
Abstract
There are currently no therapies proven to promote early recovery of consciousness in patients with severe brain injuries in the intensive care unit (ICU). For patients whose families face time-sensitive, life-or-death decisions, treatments that promote recovery of consciousness are needed to reduce the likelihood of premature withdrawal of life-sustaining therapy, facilitate autonomous self-expression, and increase access to rehabilitative care. Here, we present the Connectome-based Clinical Trial Platform (CCTP), a new paradigm for developing and testing targeted therapies that promote early recovery of consciousness in the ICU. We report the protocol for STIMPACT (Stimulant Therapy Targeted to Individualized Connectivity Maps to Promote ReACTivation of Consciousness), a CCTP-based trial in which intravenous methylphenidate will be used for targeted stimulation of dopaminergic circuits within the subcortical ascending arousal network (ClinicalTrials.gov NCT03814356). The scientific premise of the CCTP and the STIMPACT trial is that personalized brain network mapping in the ICU can identify patients whose connectomes are amenable to neuromodulation. Phase 1 of the STIMPACT trial is an open-label, safety and dose-finding study in 22 patients with disorders of consciousness caused by acute severe traumatic brain injury. Patients in Phase 1 will receive escalating daily doses (0.5–2.0 mg/kg) of intravenous methylphenidate over a 4-day period and will undergo resting-state functional magnetic resonance imaging and electroencephalography to evaluate the drug’s pharmacodynamic properties. The primary outcome measure for Phase 1 relates to safety: the number of drug-related adverse events at each dose. Secondary outcome measures pertain to pharmacokinetics and pharmacodynamics: (1) time to maximal serum concentration; (2) serum half-life; (3) effect of the highest tolerated dose on resting-state functional MRI biomarkers of connectivity; and (4) effect of each dose on EEG biomarkers of cerebral cortical function. Predetermined safety and pharmacodynamic criteria must be fulfilled in Phase 1 to proceed to Phase 2A. Pharmacokinetic data from Phase 1 will also inform the study design of Phase 2A, where we will test the hypothesis that personalized connectome maps predict therapeutic responses to intravenous methylphenidate. Likewise, findings from Phase 2A will inform the design of Phase 2B, where we plan to enroll patients based on their personalized connectome maps. By selecting patients for clinical trials based on a principled, mechanistic assessment of their neuroanatomic potential for a therapeutic response, the CCTP paradigm and the STIMPACT trial have the potential to transform the therapeutic landscape in the ICU and improve outcomes for patients with severe brain injuries.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
For patients with severe brain injuries in the intensive care unit (ICU), the range of possible outcomes includes death, a persistent disorder of consciousness (DoC), recovery of consciousness with functional disability, and recovery of functional independence [1]. Early recovery of consciousness in the ICU is a strong predictor of long-term outcome [2,3,4] and a critical determinant of decisions to continue or withdraw life-sustaining therapy [5]. However, there are currently no treatments proven to promote early recovery of consciousness in the ICU. Without knowing whether a patient can recover consciousness, many families—guided by intensive care clinicians—withdraw life-sustaining therapy, a decision that accounts for up to 70% of deaths in ICU patients with severe traumatic brain injury (TBI) [6, 7] and over 40% of deaths in patients with hypoxic-ischemic injury [8]. Furthermore, decisions to withdraw life-sustaining therapy often occur in the first 3 days of hospitalization [6], when prognosis is most uncertain. A new treatment that promotes early recovery of consciousness in the ICU would benefit patients and families by reducing the likelihood of premature withdrawal of life-sustaining therapy and could decrease ICU complications related to immobility, facilitate self-expression, enable autonomous decision-making, and increase access to specialized rehabilitative care.
Two barriers currently prevent the development of consciousness-promoting therapies in the ICU. First, prior to enrollment in clinical trials, patients’ injuries are not rigorously classified. Instead of selecting patients based on the precise pathophysiologic mechanism underlying coma, trials enroll patients based on behavioral measures, such as the Glasgow Coma Scale (GCS) score, that stratify brain injuries into crude categories of severity. This approach is ineffective because coma is a highly heterogeneous condition. Traumatic and hypoxic-ischemic coma, for example, are associated with variable patterns of axonal disconnections within the subcortical ascending arousal network (AAN), cortical default mode network (DMN), and other networks that contribute to consciousness [9,10,11,12,13,14]. Without precision tools to map preserved network connections in individual patients, it is not possible to identify patients whose connectomes are amenable to therapeutic modulation.
Second, in early-phase clinical trials, therapeutic responses are not routinely tested with direct measures of brain function. Rather, early-phase trials rely on indirect serologic markers of brain injury or insensitive, delayed measures of functional disability [15, 16]. Without biomarkers that directly and quantitatively measure brain function, fundamental questions about a therapy’s neurobiological effects are not being answered in Phases 1 and 2 before moving to Phase 3 trials. Clearly, a new approach to clinical trial design is of scientific and ethical import for this vulnerable population [5]. Indeed, experts in civilian and military brain injury [15,16,17], including leaders at the National Institute of Neurological Disorders and Stroke [18], are now calling for new approaches to clinical trial design for patients with coma and other DoCs. This goal is also a central component of the Curing Coma Campaign launched by the Neurocritical Care Society in 2019.
