
Abstract
The small guanosine triphosphatase RhoA has recently been implicated in the pathogenesis of epilepsy in animals. In this study, we investigated the expression of RhoA in human epilepsy. Brain tissue samples from 40 patients with intractable epilepsy and 14 histologically normal temporal lobes from control patients were used to compare RhoA immunoreactivity using immunohistochemistry, immunofluorescent staining, and western blotting. Immunohistochemical staining and western blot analysis showed that RhoA immunoreactivity was increased in the patient group (p < 0.05). Immunofluorescent staining showed that RhoA was mainly expressed on cell membranes and in the cytoplasm. Our findings demonstrate that upregulation of RhoA immunoreactivity occurs in the brains of patients with intractable epilepsy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a condition or a group of conditions in which abnormal neuronal discharge causes transient disturbance of cerebral function and recurrent seizures. However, the mechanisms that underlie the pathogenesis of epilepsy are unclear. Some studies have shown that repeated axonal discharge causes abnormal synaptic reorganization and neuronal loss (Armstrong 2005), which may have an important role in the pathology and physiology of refractory epilepsy.
In addition, seizures induce synaptic reorganization in hippocampal and cortical pathways. This reorganization is generally believed to be a result of the seizures, rather than an event that induces seizures (Frotscher et al. 2006; Sutula and Dudek 2007). RhoA is part of a larger family of related proteins known as the Ras superfamily; proteins involved in the regulation and timing of cell division. Activation of RhoA may affect neurite outgrowth and lead to the formation of new synaptic connections. This aberrant synaptogenesis generates recurrent excitatory circuits that affect brain function (Negishi and Katoh 2002; Rubenstein et al. 2007). Two Rho kinase (ROCK) inhibitors, Y-27632 and fasudil, diminish the onset of myoclonic jerks, clonic convulsions, and tonic hindlimb extensions in an animal model of kindling induced by pentylenetetrazol (Narumiya et al. 2000; Inan and Buyukafsar 2008). Furthermore, our recent gene chip study on brain tissue from patients with intractable epilepsy (IE) showed that RhoA mRNA is highly expressed in the central nervous system (CNS) of adult patients (Liu et al. 2007; Xiao et al. 2008). These data suggest that RhoA is involved in epileptogenesis and that the small guanosine triphosphatase RhoA could be a new treatment target.
Temporal lobe epilepsy (TLE), a common form of focal epilepsy, is often refractory to antiepileptic drugs. In human TLE and a rat model of TLE, seizures induce synaptic reorganization in hippocampal and cortical pathways (Lin et al. 2007; Holopainen 2008). Moreover, RhoA is involved in epileptogenesis, as described above. Therefore, we hypothesized that the small guanosine triphosphatase RhoA contributes to the induction of TLE. This study was done to determine the RhoA immunoreactivity in adult patients with intractable TLE.
Materials and Methods
Patients and Clinical Data
Brain tissue samples from 40 patients who had undergone neurosurgery for IE (11−57 years old; mean age ± standard error, 26.98 ± 10.15 years) were randomly obtained from our epilepsy brain bank. Seizure types were classified according to the 1981 International Classification of Epileptic Seizures of the International League Against Epilepsy (International League Against Epilepsy 1981). Pre-surgical assessment consisted of obtaining a detailed history, neurological examination, interictal and ictal EEG tests, and neuroradiological testing. The epilepsy group included patients who had undergone surgical removal of the epileptogenic zone for the treatment of seizures. The typical surgical method was anterior temporal neocortex resection. Briefly, the anterior 3.5−4.0 cm of the lateral temporal lobe was resected en bloc from the superior temporal gyrus to the collateral fissure. The clinical characteristics of the patients are summarized in Table 1.
For comparison, 14 histologically normal temporal lobe tissue samples selected at random from our brain bank were used as controls. Four specimens were autopsy samples from adult patients without neurological disorders. The postmortem intervals ranged from 5-24 h. The other ten specimens were histologically normal anterior temporal neocortex samples from patients treated for increased intracranial pressure because of head trauma. These subjects had no history of epilepsy and were not exposed to antiepileptic drugs.
This research was conducted in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) (Rickham 1964). All experimental protocols in this study were approved by the National Institutes of Health and the Committee on Human Research at the Chongqing Medical University. Informed consent was obtained from each patient for the use of brain tissue and for access to medical records for research purposes.
