG repeats in PD (Eerola et al. 2010). Similar results have been obtained by Anvret et al. (2010) in Swedish PD patients. These authors reported a significant association between the non-10/11Q repeats with PD (Anvret et al. 2010). These findings may be attributable to a beneficial function of the 10/11Q repeat protein or to linkage disequilibrium between the 10Q allele and another variation within POLG1 (Eerola et al. 2010). However, further large case-control studies are strongly needed, as well as analyses on functional differences of POLG1 poly-Q variants.
In humans, POLG1 mutations account for a substantial proportion of patients with mitochondrial myopathy and mtDNA deletions or depletion, and may cause both clinically and genetically heterogeneous disorders (Filosto et al. 2003). The case of a PD patient with some signs or symptoms suggestive of mitochondrial disease (i.e., ptosis, myopathy, neuropathy) is a relatively common event in the neurological practice. In this article, we do not revise the mitochondrial hypothesis of sporadic, idiopathic PD, extensively discussed elsewhere (Fukae et al. 2007), but we review POLG1-related parkinsonism and other well-defined forms of “mitochondrial parkinsonisms”, with mtDNA mutations or rearrangements. At last, we try to introduce a possible diagnostic approach for patients with parkinsonism and suspected mitochondrial disorder.
Mitochondrial Disorders
Mitochondrial disorders (MDs) are a group of disorders caused by impairment of the mitochondrial respiratory chain (DiMauro and Schon 2003). The effects of mutations affecting the respiratory chain may be multisystemic, with involvement of visual and auditory pathways, heart, central nervous system, and skeletal muscle (DiMauro and Schon 2003). The estimated prevalence of MDs is 1–2 in 10,000 (Schaefer et al. 2008). MDs are, therefore, one of the commonest inherited neuromuscular disorders. The genetic classification of MDs distinguishes disorders due to defects in mtDNA from those due to defects in nuclear DNA (nDNA; DiMauro and Schon 2003). The first ones are inherited according to the rules of mitochondrial genetics (maternal inheritance, heteroplasmy and the threshold effect, mitotic segregation; DiMauro and Schon 2003). Each cell contains multiple copies of mtDNA (polyplasmy), which in normal individuals are identical (homoplasmy; DiMauro and Schon 2003). Heteroplasmy refers to the coexistence of two populations of mtDNA, normal and mutated. Mutated mtDNA in a given tissue has to reach a minimum critical number before oxidative metabolism is impaired severely enough to cause dysfunction (threshold effect; DiMauro and Schon 2003). Differences in mutational loads surpassing the pathogenic threshold in some tissues but not in others may contribute to the heterogeneity of phenotypes. Because of the mitotic segregation, the mutation load can change from one cell generation to the next and, with time, it can either surpass or fall below the pathogenic threshold (DiMauro and Schon 2003). Further, the pathogenic threshold varies from tissue to tissue, according to the relative dependence of each tissue on oxidative metabolism (DiMauro and Schon 2003). For instance, central nervous system, skeletal muscle, heart, endocrine glands, the retina, the renal tubule, and the auditory sensory cells are highly dependent on oxidative metabolism for energy generation. Moreover, it is important to notice that the occurrence of a single large-scale deletion, common cause of progressive external ophthalmoplegia (PEO), is almost sporadic (DiMauro and Schon 2003). Therefore, a negative family history does not rule out a possible diagnosis of MD.
MD related to nDNA are caused by mutations in structural components or ancillary proteins of the respiratory chain, by defects of the membrane lipid milieu, of Coenzyme Q10 biosynthetic genes, and by defects in intergenomic signaling (associated with mtDNA depletion or multiple deletions; DiMauro and Schon 2003). Mutations in nuclear genes adenine nucleotide translocator 1 (ANT1), twinkle mitochondrial helicase (PEO1), and mitochondrial catalytic subunit of DNA polymerase (POLG1) result in MD with mtDNA multiple deletions—commonly with a PEO phenotype—or mtDNA depletion with Alpers’ syndrome phenotype (Filosto et al. 2003; Gonzalez-Vioque et al. 2006; Copeland 2010). More recently, OPA1 gene has been also associated with MD and mtDNA multiple deletions (Stewart et al. 2008). In a family with autosomal dominant PEO and parkinsonism, a heterozygous PEO1 mutation has been reported to segregate with the disease phenotype (Baloh et al. 2007). No pathogenic mutations were present in POLG1 or ANT1 (Baloh et al. 2007). Very rarely, nuclear mutations in structural components of the respiratory chain, such as NDUFV2 (Nishioka et al. 2010), may also cause familial PD, but further studies are still needed.