To address the call for clinical trial design innovation, we propose the Connectome-based Clinical Trial Platform (CCTP), a new mechanistic paradigm for developing and testing targeted therapies that promote early recovery of consciousness in the ICU. The CCTP incorporates two innovations: (1) predictive biomarkers to enroll patients in clinical trials based on connectomes that can be targeted by new therapies; and (2) pharmacodynamic biomarkers to measure how targeted therapies modulate networks, reactivate the cerebral cortex and restore consciousness. By enrolling patients in clinical trials based on a principled, mechanistic assessment of their neuroanatomic potential for a therapeutic response, the CCTP will generate enhanced study samples of patients with similar connectomes, thereby increasing treatment effect sizes, decreasing sample sizes needed to power trials, and ultimately reducing trial duration, cost and risk to subjects [16]. Most importantly, by enhancing the scientific rigor of early-phase clinical trials, we hypothesize that the CCTP will improve the success rate of late-phase trials so that investigators can bring new therapies to clinical practice in the ICU.
To demonstrate the feasibility and utility of implementing the CCTP in the ICU, we report the protocol for STIMPACT (Stimulant Therapy Targeted to Individualized Connectivity Maps to Promote ReACTivation of Consciousness), a CCTP-based clinical trial in which intravenous methylphenidate (IV MPH) will be used to promote recovery of consciousness in severely brain-injured ICU patients. STIMPACT will begin with a Phase 1, open-label, safety and dose-finding study of IV MPH in patients with DoC caused by acute severe TBI (ClinicalTrials.gov NCT03814356). The findings from the primary and secondary outcomes of Phase 1 of STIMPACT will dictate whether we will proceed to Phase 2A. Below, we describe the conceptual framework of the CCTP, and we present the STIMPACT Phase 1 trial protocol as well as the prespecified criteria for proceeding to Phases 2A and 2B.
Conceptual and Empiric Basis for the CCTP
Current theories propose that consciousness requires the integrated function of multiple brain networks, not any single node or connection [19, 20]. This network-based model of consciousness is supported by histologic and radiologic data demonstrating that coma can be caused by variable network disconnections [9, 10, 21,22,23] and that consciousness requires dynamic network interactions [24]. For a patient to recover consciousness, it is believed that the subcortical AAN, which modulates wakefulness [25, 26], must reconnect with the DMN and other cortical networks that mediate awareness [13, 27]. However, the precise mechanisms underlying this reintegration of subcortical and cortical networks are poorly understood. Furthermore, it may be weeks, months, or even years before these recovery mechanisms allow preserved AAN connections to reactivate the cerebral cortex [28,29,30]. For patients whose families face time-sensitive, life-or-death decisions in the ICU, treatments that promote recovery are needed within days. The CCTP will accelerate recovery of consciousness via targeted therapeutic modulation of structurally preserved brain network connections. The ultimate goal of the CCTP is to provide ICU clinicians with an armamentarium of targeted therapies that promote recovery of consciousness in patients with a broad range of DoC etiologies.
Testing the CCTP: Patient and Target Selection for STIMPACT
STIMPACT will test the CCTP in patients with acute severe TBI who are being treated in the ICU. We selected this patient population for our inaugural CCTP trial because prior evidence suggests that components of the AAN may be spared, and therefore serve as therapeutic targets, in patients with severe TBI. Histopathological studies show that although the AAN is invariably injured in animals and humans with coma caused by severe TBI [10, 31], not all AAN nuclei are lesioned and not all axons are disconnected. For example, both histopathological and MRI studies reveal that the ventral midbrain, home to the dopaminergic ventral tegmental area (VTA), is heterogeneously injured by severe TBI, remaining partially intact in some patients [22, 32,33,34]. This phenomenon pertains not only to VTA neuronal cell bodies in the midbrain, but also to VTA axons that connect with the diencephalon, basal forebrain, and cortex (Figs. 1, 2). Consistent with these observations, nuclear imaging studies show that signal transmission in dopaminergic circuits is variably altered by severe TBI [35,36,37]. These converging lines of evidence support the hypothesis that preserved VTA connections are a potential target for therapeutic modulation in ICU patients with severe TBI.

Personalized connectome mapping in the ICU reveals preserved ventral tegmental area (VTA) connections. VTA tracts are shown from a left lateral view in a 33-year-old healthy male control and a 29-year-old man with acute severe TBI. The patient was comatose on arrival to the hospital and in a minimally conscious state at the time of this scan on post-injury day 7, as determined by a Coma Recovery Scale-Revised assessment. Tractography analysis was performed using TrackVis, as previously described [26]. All tracts are color-coded by sites of VTA connectivity: turquoise with dorsal raphe (DR), blue with locus coeruleus (LC), green with median raphe (MR), and pink with cortex, thalamus (Th), hypothalamus (Hy), or basal forebrain (BF). Multiple VTA connections are preserved in the patient, including with the medial prefrontal cortex (MPFC), which is a node of the default mode network. The patient recovered consciousness and functional independence by 6 months. Even in a patient with acute severe traumatic brain injury, the VTA may be a hub through which multiple brainstem AAN nodes connect with the Th, Hy, BF and cerebral cortex

Ventral tegmental area (VTA) axonal connections are spared in a subset of patients. We analyzed VTA connectivity with the hypothalamus (Hy), thalamus (Th), and basal forebrain (BF) in 16 patients, as well as in 16 matched controls. We calculated a connectivity probability (CP) between the VTA and each target ascending arousal network (AAN) node using previously described methods [9, 48, 84]. On a group level, median CP values were lower between the VTA and each AAN node in patients compared with controls. However, there is significant variance in control-group CP values, and many patients fall within the control-group interquartile range (IQR). These results indicate relative sparing of VTA connections in a subset of patients with acute severe TBI. See the Supplementary Material for a detailed description of the CP calculation algorithm
Testing the CCTP: Therapy Selection for STIMPACT
We selected IV MPH as the therapeutic agent for STIMPACT because of its well-established mechanism of action on the VTA, compelling rodent data suggesting its role in promoting consciousness, and decades of data indicating its safe use in humans. IV MPH is a dopamine reuptake inhibitor that potentiates dopaminergic neurotransmission by VTA neurons [38, 39]. Although multiple neurotransmitters contribute to arousal [25], empiric evidence strongly supports the role of dopamine in therapeutic modulation of consciousness. In rodent models, it has been shown that a dopamine D1 receptor agonist (but not a D2 agonist) restores consciousness from anesthetic coma [40]. Furthermore, optogenetic [41] and electrical stimulation [42] of dopaminergic VTA neurons promotes recovery of consciousness in anesthetized rodents. This effect was strongly inhibited by a D1 receptor antagonist, suggesting that the arousal effects of VTA dopaminergic stimulation are D1 receptor-dependent. Collectively, these observations suggest that IV MPH promotes reemergence of consciousness by stimulating dopaminergic VTA neuronal signaling via D1 receptors [38, 42].