Tissue Processing
In our brain bank, specimens are divided into two pieces. One portion of the resected brain tissue from each subject (both the epilepsy group and the control group was immediately placed in a cryovial, incubated in buffered diethylpyrocarbonate (1:1,000) for 24 h, and then stored in liquid nitrogen for use in western blotting. The other portion was fixed in 10% buffered formalin for 48 h, embedded in paraffin, sectioned at 5 μm for immunohistochemistry or 8-10 μm for immunofluorescence analysis, and then mounted onto polylysine-coated slides. Two sections from each specimen were processed for hematoxylin-eosin staining. Control tissues were shown to be normal by neuropathological examination.
Immunohistochemistry
All paraffin-embedded tissue sections were first deparaffinized in xylene and rehydrated in graded ethanol. After treatment with 3% H2O2 for 5 min to quench endogenous peroxidase activity, antigen recovery was performed by heating the sections in 10 mmol/L boiling sodium citrate buffer (pH 6.0) for 10 min at 94−98°C. Sections were blocked in 10% normal rat serum in 0.3% Triton X-100 and 0.1 M phosphate buffered saline (PBS; pH 7.2) at room temperature for 30 min to reduce non-specific binding and then were incubated at 4°C overnight in primary antibody against RhoA (1:50; mouse monoclonal antibody, Santa Cruz Biotechnology, Santa Cruz, CA, USA). After extensive washing in PBS (pH 7.2) for 5 min each wash, sections were incubated in biotinylated secondary antibody from the streptavidin-biotin complex kit (Santa Cruz Biotechnology) for 30 min. The sections were washed and then treated with avidin-biotin complex solution at 30°C for 30 min, washed with PBS, and incubated in 3,3′-diaminobenzidine (DAB, Santa Cruz Biotechnology) for 10 min. Counterstaining was done with the Harris hematoxylin stain (Santa Cruz Biotechnology). As negative controls, the primary or secondary antibody was omitted and replaced with PBS to evaluate the experimental procedure and reagent quality. Four visual fields were randomly chosen, imaged on every section using the macroprogram Image Pro-Plus 5.0 (Media Cybernetics, Silver Spring, MD, USA), and automatically analyzed using a Motic Med 6.0 CMIAS pathology image analysis system. The gray value, ranging from 0 (black) to 1.0 (white), corresponds to the intensity of the labeling. The averaged optical density (mean OD) obtained by the immunohistochemical assay indicates the average intensity of staining and the average amount of stained material in that area. The cytoplasm of positive cells was yellowish-brown, and negative cells were unstained. Data are expressed as the mean ± standard error as assessed by a t test. A p < 0.05 value was considered significant.
Double Immunofluorescence and Confocal Microscopy
Sections were deparaffinized and antigen recovery was done as described above. Tissue sections were incubated in 0.3% calf serum for 1 h and then in 0.5% normal rabbit serum (Zhongshan Golden Bridge Inc, Beijing, China) for 30 min. The sections were incubated at 4°C overnight in anti-RhoA (1:100) and in anti-neuron-specific enolase (NSE) (Zhongshan Golden Bridge Inc, both at 1:200). After washing in PBS, the sections were incubated in tetramethylrhodamine isothiocyanate (TRITC)-conjugated anti-rabbit and fluorescein isothiocyanate (FITC)-conjugated anti-mouse secondary antibodies (Zhongshan Golden Bridge Inc, 1:100) in the dark. The sections were coverslipped, sealed, and dried overnight. There was no cross-contamination of the fluorescent antibodies among the different species. Fluorescent-stained sections were examined by confocal laser scanning microscopy (Leica Microsystems Heidelberg GmbH, Germany), and the images were collected and processed using Olympus Micro image (version 4.0).