“Mitochondrial Parkinsonisms”
As mentioned above, there is increasing evidence that impairment of mitochondrial function and oxidative damage are contributing factors to the pathophysiology of sporadic PD. In some cases, mitochondrial genetic abnormalities can directly cause parkinsonisms (Mancuso et al. 2008a).
mtDNA point mutations causing parkinsonism have been reported only rarely, typically as one feature of a larger syndrome, i.e., with sensorineural deafness and neuropathy (Thyagarajan et al. 2000), MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes; De Coo et al. 1999) or Leber’s hereditary optic neuropathy (Nikoskelainen et al. 1995). A family with maternally inherited, adult-onset multisystem degeneration including prominent parkinsonism was reported in 1999 (Simon et al. 1999). The parkinsonism was levodopa responsive and was associated with the loss of pigmented neurons in the substantia nigra in at least one patient. In this family, a mutation of the mitochondrial ND4 gene of complex I (previously reported in Leber’s disease) was found (Simon et al. 1999).
Horvath et al. (2007) observed the A8344G “MERRF” (myoclonic epilepsy with ragged red fibers) mutation on the tRNALys gene of the mtDNA in a 66-year-old man with dopa-responsive parkinsonism, reduced muscle strength, and ragged red fibers (pathological hallmark of mitochondrial myopathy). No other clinical signs typical of MERRF were noted. The A8344G mutation was present in a virtually homoplasmic state in the patient’s muscle, and 80% mutant was detected in blood DNA (Horvath et al. 2007). In order to evaluate if this mutation could represent a common cause of sporadic PD, our group subsequently analysed 159 Italian patients with PD (Mancuso et al. 2008b). None of the patients carried the A8344 mutation in DNA extracts from peripheral blood lymphocytes. Thus, the screening for this mutation should be restricted to PD patients with other canonical features of mitochondrial encephalomyopathies (i.e., myopathy, ophthalmoplegia, neuropathy, cerebellar ataxia; Mancuso et al. 2008b).
More frequently parkinsonism has been associated with large-scale rearrangements in mtDNA (Siciliano et al. 2001). Further, co-existence of parkinsonism and mutations in the mitochondrial polymerase gamma nDNA gene (POLG1) with mtDNA multiple deletions has been reported in several families, suggesting that when defective, this gene can be responsible for some of the mendelian transmission of Parkinsonism (Mancuso et al. 2008a). POLG1-related parkinsonisms will be considered in the next paragraph.
mtDNA mutations may also cause movement disorders other from parkinsonism, i.e., our group reported the case of a 36-year-old man who had severe postural and kinetic tremor of the arms since the age of 17 years and a mild myopathy. The levodopa test resulted negative, and DATSCAN showed a normal dopamine transporters concentration in the striatum, suggesting a diagnosis of essential tremor (Mancuso et al. 2008c). Southern Blot analysis revealed the presence of a single deletion of 7.7-kb interesting 58% of mtDNA in the skeletal muscle (Mancuso et al. 2008c).
Interestingly, Simon et al. (2003) reported a mtDNA mutation in the gene encoding the ND1 subunit of mitochondrial complex I in a patient with adult-onset dystonia, spasticity, and myopathy. The same mutation subsequently was identified in two additional unrelated patients with adult-onset dystonia (Simon et al. 2003). Subsequently, two independent patients presenting with progressive generalized dystonia and bilateral striatal necrosis with a mutation in the mitochondrial ND6 gene were reported (Solano et al. 2003). More recently, “mitochondrial dystonia” has also been associated with mitochondrial ND3 gene (Sarzi et al. 2007; Wang et al. 2009).