In humans, multiple enteral and subcutaneous therapies that stimulate dopaminergic neurotransmission have been shown to benefit patients with subacute or chronic TBI, including enteral MPH [43, 44]. Moreover, enteral amantadine, the only therapy shown to promote recovery of consciousness in a randomized controlled trial of patients with subacute TBI [45], increases dopamine release and blocks dopamine reuptake, thereby increasing synaptic levels of dopamine. Prior clinical trials of MPH investigated the enteral formulation, which is approved by the FDA to treat attention deficit disorder [39] and prescribed “off-label” to promote recovery in patients with TBI [46, 47]. However, positron emission tomography studies show that the IV formulation of MPH has a far more rapid uptake in the human brain than does the enteral formulation (~ 10 min versus ~ 90 min) [39]. These positron emission tomography data also indicate that the IV formulation of MPH generates higher levels of dopamine signaling than does the enteral formulation. Collectively, prior animal and human data thus provide a biological basis for using IV MPH to upregulate structurally intact, functionally dormant dopaminergic VTA circuits in ICU patients with severe TBI.
Testing the CCTP: Predictive Biomarker Selection for STIMPACT
To identify patients with preserved VTA connections, we will map the structural AAN connectome on a clinical MRI scanner. We will use high angular resolution diffusion imaging (HARDI) tractography, an MRI technique that maps axonal connectivity based on directional water diffusion [48]. We have previously shown that HARDI tractography can map the AAN connectome in the ex vivo human brain [26], detect acute AAN disconnections ex vivo [10], and detect disruptions in vivo in ICU patients with acute severe TBI [9].
Given that the VTA and cerebral cortex are connected both monosynaptically and polysynaptically [26, 49] (Fig. 3), it is unlikely that a single preserved VTA connection will predict a patient’s response to IV MPH. Rather, we will use a graph theoretical analysis measure, VTA hub strength (SVTA), as a predictive biomarker for the STIMPACT trial, because this biomarker measures both direct (monosynaptic) and indirect (polysynaptic) VTA connections. We selected subcortical AAN and cortical DMN nodes as targets in the SVTA measurement because functional connectivity studies suggest that DMN reactivation is essential for recovery of consciousness after severe brain injury [12, 50,51,52,53] and that the VTA activates the DMN via the AAN [54].

Predictive and pharmacodynamic biomarkers. Left panel: This type of individualized ascending arousal network (AAN) connectome map will be used as a biomarker to predict patient responses to therapy. In this proposed model of a “connectogram,” brainstem nodes are shown on the outside, while hypothalamic, thalamic, and basal forebrain nodes are shown in the middle. Line thickness is proportional to the connectivity probability (CP; see Supplementary Material for how this value is measured) for each node–node pair. Nodal gray shading is proportional to the percentage of each node occupied by a traumatic lesion (bottom right bar). These structural connectivity data, along with structural connectivity measures between the VTA and default mode network (DMN), are used to calculate SVTA. Connectogram artwork by Kimberly Main Knoper. Right panel: Resting-state functional MRI (rs-fMRI) maps illustrating concurrent recovery of consciousness and reactivation of the DMN. Hot colors indicate correlated activity within the DMN. Cool colors indicate regions anti-correlated with the DMN (inset). Functional connectivity between the VTA and DMN (ZVTA-DMN) is an rs-fMRI pharmacodynamic biomarker that will be used to determine the neurobiological effects of intravenous methylphenidate in patients with acute severe traumatic brain injury. Abbreviations: nucleus basalis of Meynert/substantia innominata (BNM/SI), diagonal band of Broca (DBB), dorsal raphe (DR), intralaminar nuclei of the thalamus (IL), lateral hypothalamic area (LHA), laterodorsal tegmental nucleus (LDTg), locus coeruleus (LC), median raphe (MnR), mesencephalic reticular formation (mRt), parabrachial complex (PBC), paraventricular nucleus of the thalamus (PV), pedunculotegmental nucleus (PTg), periaqueductal gray (PAG), pontis oralis (PnO), reticular nuclei of the thalamus (Ret), tuberomammillary nucleus of the hypothalamus (TMN), ventral tegmental area (VTA)
STIMPACT Hypothesis
The overarching hypothesis of the STIMPACT trial is that preservation of VTA connections within the AAN connectome predicts a response to IV MPH in patients with acute severe TBI. We hypothesize that for the subset of patients with preserved VTA connections, IV MPH will reconnect the cerebral cortex, accelerate reemergence of consciousness, and transform the course of their recovery.