Western Blotting
Western blot analysis was performed to compare the RhoA immunoreactivity levels in the TLE group and the control group. Tissue samples were cut into small pieces, homogenized in buffer [0.05 mol/L Tris-Hcl (pH 7.2),0.15 mol/L NaCl 1% Triton ×100, 1% (w/v) sodium deoxycholate and 4% (w/v) sodium dodecyl sulfate; SDS] including protease inhibitors (l5 μg/ml aprotinin and 1 mM phenylmethylsulfonyl fluoride), and centrifuged at 16,000×g at 4°C for 5 min. This crude nuclear pellet was lysed in a lysis buffer (Pierce Biotechnology, USA) for 30 min and centrifuged at 16,000×g at 4°C for 15 min. The protein concentration of the lysates was determined using a BCA protein assay kit (Pierce Biotechnology, USA). The extracts (40 μg) were resolved by 10% SDS-polyacrylamide gel electrophoresis and electrotransferred to a polyvinylidene difluoride (PVDF) membrane (Millipore Biotechnology, USA). PVDF membranes were divided into two parts according to the location of molecular weight markers to detect RhoA (24 kDa) and β-actin (42 kDa), which was used as a loading control. The PVDF membrane was blocked with 3% BSA in PBS (pH 7.2) then incubated for 2 h at room temperature. After extensive washing with PBS, the membranes were incubated in RhoA (1:100, Mouse monoclonal antibody, Abcam, UK) and β-actin (mouse monoclonal IgG1, 200 µg/ml, 1:1,000 dilution; Santa Cruz Biotechnology) antibodies for 1 h. After incubation with a horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody (1:5,000 dilution; Santa Cruz Biotechnology) for 1 h at 37°C, the immunoreactive bands were visualized by enhanced chemiluminescence (ECL). Relative quantification of the ECL signal on X-ray film was analyzed using Labworks Analysis Software (USA). The ratio of the RhoA and β-actin band intensities (RhoA/β-actin) from the same time of electrophoresis was analyzed.
Results
Comparison of Clinical Characteristics
Table 1 summarizes the clinical characteristics of the subjects. There were no significant differences in age or gender between the epilepsy patients and control subjects. In our study, 65% of the subjects with epilepsy (26/40, Table 1) had a clinical history of more than 10 years. In addition, there were no significant correlations with TLE and variables including age, gender, and family history in the TLE patients.
Changes in RhoA Staining in the Brain Tissue of Patients with Intractable Epilepsy
Brain tissue sections from TLE and control subjects were immunohistochemically stained. In the sections from TLE patients, strong RhoA immunoreactivity was observed, whereas only a faint distribution of RhoA immunoreactivity was observed in sections from controls subjects (Fig. 1). The mean optical density of RhoA expression in IE tissues was 0.2668 ± 0.0580 and in the control group was 0.0895 ± 0.0180 (p < 0.05). No immunoreactivity was seen in negative controls in which the primary antibody was omitted. RhoA immunofluorescent staining was observed on cell membranes and in the cytoplasm. Moreover, NSE (red) and RhoA (green) were co-expressed in neurons (Fig. 2). The results indicate that RhoA was mainly expressed in neurons.

Immunohistochemistry of RhoA expression in the temporal lobe of epilepsy patients and in controls. Expression of RhoA is shown in the brain of a TLE patient (a) and in the brain of a normal control subject (b). Arrows indicate positively labeled cells. Scale bar in a = 100 µm, scale bar in b = 100 µm

Double immunofluorescent staining for RhoA (green) and NSE (red) in neurons from the temporal lobe of an epilepsy patient (a-c) and from the neocortex of a control subject (d-f); c and f show RhoA and NSE coexpression. FITC staining for NSE (a, d); TRITC staining for RhoA (b, e). Arrows indicate double-positive neurons. Scale bar, 75 µm for all panels. Original magnification, ×400
Changes in RhoA Levels in the Brain Tissue of Patients with IE using Western Blotting
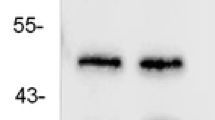
To further confirm the changes in RhoA staining of IE brain tissue, we examined the expression of RhoA immunoreactivity using western blotting. Immunoblotting was performed with two mouse monoclonal antibodies that detect RhoA (24 kDa) and β-actin (42 kDa), an internal control. The mean optical density value of the TLE samples was 17.68 ± 5.51 as compared with 9.76 ± 1.57 from the control samples (p < 0.05). Compared with the control group, strong upregulation of RhoA immunoreactivity was present in all samples from TLE patients. β-actin was expressed at similar levels in each sample (Fig. 3). The variance between the two groups of patients was examined by calculating the ratio of the optical density of RhoA to β-actin (Fig. 3).

Semi-quantitative western blot analysis of RhoA expression in the brain tissue of TLE patients. Lanes 1 and 2 indicate protein levels from TLE patients, and lanes 3 indicate protein levels in control tissues. Immunoblot signals were developed with enhanced chemiluminescence. Lower graph shows the average ratio of RhoA to β-actin for each group of patients. Error bars are the SD
Taken together, our results demonstrate that the expression of RhoA immunoreactivity is significantly higher in IE brain tissue samples as compared with control specimens (p < 0.05).