POLG1 in Human Disease
POLG1 mutations can cause dominant or recessive disorders, frequently associated with severe and multisystem involvement. These forms of MD are associated with secondary accumulation of multiple deletions in mtDNA (Gonzalez-Vioque et al. 2006). Mutations in the POLG1 gene have emerged as one of the most common causes of inherited MDs in children and adults (Wong et al. 2008). Functional genetic variants of POLG1 are present in up to 0.5% of the general population, and pathogenic mutations have been described in most exons of the gene (Hudson and Chinnery 2006). The most severe manifestations have been associated with mutations of the “spacer” region of POLG1 (Luoma et al. 2005). In a large study, the clinical presentation ranged from the neonatal period to late adult life, with an overlapping phenotypic spectrum from severe encephalopathy and liver failure to late-onset PEO, ataxia, myopathy and isolated muscle pain or epilepsy (Horvath et al. 2006). A high incidence of psychiatric disease and primary gonadal failure have also been documented in some families transmitting dominant POLG1 mutations (Hudson and Chinnery 2006). Very recently, a new nosological entity has been also suggested—“POLG1-related multiple sclerosis-like illness” (Echaniz-Laguna et al. 2010; Harris et al. 2010).
The most common POLG1 disease mutation (467A > T) has been found in all of the major POLG1-related diseases, i.e., Alpers syndrome, ataxia-neuropathy syndromes and PEO (Chan and Copeland 2009). 467A > T mutation is generally reported as a recessive mutation; however, it may give rise to a mild dominant phenotype with late-onset ptosis (Chan and Copeland 2009). This mutation was shown to lead to a severe catalytic defect, disrupting physical association between POLG catalytic and accessory subunits, with great reduction of both polymerase and exonuclease POLG activities (Chan et al. 2005).
Moreover, mutations in POLG1 cause a recessively inherited syndrome with episodic features and progressive ataxia (Winterthun et al. 2005). Hakonen et al. (2005) reported the high carrier frequency of the 748 W > S POLG1 substitution in control subjects in Finland, explaining the high prevalence of mitochondrial recessive ataxia syndrome (MIRAS) in Scandinavia. However, although 467A > T and 748 W > S are a common cause of ataxia in Scandinavia, they are rare in other European regions, such as the United Kingdom and Italy (Craig et al. 2007). It is therefore likely that the high prevalence of MIRAS in Finland and Norway is due to a founder effect. Very recently, it has been reported that POLG1 mutations are rather common in Central European ataxia patients, and cause ataxia with PEO (47%), and, more rarely, with psychiatric comorbidities (20%) and epilepsy (14%; Schicks et al. 2010).
POLG1-Associated Parkinsonism
The first family in which parkinsonism was a prominent manifestation together with PEO and multiple mtDNA deletions, caused by a novel POLG1 mutation, was reported in 2004 (Mancuso et al. 2004). A missense mutation in the POLG1 gene (2492A > G) was found in the proband (a woman with PEO, mild axonal neuropathy and parkinsonism since the age of 47, with slowly progressive resting hand tremor, slowing of her gait, bradykinesia, rigidity, and slurred speech) and in her affected siblings (Mancuso et al. 2004). The patient’s brother developed parkinsonism in his early 40s. The proband’s muscle biopsy showed numerous ragged-red fibers and cytochrome c oxidase-negative fibers, with multiple deletions of mtDNA (Mancuso et al. 2004). This mutation, however, has also been found in Europe in healthy population. Therefore, its pathogenetic role is still unclear.
In the same period, Luoma et al. (2004) studied patients with POLG1-related PEO and their healthy relatives in seven families of various ethnic origins, by clinical, biochemical, morphological, and molecular characterisation, and by positron emission tomography (PET) studies. A significant cosegregation of parkinsonism with POLG1 mutations was observed, and PET findings were consistent with dopaminergic neuron loss (Luoma et al. 2004). Post-mortem examination in two individuals showed loss of pigmented neurons and pigment phagocytosis in the substantia nigra, without Lewy bodies (Luoma et al. 2004). Furthermore, most women with PEO had early menopause (before age 35 years). The POLG1 gene defect resulted in secondary accumulation of mtDNA deletions in patients’ tissues (Luoma et al. 2004).