STIMPACT Design Overview for Phases 1, 2A and 2B
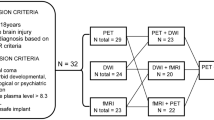
The Phase 1 STIMPACT protocol described here focuses on safety (primary outcome measure). IV MPH will be administered as daily boluses (via IV push at a rate not to exceed 20 mg/min) on Days 1–4: 0.5 mg/kg on Day 1, 1.0 mg/kg on Day 2, 2.0 mg/kg on Day 3, and the maximum tolerated dose on Day 4 (see Supplementary Material for a detailed rationale of dose selection). In Phase 1, we will also assess MPH pharmacokinetics and pharmacodynamic biomarkers of MPH responses (secondary outcome measures; see Fig. 4 for study schematic). Inclusion and exclusion criteria for STIMPACT Phase 1 are reported on ClinicalTrials.gov (NCT03814356) and in the Supplementary Material. Because the STIMPACT Phase 1 trial is focused on safety and dose-finding, all patients will receive IV MPH, regardless of their SVTA measures.

STIMPACT study design schematic. Each patient in Phase 1 of the STIMPACT Trial will undergo 5 days of data acquisition, with predictive biomarker data and baseline pharmacodynamic biomarker data collected on Day 0, and treatment-related biomarker data collected on Days 1–4. Hence, each patient’s biomarker responses to IV MPH will be measured against his/her own baseline biomarker variance. BP blood pressure, CRS-R Coma Recovery Scale-Revised, HR heart rate, IV MPH intravenous methylphenidate, SVTA ventral tegmental area hub strength, ZVTA-DMN functional connectivity between the ventral tegmental area and the default mode network
The decision to proceed from Phase 1 to Phase 2A will depend upon the safety data and pharmacodynamic biomarker data obtained in Phase 1. Pharmacokinetic data from Phase 1 will inform the inter-dose intervals for Phase 2A, which is being planned as a double-blinded, placebo-controlled crossover design trial. Predictive biomarker data acquired in Phase 1 will inform the power calculation for the number of patients needed in Phase 2A to detect a difference in pharmacodynamic responses to MPH between patients with preserved versus lost VTA connectivity (i.e., high SVTA versus low SVTA). If our hypothesis that SVTA predicts pharmacodynamic responses to IV MPH is supported in Phase 2A, then in Phase 2B we will select patients based on the SVTA predictive biomarker, thus realizing the individualized, precision medicine goal of the CCTP.
Rationale for a STIMPACT Phase 1 Trial
IV MPH has been administered to over 1700 human subjects since 1957 [55,56,57,58,59,60,61,62,63,64,65,66,67,68,69]. In these prior studies, mostly conducted in healthy subjects and patients with psychiatric disease, no serious adverse events (SAEs) were reported. Common drug-related adverse events (AEs) included hypertension, tachycardia, agitation, and nausea [70], which will be readily manageable in the ICU environment of the STIMPACT trial.
Yet despite its reassuring safety record in human subjects, IV MPH has never been administered to a patient with acute severe TBI. Experience with IV MPH in comatose humans is limited to small case series of patients with barbiturate overdoses [61, 71, 72]. Because patients with acute severe TBI have increased hepatic drug metabolism and clearance, we cannot rely on prior dose-finding and pharmacokinetic studies of healthy humans [39, 70]. Rather, it is essential to determine the maximum tolerated dose and pharmacokinetics of IV MPH in our population of interest. Thus, we believe that it is scientifically and ethically appropriate to begin STIMPACT with a Phase 1 safety and dose-finding study.
Methods
Primary Outcome Measure
The primary outcome measure of STIMPACT Phase 1 is the number of drug-related AEs at each of three IV MPH doses: 0.5, 1.0 and 2.0 mg/kg. The incidence of drug-related AEs (including SAEs) will be calculated as the ratio of the number of events divided by the number of administrations of IV MPH at each dose. The percentage of subjects experiencing each documented AE and SAE will be reported for each dose. AEs and SAEs will be classified according to the Medical Dictionary for Regulatory Activities (https://www.meddra.org).
In Phase 1, we aim to detect, with high probability, a drug-related SAE that would prevent STIMPACT from moving to Phase 2a. We therefore designed the Phase 1 trial to have a 90% chance of detecting any drug-related SAE that occurs with a frequency of ≥ 10% at any given dose. Based on these criteria, we used the binomial equation to calculate our sample size: 1 − (1 − p)n = y, where p = drug-related SAE rate, n = number of subjects, and y = chance of detecting the SAE. Using a drug-related SAE rate of 10% (p = 0.10) and requiring a 90% probability of detecting the SAE at least once (y = 0.90) at each dose, we calculate that a 22-subject cohort will be necessary to enroll in Phase 1.
If two drug-related SAEs occur at a dose (frequency ~ 10%), that dose and all higher doses will be removed from the trial. If two drug-related SAEs occur at the lowest dose, the trial will be stopped and Phase 1 will be repeated at a lower dose range. Any patient who experiences a drug-related AE at a dose of 1.0 or 2.0 mg/kg will continue in the study at the previously tolerated dose. If a drug-related AE occurs at the lowest dose of 0.5 mg/kg, the patient will be withdrawn from the study. A patient who experiences an SAE at any dose will be withdrawn from the study. Additional details regarding the definitions and criteria for AEs and SAEs are provided in the Supplementary Material.