Discussion
This study demonstrates an increase in RhoA immunoreactivity in the brains of TLE patients as compared with control subjects. To our knowledge, this is the first report to study the expression of the small GTPase RhoA in human epilepsy.
In the CNS, the small guanosine triphosphatase RhoA participates in many normal and pathophysiological processes, such as axonal and dendritic growth of neurons, neuronal differentiation, axon guidance, dendritic remodeling, and synapse formation and plasticity in the CNS (Hall 1998; Lehmann et al. 1999; Brabeck et al. 2003). As shown by recent in vitro and in vivo studies, obvious changes in the morphology, motility, and pathfinding of axons have been observed after perturbation of members of the Rho family of GTPases (Lehmann et al. 1999; Negishi and Katoh 2002; Wang et al. 2003). RhoA is a negative regulator that inhibits the collapse of growth cones through its converging actions on myosin (Wang et al. 2003; Fournier et al. 2003, 2003). In vitro studies have verified that RhoA is a key mediator of axon guidance that regulates the cytoskeletal rearrangement that underlies directed axon growth inhibition and growth cone collapse (Fournier et al. 2003; Wang et al. 2003; Yuan et al. 2003). Other studies have shown that Y-27632, a widely used ROCK inhibitor, can induce marked attractive turning of the growth cone (Narumiya et al. 2000; Dergham et al. 2002). These data provide strong evidence that the small guanosine triphosphatase RhoA plays a major role in the inhibition of neuronal regeneration and remodeling of synapses and may even influence the structural framework of the neural network during a pathological state such as epilepsy.
Neuron loss is the main pathological change in patients with IE. The proteins c-fos, matrix metalloproteinase-9, and microtubule-associated protein kinase p38 are also involved in neuronal cell death in the hippocampus after seizures (Kim et al. 2004; Armstrong 2005), and RhoA signals to or regulates all three proteins (Wong et al. 2001; Benitah et al. 2004). This suggests an important role for small guanosine triphosphatase RhoA-dependent cell loss in human IE. Because TLE is associated with neural network remodeling and neuron loss, we hypothesized that TLE patients have abnormal expression of RhoA. Our results support this idea and are consistent with the experiments of Dubreuil and colleagues, in which increased levels of RhoA were observed in the brain tissue of rats with kainic acid-induced seizures (Dubreuil et al. 2006).
Common characteristics of patients in our study included (1) temporal lobe epilepsy and (2) seizures that were not controlled by antiepileptic drugs. Therefore, our results demonstrate that abnormal expression of RhoA occurs in the temporal cortex of patients with intractable TLE. The TLE samples were, however, derived from patients with various ages at seizure onset, frequency of seizures, and course of epilepsy. Any of these parameters could influence RhoA immunoreactivity, yet attempting to draw a conclusion from such a small sample size in each subgroup would not be valid.
The underlying mechanism of RhoA in the formation of TLE could not be examined here because of the limitations of a human study. A few questions cannot be answered, including the following: (1) the selected patients have intractable epilepsy and experienced multiple seizures before surgery. Therefore, it is unclear whether upregulated RhoA immunoreactivity is a result of the seizures or is associated with the generation of epilepsy. (2) It is unknown whether RhoA mediates synapse formation and neuron loss in TLE, triggering seizures. It is also not known if the RhoA/ROCK signaling pathway is involved in epilepsy. These hypotheses require further investigation using animal models and in vitro experiments to evaluate RhoA as a potential therapeutic target in intractable TLE patients.