Tzoulis and co-workers studied 26 patients belonging to 20 families with a disorder caused by mutations in the POLG1 gene (Tzoulis et al. 2006), homozygous for 1399 G > A or 2243 G > C or compound heterozygotes for these two mutations. Irrespective of genotype, the patients exhibited a progressive neurological disorder usually starting in their teens and characterized by epilepsy, headache, ataxia, neuropathy, myoclonus and late-onset PEO. Compound heterozygotes had a significantly shorter survival time. Epilepsy occurred in 22 of the 26 patients and in the majority of these there was an occipital EEG focus (Tzoulis et al. 2006). Patients with this disorder are at high risk of death from status epilepticus and from liver failure, if exposed to sodium valproate. In this group was included a heterozygous woman who developed epilepsy at the age of 55 years. When examined at age 72, she had axonal peripheral neuropathy, ataxia, mild ptosis/PEO, and features of mild parkinsonism (unilateral bradykinesia, tremor and rigidity; Tzoulis et al. 2006).
The group of Chinnery studied a large family with autosomal dominant PEO and parkinsonism, with multiple mitochondrial DNA deletions (Hudson et al. 2007). These authors identified two novel heterozygous POLG1 exon substitutions. Autosomal-dominant progressive PEO segregated with 1532 G > A mutation in exon 8, predicted to alter a highly conserved serine to asparagine in the linker region of POLG. The one patient with parkinsonism had an additional heterozygous substitution in exon 7 (1389 G > T), in trans as regards the other mutation. This mutation, present only in the patient with parkinsonism, was predicted to alter a highly conserved leucine to phenylalanine and, based on this pedigree, it seemed unlikely that it could cause disease on its own (Hudson et al. 2007). None of the substitutions were found in a large group of healthy controls, patients with PEO without parkinsonism, and patients with sporadic PD. These two mutations did not directly affect the polymerase domain of POLG. Like other linker region mutations, they are likely to affect polymerase activity of the enzyme (Hudson et al. 2007). DATSCAN from the index case with PEO and parkinsonism showed reduced uptake in the left caudate nucleus and both putamen, consistent with a nigrostriatal dopaminergic deficiency (Hudson et al. 2007). Although his symptoms improved on a dopamine agonist, he had no tremor and the extrapyramidal rigidity was symmetric.
Pagnamenta et al. (2006) reported a family in which the proband, her mother, and her maternal grandmother presented with PEO and premature ovarian failure. The mother developed parkinsonian-like resting tremor and reduced rapid alternating movements affecting her left arm and leg, and mild bradykinesia, in her sixth decade. The tremor responded to levodopa treatment. Sequence analysis of POLG1 revealed a dominant 955Y > C mutation segregating with the disease. Southern blot analysis demonstrated mtDNA depletion in fibroblasts (43% of controls), whereas multiple rearrangements of mtDNA were seen in skeletal muscle (Pagnamenta et al. 2006). Mouse and yeast models with this mutation show enhanced amounts of oxidative lesions (Graziewicz et al. 2007). The 955Y > C mutated POLG displays relaxed discrimination when incorporating oxidatively damaged nucleotides, offering a biochemical link between oxidative stress and PD, and suggesting that patients harbouring POLG1 mutations may undergo enhanced oxidative stress and mtDNA mutagenesis (Graziewicz et al. 2007).
In previous years, other patients with POLG1 gene mutation and parkinsonism have been reported (Galassi et al. 2008; Betts-Henderson et al. 2009). Two sisters with early-onset parkinsonism (dystonic toe curling, action tremor, masked face, bradykinesia, stooped posture, and rigidity), together with clinical and electrophysiological signs of sensorimotor axonal peripheral neuropathy, were reported to have ragged-red and cytochrome c oxidase-negative fibers in muscle biopsy (Davidzon et al. 2006). Multiple mitochondrial DNA deletions were found by long polymerase chain reaction, and sequencing of the POLG1 gene showed that the patients were compound heterozygous for two pathogenic mutations (Davidzon et al. 2006). Therefore, POLG1 mutations can cause early-onset parkinsonism even in the absence of PEO.