Secondary Outcome Measures
Pharmacokinetics
For analysis of IV MPH pharmacokinetics, blood samples (1 mL) will be obtained via arterial or central venous catheters at baseline (immediately pre-dose), 5 min, 15 min, 30 min, 1 h, 1.5 h, 2 h, 4 h, 8 h, 12 h, and 16 h after each dose. Samples will be stored at -70 °C until liquid chromatography/tandem mass spectrometry analysis is performed by Worldwide Clinical Trials (Austin, TX). Plasma samples will be analyzed for total MPH (ng/mL) and ritalinic acid metabolic (ng/mL) concentrations.
As pharmacokinetic and pharmacodynamic potency may differ between MPH enantiomers [73], serum samples will also be analyzed for d-threo-MPH and l-threo-MPH enantiomer concentrations (ng/mL). Non-compartmental analysis will be used to calculate pharmacokinetic parameters using Pheonix WinNonLin (Version 8.1; Certara, L.P; Princeton, NJ) to determine time to maximum concentration (Tmax) and elimination half-life (T½), as well as maximum concentration (Cmax) and area under the concentration–time curve (AUCinf). Pharmacokinetic data will be summarized using descriptive statistics.
Pharmacodynamics
The pharmacodynamic outcome measures in STIMPACT Phase 1 are the effect of IV MPH on resting-state functional MRI (rs-fMRI) and EEG biomarkers of brain function, which will provide a direct assessment of IV MPH on its target brain network. On Days 1–3, we will record EEG continuously to measure the EEG biomarker response to each dose. On Day 4, each patient will receive his/her maximum tolerated dose during an rs-fMRI scan to measure the rs-fMRI biomarker response to this dose. The rs-fMRI scan will begin 10 min before administration of IV MPH and will continue for 30 min after IV MPH administration. Based upon prior positron emission tomography data [39], we anticipate that the peak MPH response in the brain will occur approximately 10 min after MPH administration. Thus, we expect to capture the peak brain concentration of MPH during the 30-min of rs-fMRI data acquisition that follow drug administration. We will use a self-controlled design, whereby each patient’s baseline, pre-dose biomarker data (i.e., obtained immediately prior to IV MPH administration) are compared to the post-dose biomarker data.
For the rs-fMRI analysis, we and others have demonstrated the feasibility of using rs-fMRI to map functional brain networks in ICU patients with acute severe TBI (Figs. 3, 5) [12, 51, 52]. Given that brainstem functional network mapping of VTA-DMN connectivity requires high spatial and temporal resolution, we implemented a simultaneous multislice [74] blood-oxygen level dependent (BOLD) sequence on the 3 Tesla Siemens Skyra MRI scanner in the Massachusetts General Hospital Neurosciences ICU (2 mm isotropic voxels, TR = 1.25 s). To control for the effects of physiological fluctuations on the BOLD signal [75], we measure and statistically regress cardiac and respiratory activity during rs-fMRI (Fig. 6 and Supplementary Figures 1 and 2). Additional details regarding rs-fMRI sequence parameters, as well as physiologic data acquisition and analysis, are provided in the Supplementary Material.

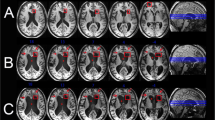
Functional connectivity between the ventral tegmental area (VTA) and default mode network (DMN) reemerges during recovery. Eight patients with acute severe TBI underwent resting-state functional MRI (rs-fMRI) in the ICU and at 6-month follow-up, by which time all had recovered consciousness. Mean group-level VTA functional connectivity maps are shown, based on Fisher Z-transformed correlations in the BOLD signal (color bar). Correlations are adjusted for significance with p < 0.01 height-level and p < 0.05 false discovery rate (FDR)-corrected cluster-level thresholds. There were no VTA-DMN connections acutely, but VTA connections with the medial prefrontal cortex (MPFC) node of the DMN reemerged during recovery of consciousness (red arrow). The statistical threshold used here is more liberal than in our recent studies of DMN connectivity [12, 51], which underscores the need for the higher-resolution rs-fMRI sequence that we developed for the CCTP

Wavelet transform coherence analysis of a representative control subject (upper panel) and a representative patient (lower panel). Time series of respiratory variation and change in blood-oxygen level dependent (ΔBOLD) signal in the left ventral diencephalon are shown in the left column. The dynamic interaction between respiratory variation and ΔBOLD is demonstrated by the squared wavelet coherence map between the time series of respiratory variation and ΔBOLD shown in the middle column. The magnitude of coherence ranges between 0 and 1, where warmer color represents stronger coherence and cooler color represents weaker coherence. Significant coherence between respiratory variation and ΔBOLD occurs in the area defined by the thick contour of the unfaded region. The x-coordinate of the area provides information on the duration of the oscillating cycle when respiratory variation interacts with ΔBOLD, and the y-coordinate shows the time when this interaction occurs over the resting state fMRI scan. The simplified format of coherence between respiratory variation and ΔBOLD is shown in the right column, with the features of oscillations displayed in terms of frequency. While increased coherence is found between respiratory variation and ΔBOLD at the frequency range of 0.008-0.063 Hz in the healthy subject, the coherence between respiratory variation and ΔBOLD in the same frequency range is diminished in the patient with acute severe TBI. Compared with the healthy subject, the resting state BOLD signal changes are less influenced by respiratory variation in the patient with acute severe TBI. A detailed interpretation of the wavelet coherence findings is included in Supplementary Material
To test for an rs-fMRI biomarker response to the maximum tolerated IV MPH dose, we will perform a patient-specific, change-point analysis [76, 77] that detects whether time-series values deviate between two epochs of data (i.e., pre- versus post-dose). First, we will generate averaged rs-fMRI time-series in VTA and DMN regions of interest and regress physiological heart rate and respiration data from these time-series (see Supplementary Material). Then, we will use dynamic functional connectivity (DFC) [24, 78] to calculate a correlation value (r) between the VTA and DMN for every 10 s of rs-fMRI data. We will Fisher-Z transform the r values into Z values to generate 60 ZVTA-DMN (pre IV MPH) values across the 10 min of rs-fMRI data acquisition before IV MPH is administered and 180 ZVTA-DMN (post IV MPH) values across the 30 min of rs-fMRI data acquisition after IV MPH is administered. We will analyze the time-series, composed of ZVTA-DMN (pre IV MPH) and ZVTA-DMN (post IV MPH) using the R change-point program [79]. A patient will be considered a responder to IV MPH if the change-point analysis identifies a deviation in the ZVTA-DMN (post IV MPH) time-series compared to ZVTA-DMN (pre IV MPH) time-series.