References
Armstrong DD (2005) Epilepsy-induced microarchitectural changes in the brain. Pediatr Dev Pathol 8:607–14, G.L. Li, N. Zhang, J. Li, D.T. Zhu
Benitah SA, Valerón PF, van Aelst L, Marshall CJ, Lacal JC (2004) Rho GTPases in human cancer: an unresolved link to upstream and downstream transcriptional regulation. Biochim Biophys Acta 1705(2):121–32
Brabeck C, Mittelbronn M, Bekure K, Meyermann R, Schluesener HJ, Schwab JM (2003) Effect of focal cerebral infarctions on lesional RhoA and RhoB expression. Arch Neurol 60(9):1245–1249
Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L (2002) Rho signaling pathway targeted to promote spinal cord repair. J Neurosci 22:6570–6577
Dubreuil CI, Marklund N, Deschamps K, McIntosh TK, McKerracher L (2006) Activation of Rho after traumatic brain injury and seizure in rats. Exp Neurol 198:361–369
Fournier AE, Takizawa BT, Strittmatter SM (2003) Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci 23(4):1416–1423
Frotscher Michael, Jonas Peter, Sloviter RS (2006) Synapses formed by normal and abnormal hippocampal mossy fibers. Cell Tissue Res 326:361–7
Hall A (1998) Rho GTPases. J Biol Chem 273:20685–20688
Holopainen IE (2008) Seizures in the developing brain: cellular and molecular mechanisms of neuronal damage, neurogenesis and cellular reorganization. Neurochem Int 52:935–47
Inan S, Buyukafsar K (2008) Antiepileptic effects of two Rho-kinase inhibitors, Y-27632 and fasudil, in mice. Br J Pharmacol 155:44–51
International League Against Epilepsy (1981) Proposal for revised clinical and electroencephalographic classification of epileptic seizures. From the Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 22(4):489–501
Kim SW, Yu YM, Piao CS, Kim JB, Lee JK (2004) Inhibition of delayed induction of p38 mitogen-activated protein kinase attenuates kainic acid-induced neuronal loss in the hippocampus. Brain Res 1007:188–91
Lehmann M, Fournier A, Selles-Navarro I, Dergham P, Sebok A, Leclerc N, Tigyi G, McKerracher L (1999) Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci 19:7537–7547
Lin H, Wu LW, Huang YG, Chen YC, Chen YC, Wen XN (2007) Correlation between hippocampal mossy fiber sprouting and synaptic reorganization and mechanisms of temporal lobe epilepsy. Zhonghua Yi Xue Za Zhi 87:341–4
Liu FY, Wang XF, Li MW, Li JM, Xi ZQ, Luan GM, Zhang GM, Zhang JG, Wang YP, Sun JJ, Li YL (2007) Upregulated expression of postsynaptic density-93 and n-methyl-d-aspartate receptors subunits 2B mRNA in temporal lobe tissue of epilepsy. Biochem Biophys Res Commun 358:825–30
Narumiya S, Ishizaki T, Uehata M (2000) Use and properties of ROCK- specific inhibitor Y-27632. Methods Enzymol 325:273–84
Negishi Manabu, Katoh Hironori (2002) Rho family GTPases as key regulators for neuronal network formation. J Biochem 132:157–166
Rickham PP (1964) Human experimentation code of ethics of the World Medical Association, Declaration of Helisinki. Br Med J 2:177–179
Rubenstein NM, Callahan JA, Lo DH, Firestone GL (2007) Selective glucocorticoid control of Rho kinase isoforms regulate cell-cell interactions. Biochem Biophys Res Commun 354:603–7
Sutula TP, Dudek A (2007) Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog Brain Res 163:541–63
Wang H-R, Zhang V, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL (2003) Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 302:1690–1
Wong K, Ren XR, Huang YZ, Xie Y, Liu G, Saito H, Tang H, Wen L, Brady-Kalnay SM, Mei L, Wu JY, Xiong WC, Rao Y (2001) Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the slit-robo pathway. Cell 107:209–21
Xiao F, Wang XF, LI JM, Xi ZQ, Lu Y, Wang L, Zeng Y, Guan LF, Yuan J (2008) Overexpression of N-WASP in the brain of human epilepsy. Brain Res 1233:168–75
Yuan XB, Jin M, Xu X, Song YQ, Wu CP, Poo MM, Duan S (2003) Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat Cell Biol 5:38–45
Acknowledgments
The authors sincerely thank the patients and their families for their participation in this study. We thank Tiantan Hospital and Xianwu Hospital of the Capital University of Medical Sciences and Xinqiao Hospital of the Third Military Medical University for their support in brain tissue procurement, and the local ethics committee and the National Board of the Medical Affairs for their support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yuan, J., Wang, Ly., Li, Jm. et al. Altered Expression of the Small Guanosine Triphosphatase RhoA in Human Temporal Lobe Epilepsy. J Mol Neurosci 42, 53–58 (2010). https://doi.org/10.1007/s12031-010-9330-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-010-9330-4