Remes et al. (2008) reported the 748 W > S mutation (one of the most common mutations in this gene, found to be a frequent cause of autosomal recessive ataxia in adults and of Alpers syndrome in children) in a 65-year-old man with a late-onset syndrome consisting of ataxia, parkinsonism, PEO, peripheral neuropathy, and sensorineural hearing loss (Remes et al. 2008). Interestingly, Invernizzi et al. (2008) reported a compound heterozygous patient with two novel mutations in POLG1, who had autosomal recessive PEO, followed by the development of pseudo-orthostatic tremor evolving into levodopa-responsive parkinsonism.
Conclusion
In some cases, mtDNA primary genetic abnormalities, or more commonly secondary mtDNA rearrangements due to POLG1 gene mutation, can directly cause parkinsonism. Mitochondrial parkinsonisms do not have distinctive features allowing an immediate diagnosis. Usually, age at onset is about 50 years. Reduced dopamine uptake in the corpus striatum and good response to levodopa or dopamine agonists (Synofzik et al. 2010) have been well documented. Levodopa-induced dyskinesias and motor fluctuations may also occur (Wilcox et al. 2007). POLG1 mutations should be considered in the differential diagnosis of parkinsonism, especially in families with autosomal dominant transmission. However, a negative family history does not rule out a possible diagnosis of MD.
At present, diagnosis of MD requires a complex approach, including clinical and family data, measurement of serum lactate, exercise testing, electromyography, muscle histology, and enzymology, and genetic analysis (Fig. 1). Ragged red fibers, ragged blue fibers, and cytochrome c oxidase-negative muscle fibers are pathological hallmarks. However, reliable biomarkers for these disorders are still needed. “Red flags” for MD are short stature, neurosensory hearing loss, ptosis, ophthalmoplegia, axonal neuropathy, diabetes mellitus, hypertrophic cardiomyopathy, renotubular acidosis, migraine-like headache (Mancuso et al. 2009). Therapy of MD is still inadequate, despite great progress in the molecular understanding of these disorders. Therapies that have been attempted include respiratory chain cofactors, other metabolites secondarily decreased in MD, antioxidants and agents acting on lactic acidosis, but their role in the treatment of the majority of MD remains unclear (Orsucci et al. 2009).

Diagnostic approach to mitochondrial disorders (MD) in patients with parkinsonism. This figure is conceived as a flow chart, but the real diagnostic process depends on the specific clinical presentation of the patient, and is not a rigid process. COX cytochrome c oxidase, EMG electromyography, mtDNA mitochondrial DNA, RRF ragged red fibers
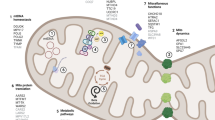
It is important to consider a possible diagnosis of mitochondrial parkinsonism in PD patients with other features of MD (Fig. 2), and especially of POLG1-related mitochondrial encephalomyopathies (i.e., PEO, myopathy, isolated muscle pain, dysphagia, neuropathy, ataxia, psychiatric disorders, hypogonadism, epilepsy). POLG1 mutations result in extremely heterogenous phenotypes which often have overlapping clinical findings, making it difficult to categorize patients into syndromes. The lack of signs of mitochondrial dysfunction in the muscle biopsy does not exclude a POLG1-related disease (Milone and Massie 2010). Analysis of mtDNA of clinically affected tissues is often informative, but not always. The clinical features of the patient are more important to select putative POLG1 mutation carriers than the presence of mtDNA deletions or muscle oxidative phosphorylation activity defects (Blok et al. 2009). Molecular analysis of POLG1 is essential when POLG1-related disease is suspected. In these patients, sodium valproate should be avoided because of the risk of liver failure.