With respect to the EEG pharmacodynamic data, we aim to identify a resting-state EEG biomarker that measures the effect of IV MPH on cortical circuit function. Crucial to this effort is the development of statistical methods that quantify EEG oscillatory dynamics. Patients with DoC experience frequent fluctuations in arousal [80], and the dynamic range and robustness of EEG biomarkers during these fluctuations in the ICU environment is unknown. Therefore, to minimize confounders and optimize the signal-to-noise ratio of the EEG data, we will again use a self-controlled design in which we compare post-dose EEG data to each patient’s immediate pre-dose EEG data. This design allows therapeutic responses to be compared to each patient’s baseline biomarker data on the same clinical medication regimen.
We considered a broad range of candidate EEG biomarkers for the EEG pharmacodynamic biomarker analysis (see Supplementary Material). Recent studies of patients with chronic DoC suggest that spectral measures perform better than other EEG-based cortical measures as biomarkers of consciousness [81]. Our preliminary results suggest that ICU patients with severe TBI experience wide-ranging fluctuations in frequency band-specific spectral measures over the course of a clinical recording (Fig. 7). Nevertheless, the ratio of alpha to delta power appears to reliably distinguish ICU patients with acute DoC from patients who have recovered consciousness. We therefore selected alpha-delta ratio as the EEG-based pharmacodynamic biomarker for STIMPACT Phase 1. Details of EEG data acquisition and processing are provided in the Supplementary Material.

Alpha-delta ratio as a biomarker of recovery of consciousness. We computed the dynamic range of the resting-state alpha-delta ratio in EEG recordings of patients with severe traumatic brain injury and controls. The plots show comparisons of alpha-delta ratio measures in 12 ICU patients with acute severe traumatic brain injury (left), these same 12 patients at 6-month follow-up (middle), and 16 healthy controls (right). Each column represents a single subject. The three red lines within each subject’s alpha-delta ratio plot represent the interquartile range (outer red lines) and the median (middle red line). These results suggest that (1) alpha-delta ratio is generally lower in unconscious ICU patients (coma and vegetative state) than in conscious ICU patients (minimally conscious state and post-traumatic confusional state); (2) in ICU patients who recover consciousness by 6 months, alpha-delta ratio typically increases to values similar to those of controls; and (3) there is substantial intra-subject variance in alpha-delta ratio during a single EEG recording
The analytic approach for the EEG alpha/delta pharmacodynamic biomarker data will parallel the approach for the rs-fMRI ZVTA-DMN pharmacodynamic biomarker described above. To test for an EEG biomarker response at the maximum tolerated IV MPH dose, we will generate a time-series composed of the alpha/delta statistic calculated from 10-s time windows before (alpha/delta(pre IV MPH)) and after (alpha/delta(post IV MPH)) MPH administration. A patient will be considered a responder to IV MPH based on EEG data if the change-point analysis identifies a deviation in the alpha/delta(post IV MPH) time-series compared to alpha/delta(pre IV MPH) time-series.
Criteria for Proceeding from Phase 1 to Phase 2A
We will proceed from Phase 1 to Phase 2a if predetermined safety and pharmacodynamic biomarker criteria are met. The safety criterion is that an IV MPH dose will be used in Phase 2A only if there are fewer than 2 drug-related SAEs at that dose in Phase 1. The pharmacodynamic biomarker criterion is that there must be a pharmacodynamic response in ≥ 10% of patients, in either the rs-fMRI biomarker or the EEG biomarker, at the maximum tolerated dose. Without such a response, we will repeat Phase 1 at a higher dose range. The rationale for this second criterion is that even though our dose range of 0.5–2.0 mg/kg is broader than the 0.5–0.9 mg/kg dose range used in most prior human studies [39, 70], our dose range may still be insufficient to detect a pharmacodynamic response in patients with severe TBI.
The safety and pharmacodynamic criteria for proceeding to Phase 2A yield three possible outcomes for STIMPACT Phase 1 (Supplementary Fig. 3): (1) IV MPH is safe and ≥ 10% of the 22 patients show a response in ZVTA-DMN or alpha/delta → we will proceed to Phase 2a at the maximum tolerated dose; (2) IV MPH is not safe, even at the lowest dose → we will repeat Phase 1 in 22 new patients at a lower dose range; (3) IV MPH is safe, but < 10% show a response → we will explore alternative rs-fMRI [12, 24] and EEG analytic biomarkers [81, 82], as well as different statistical models of the pharmacodynamic response (see Supplementary Material), and will consider repeating Phase 1 at a higher dose range. Recognizing that rs-fMRI and EEG biomarker development is an active area of investigation in the field of DoC, the CCTP provides alternate approaches at each stage of trial design while maintaining the central focus on connectome-based measures of therapeutic responses.