“Red flags” for mitochondrial disorders and particularly for POLG1-related disorders. It is important to consider a possible diagnosis of mitochondrial parkinsonism in patients/families with Parkinson disease and some of these adjunctive features. WPW Wolff–Parkinson–White
The case of a PD patient with some signs or symptoms suggestive of mitochondrial disease is a relatively common event in neurological practice. Conversely, patients with a previous diagnosis of MD may show some features of parkinsonism. Further studies are needed in order to study the prevalence of MD among PD patients, and conversely of movement disorders among MD patients. Furthermore, they should aim to better understand the molecular basis underlying the phenotypic variability among these patients. Large, multicenter studies are strongly needed to better characterize the phenotype and natural history of MD, in order to identify some countermeasures (i.e., pharmacological, physical, or others) capable of benefit patients with these chronic, still incurable disorders.
References
Anvret A, Westerlund M, Sydow O et al (2010) Variations of the CAG trinucleotide repeat in DNA polymerase gamma (POLG1) is associated with Parkinson’s disease in Sweden. Neurosci Lett 485:117–120
Autere J, Moilanen JS, Finnila S et al (2004) Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Hum Genet 115:29–35
Baloh RH, Salavaggione E, Milbrandt J, Pestronk A (2007) Familial parkinsonism and ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch Neurol 64:998–1000
Batabyal D, McKenzie JL, Johnson KA (2010) Role of histidine-932 of the human mitochondrial DNA polymerase in nucleotide discrimination and inherited disease. J Biol Chem 285:34191–34201
Betts-Henderson J, Jaros E, Krishnan KJ et al (2009) Alpha-synuclein pathology and Parkinsonism associated with POLG1 mutations and multiple mitochondrial DNA deletions. Neuropathol Appl Neurobiol 35:120–124
Blok MJ, van den Bosch BJ, Jongen E et al (2009) The unfolding clinical spectrum of POLG mutations. J Med Genet 46:776–785
Bueler H (2009) Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp Neurol 218:235–246
Chan SS, Copeland WC (2009) DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta 1787:312–319
Chan SS, Longley MJ, Copeland WC (2005) The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J Biol Chem 280:31341–31346
Copeland WC (2010) The mitochondrial DNA polymerase in health and disease. Subcell Biochem 50:211–222
Craig K., Ferrari G., Tiangyou W., et al. (2007) The A467T and W748S POLG substitutions are a rare cause of adult-onset ataxia in Europe. Brain 130, E69; author reply E70.
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909
Davidzon G, Greene P, Mancuso M et al (2006) Early-onset familial parkinsonism due to POLG mutations. Ann Neurol 59:859–862
De Coo IF, Renier WO, Ruitenbeek W et al (1999) A 4-base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann Neurol 45:130–133
DiMauro S, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348:2656–2668
Echaniz-Laguna A, Chassagne M, de Seze J et al (2010) POLG1 variations presenting as multiple sclerosis. Arch Neurol 67:1140–1143
Eerola J, Luoma PT, Peuralinna T et al (2010) POLG1 polyglutamine tract variants associated with Parkinson’s disease. Neurosci Lett 477:1–5
Filosto M, Mancuso M, Nishigaki Y et al (2003) Clinical and genetic heterogeneity in progressive external ophthalmoplegia due to mutations in polymerase gamma. Arch Neurol 60:1279–1284
Fukae J, Mizuno Y, Hattori N (2007) Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 7:58–62
Galassi G, Lamantea E, Invernizzi F et al (2008) Additive effects of POLG1 and ANT1 mutations in a complex encephalomyopathy. Neuromuscul Disord 18:465–470
Ghezzi D, Marelli C, Achilli A et al (2005) Mitochondrial DNA haplogroup K is associated with a lower risk of Parkinson’s disease in Italians. Eur J Hum Genet 13:748–752
Gonzalez-Vioque E, Blazquez A, Fernandez-Moreira D et al (2006) Association of novel POLG mutations and multiple mitochondrial DNA deletions with variable clinical phenotypes in a Spanish population. Arch Neurol 63:107–111
Graziewicz MA, Bienstock RJ, Copeland WC (2007) The DNA polymerase gamma Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7, 8-dihydro-8-oxo-2′-deoxyguanosine. Hum Mol Genet 16:2729–2739
Gu M, Cooper JM, Taanman JW, Schapira AH (1998) Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann Neurol 44:177–186
Hakonen AH, Heiskanen S, Juvonen V et al (2005) Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet 77:430–441
Harris MO, Walsh LE, Hattab EM, Golomb MR (2010) Is it ADEM, POLG, or both? Arch Neurol 67:493–496
Horvath R, Hudson G, Ferrari G et al (2006) Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain 129:1674–1684
Horvath R, Kley RA, Lochmuller H, Vorgerd M (2007) Parkinson syndrome, neuropathy, and myopathy caused by the mutation A8344G (MERRF) in tRNALys. Neurology 68:56–58
Hudson G. and Chinnery P.F. (2006) Mitochondrial DNA polymerase-gamma and human disease. Hum Mol Genet 15 Spec No 2, R244–252.