Overview of Phase 2A and Phase 2B
The study design for Phase 2A will be finalized upon completion of Phase 1. STIMPACT Phase 2A is being planned as a double-blind, placebo-controlled, crossover trial to test the primary hypothesis that pharmacodynamic biomarkers respond to IV MPH more than to placebo. The secondary hypothesis of Phase 2A is that preserved SVTA predicts a pharmacodynamic biomarker response to IV MPH relative to placebo. As in Phase 1, patients will be enrolled in Phase 2A regardless of their SVTA measures. If the secondary hypothesis of Phase 2A is supported by the pharmacodynamic biomarker data, suggesting that SVTA predicts a response to IV MPH, we will select patients for enrollment in Phase 2B based upon their SVTA measures.
In Phase 2A, we will also investigate the effect of IV MPH versus placebo on the change in level of consciousness on behavioral assessment with the Coma Recovery Scale-Revised (CRS-R) [83]. CRS-R data will be used to inform the power calculation for Phase 2B. The primary hypothesis in Phase 2B will be that there is a greater behavioral response to IV MPH relative to placebo, as assessed by the CRS-R. Phase 2B thus represents a realization of the fundamental premise of the CCTP—that a principled, mechanistic approach to study design in early phase clinical trials leads to enriched study samples in late phase trials that are more likely to show a drug effect.
Trial Oversight
Between July 2018 and February 2020, we convened a Clinical Oversight Committee, led by an experienced Independent Medical Monitor (Dr. Thomas P. Bleck), which approved the protocol for the Phase 1 STIMPACT trial. To ensure that the TBI community’s interests are represented, we also convened a Patient and Family Advisory Board, which reviewed the research plan and made recommendations regarding recruitment, study activities, and data dissemination. The investigational new drug (IND) application was approved by the FDA in August, 2018 (IND# 140675), and the study protocol was approved by the Partners Healthcare Institutional Review Board in May, 2019.
Summary
We propose a mechanistic clinical trial paradigm, the Connectome-based Clinical Trial Platform (CCTP), to predict and measure responses to new targeted therapies in the ICU. We also describe a CCTP-based trial, STIMPACT, which will test the safety of IV MPH in Phase 1, and then in later phases will ultimately test the hypothesis that IV MPH promotes recovery of consciousness in patients who have preserved VTA connections within their AAN connectomes. The CCTP will allow clinicians to provide targeted treatments that are personalized to each patient’s connectome, ensuring that each patient is given the best possible chance to recover consciousness in the ICU. Ultimately, we envision multidisciplinary discussions about optimal treatment selection at a Coma Board, akin to the Tumor Board at which therapeutic decisions are made by a multidisciplinary team for patients with cancer. By providing a clinical trial platform to test targeted pharmacologic and electrophysiologic therapies that reactivate the injured human connectome, the CCTP has the potential to transform the therapeutic landscape and improve outcomes for patients with DoC.
References
Giacino JT, Fins JJ, Laureys S, et al. Disorders of consciousness after acquired brain injury: the state of the science. Nat Rev Neurol. 2014;10:99–114.
Giacino JT, Kalmar K. The vegetative and minimally conscious states: a comparison of clinical features and functional outcome. J Head Trauma Rehabil. 1997;12:36–51.
Claassen J, Doyle K, Matory A, et al. Detection of brain activation in unresponsive patients with acute brain injury. N Engl J Med. 2019;380:2497–505.
Faugeras F, Rohaut B, Valente M, et al. Survival and consciousness recovery are better in the minimally conscious state than in the vegetative state. Brain Inj. 2018;32:72–7.
Fins JJ. Rights come to mind: brain injury, ethics, and the struggle for consciousness. New York: Cambridge University Press; 2015.
Turgeon AF, Lauzier F, Simard JF, et al. Mortality associated with withdrawal of life-sustaining therapy for patients with severe traumatic brain injury: a Canadian multicentre cohort study. CMAJ. 2011;183:1581–8.
Izzy S, Compton R, Carandang R, et al. Self-fulfilling prophecies through withdrawal of care: do they exist in traumatic brain injury, too? Neurocrit Care. 2013;19:347–63.
Peberdy MA, Kaye W, Ornato JP, et al. Cardiopulmonary resuscitation of adults in the hospital: a report of 14720 cardiac arrests from the National Registry of Cardiopulmonary Resuscitation. Resuscitation. 2003;58:297–308.
Snider SB, Bodien YG, Bianciardi M, et al. Disruption of the ascending arousal network in acute traumatic disorders of consciousness. Neurology. 2019;93:e1281–7.
Edlow BL, Haynes RL, Takahashi E, et al. Disconnection of the ascending arousal system in traumatic coma. J Neuropathol Exp Neurol. 2013;72:505–23.
Rosenblum WI. Immediate, irreversible, posttraumatic coma: a review indicating that bilateral brainstem injury rather than widespread hemispheric damage is essential for its production. J Neuropathol Exp Neurol. 2015;74:198–202.
Threlkeld ZD, Bodien YG, Rosenthal ES, et al. Functional networks reemerge during recovery of consciousness after acute severe traumatic brain injury. Cortex. 2018;106:299–308.
Demertzi A, Antonopoulos G, Heine L, et al. Intrinsic functional connectivity differentiates minimally conscious from unresponsive patients. Brain. 2015;138:2619–31.