Hudson G, Schaefer AM, Taylor RW et al (2007) Mutation of the linker region of the polymerase gamma-1 (POLG1) gene associated with progressive external ophthalmoplegia and Parkinsonism. Arch Neurol 64:553–557
Huerta C, Castro MG, Coto E et al (2005) Mitochondrial DNA polymorphisms and risk of Parkinson’s disease in Spanish population. J Neurol Sci 236:49–54
Invernizzi F, Varanese S, Thomas A, Carrara F, Onofrj M, Zeviani M (2008) Two novel POLG1 mutations in a patient with progressive external ophthalmoplegia, levodopa-responsive pseudo-orthostatic tremor and parkinsonism. Neuromuscul Disord 18:460–464
Latsoudis H, Spanaki C, Chlouverakis G, Plaitakis A (2008) Mitochondrial DNA polymorphisms and haplogroups in Parkinson’s disease and control individuals with a similar genetic background. J Hum Genet 53:349–356
Luoma P, Melberg A, Rinne JO et al (2004) Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 364:875–882
Luoma PT, Luo N, Loscher WN et al (2005) Functional defects due to spacer-region mutations of human mitochondrial DNA polymerase in a family with an ataxia-myopathy syndrome. Hum Mol Genet 14:1907–1920
Luoma PT, Eerola J, Ahola S et al (2007) Mitochondrial DNA polymerase gamma variants in idiopathic sporadic Parkinson disease. Neurology 69:1152–1159
Mancuso M, Filosto M, Oh SJ, DiMauro S (2004) A novel polymerase gamma mutation in a family with ophthalmoplegia, neuropathy, and Parkinsonism. Arch Neurol 61:1777–1779
Mancuso M, Filosto M, Orsucci D, Siciliano G (2008a) Mitochondrial DNA sequence variation and neurodegeneration. Hum Genomics 3:71–78
Mancuso M, Nesti C, Petrozzi L et al (2008b) The mtDNA A8344G “MERRF” mutation is not a common cause of sporadic Parkinson disease in Italian population. Parkinsonism Relat Disord 14:381–382
Mancuso M, Orsucci D, Gori S, Ceravolo R, Siciliano G (2008c) Mitochondrial DNA single deletion in a patient with postural tremor. Mov Disord 23:2098–2100
Mancuso M, Orsucci D, Coppede F, Nesti C, Choub A, Siciliano G (2009) Diagnostic approach to mitochondrial disorders: the need for a reliable biomarker. Curr Mol Med 9:1095–1107
Milone M, Massie R (2010) Polymerase gamma 1 mutations: clinical correlations. Neurologist 16:84–91
Nikoskelainen EK, Marttila RJ, Huoponen K et al (1995) Leber’s “plus”: neurological abnormalities in patients with Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry 59:160–164
Nishioka K, Vilarino-Guell C, Cobb SA et al (2010) Genetic variation of the mitochondrial complex I subunit NDUFV2 and Parkinson disease. Parkinsonism Relat Disord 16:686–687
Orsucci D, Filosto M, Siciliano G, Mancuso M (2009) Electron transfer mediators and other metabolites and cofactors in the treatment of mitochondrial dysfunction. Nutr Rev 67:427–438
Pagnamenta AT, Taanman JW, Wilson CJ et al (2006) Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum Reprod 21:2467–2473
Parker WD Jr, Boyson SJ, Parks JK (1989) Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol 26:719–723
Pyle A, Foltynie T, Tiangyou W et al (2005) Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann Neurol 57:564–567
Remes AM, Hinttala R, Karppa M et al (2008) Parkinsonism associated with the homozygous W748S mutation in the POLG1 gene. Parkinsonism Relat Disord 14:652–654
Sarzi E, Brown MD, Lebon S et al (2007) A novel recurrent mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am J Med Genet A 143:33–41
Schaefer AM, McFarland R, Blakely EL et al (2008) Prevalence of mitochondrial DNA disease in adults. Ann Neurol 63:35–39
Schicks Md J, Synofzik Md M, Schulte C, Schols Md L (2010) POLG, but not PEO1, is a frequent cause of cerebellar ataxia in Central Europe. Mov Disord 25:2678–2682
Siciliano G, Mancuso M, Ceravolo R, Lombardi V, Iudice A, Bonuccelli U (2001) Mitochondrial DNA rearrangements in young onset parkinsonism: two case reports. J Neurol Neurosurg Psychiatry 71:685–687
Simon DK, Friedman J, Breakefield XO et al (2003) A heteroplasmic mitochondrial complex I gene mutation in adult-onset dystonia. Neurogenetics 4:199–205
Simon DK, Pulst SM, Sutton JP, Browne SE, Beal MF, Johns DR (1999) Familial multisystem degeneration with parkinsonism associated with the 11778 mitochondrial DNA mutation. Neurology 53:1787–1793
Simon DK, Pankratz N, Kissell DK et al (2010) Maternal inheritance and mitochondrial DNA variants in familial Parkinson’s disease. BMC Med Genet 11:53
Solano A, Roig M, Vives-Bauza C et al (2003) Bilateral striatal necrosis associated with a novel mutation in the mitochondrial ND6 gene. Ann Neurol 54:527–530
Stewart JD, Hudson G, Yu-Wai-Man P et al (2008) OPA1 in multiple mitochondrial DNA deletion disorders. Neurology 71:1829–1831
Synofzik M, Asmus F, Reimold M, Schols L, Berg D (2010) Sustained dopaminergic response of parkinsonism and depression in POLG-associated parkinsonism. Mov Disord 25:243–245
Thyagarajan D, Bressman S, Bruno C et al (2000) A novel mitochondrial 12SrRNA point mutation in parkinsonism, deafness, and neuropathy. Ann Neurol 48:730–736
Trifunovic A, Wredenberg A, Falkenberg M et al (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417–423
Tzoulis C, Engelsen BA, Telstad W et al (2006) The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain 129:1685–1692
van der Walt JM, Nicodemus KK, Martin ER et al (2003) Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet 72:804–811
Wang K, Takahashi Y, Gao ZL et al (2009) Mitochondrial ND3 as the novel causative gene for Leber hereditary optic neuropathy and dystonia. Neurogenetics 10:337–345
Wilcox RA, Churchyard A, Dahl HH, Hutchison WM, Kirby DM, Thyagarajan D (2007) Levodopa response in Parkinsonism with multiple mitochondrial DNA deletions. Mov Disord 22:1020–1023
Winterthun S, Ferrari G, He L et al (2005) Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology 64:1204–1208
Wong LJ, Naviaux RK, Brunetti-Pierri N et al (2008) Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat 29:E150–E172
Acknowledgment
This study was partially supported by Telethon Grant GUP09004.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Orsucci, D., Caldarazzo Ienco, E., Mancuso, M. et al. POLG1-Related and other “Mitochondrial Parkinsonisms”: an Overview. J Mol Neurosci 44, 17–24 (2011). https://doi.org/10.1007/s12031-010-9488-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-010-9488-9