Newcombe VF, Williams GB, Scoffings D, et al. Aetiological differences in neuroanatomy of the vegetative state: insights from diffusion tensor imaging and functional implications. J Neurol Neurosurg Psychiatry. 2010;81:552–61.
Maas AIR, Menon DK, Adelson PD, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16:987–1048.
Diaz-Arrastia R, Kochanek PM, Bergold P, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma. 2014;31:135–58.
Kochanek PM, Dixon CE, Mondello S, et al. Multi-center pre-clinical consortia to enhance translation of therapies and biomarkers for traumatic brain injury: operation brain trauma therapy and beyond. Front Neurol. 2018;9:640.
Smith DH, Hicks R, Povlishock JT. Therapy development for diffuse axonal injury. J Neurotrauma. 2013;30:307–23.
Tononi G, Boly M, Massimini M, et al. Integrated information theory: from consciousness to its physical substrate. Nat Rev Neurosci. 2016;17:450–61.
Dehaene S, Changeux JP, Naccache L, et al. Conscious, preconscious, and subliminal processing: a testable taxonomy. Trends Cogn Sci. 2006;10:204–11.
Sharp DJ, Scott G, Leech R. Network dysfunction after traumatic brain injury. Nat Rev Neurol. 2014;10:156–66.
Izzy S, Mazwi NL, Martinez S, et al. Revisiting grade 3 diffuse axonal injury: not all brainstem microbleeds are prognostically equal. Neurocrit Care. 2017;27:199–207.
Jang SH, Kim SH, Lim HW, et al. Injury of the lower ascending reticular activating system in patients with hypoxic-ischemic brain injury: diffusion tensor imaging study. Neuroradiology. 2014;56:965–70.
Demertzi A, Tagliazucchi E, Dehaene S, et al. Human consciousness is supported by dynamic complex patterns of brain signal coordination. Sci Adv. 2019;5:eaat7603.
Parvizi J, Damasio A. Consciousness and the brainstem. Cognition. 2001;79:135–60.
Edlow BL, Takahashi E, Wu O, et al. Neuroanatomic connectivity of the human ascending arousal system critical to consciousness and its disorders. J Neuropathol Exp Neurol. 2012;71:531–46.
Koch C, Massimini M, Boly M, et al. Neural correlates of consciousness: progress and problems. Nat Rev Neurosci. 2016;17:307–21.
Estraneo A, Moretta P, Loreto V, et al. Late recovery after traumatic, anoxic, or hemorrhagic long-lasting vegetative state. Neurology. 2010;75:239–45.
Voss HU, Uluc AM, Dyke JP, et al. Possible axonal regrowth in late recovery from the minimally conscious state. J Clin Investig. 2006;116:2005–11.
Hammond FM, Giacino JT, Nakase Richardson R, et al. Disorders of consciousness due to traumatic brain injury: functional status ten years post-injury. J Neurotrauma. 2019;36:1136–46.
Ommaya AK, Gennarelli TA. Cerebral concussion and traumatic unconsciousness. Correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633–54.
Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59.
Gentry LR, Godersky JC, Thompson BH. Traumatic brain stem injury: MR imaging. Radiology. 1989;171:177–87.
Edlow BL, Threlkeld ZD, Fehnel KP, et al. Recovery of functional independence after traumatic transtentorial herniation with duret hemorrhages. Front Neurol. 2019;10:1077.
Donnemiller E, Brenneis C, Wissel J, et al. Impaired dopaminergic neurotransmission in patients with traumatic brain injury: a SPECT study using 123I-beta-CIT and 123I-IBZM. Eur J Nucl Med. 2000;27:1410–4.
Jenkins PO, De Simoni S, Bourke NJ, et al. Stratifying drug treatment of cognitive impairments after traumatic brain injury using neuroimaging. Brain. 2019;142:2367–79.
Fridman EA, Osborne JR, Mozley PD, et al. Presynaptic dopamine deficit in minimally conscious state patients following traumatic brain injury. Brain. 2019;142:1887–93.
Solt K, Cotten JF, Cimenser A, et al. Methylphenidate actively induces emergence from general anesthesia. Anesthesiology. 2011;115:791–803.
Swanson JM, Volkow ND. Serum and brain concentrations of methylphenidate: implications for use and abuse. Neurosci Biobehav Rev. 2003;27:615–21.
Taylor NE, Chemali JJ, Brown EN, et al. Activation of D1 dopamine receptors induces emergence from isoflurane general anesthesia. Anesthesiology. 2013;118:30–9.
Taylor NE, Van Dort CJ, Kenny JD, et al. Optogenetic activation of dopamine neurons in the ventral tegmental area induces reanimation from general anesthesia. Proc Natl Acad Sci USA. 2016;113:12826–31.
Solt K, Van Dort CJ, Chemali JJ, et al. Electrical stimulation of the ventral tegmental area induces reanimation from general anesthesia. Anesthesiology. 2014;121:311–9.
Fridman EA, Schiff ND. Neuromodulation of the conscious state following severe brain injuries. Curr Opin Neurobiol. 2014;29:172–7.
Thibaut A, Schiff N, Giacino J, et al. Therapeutic interventions in patients with prolonged disorders of consciousness. Lancet Neurol. 2019;18:600–14.
Giacino JT, Whyte J, Bagiella E, et al. Placebo-controlled trial of amantadine for severe traumatic brain injury. N Engl J Med. 2012;366:819–26.
Barra ME, Izzy S, Sarro-Schwartz A, et al. Stimulant therapy in acute traumatic brain injury: prescribing patterns and adverse event rates at 2 level 1 trauma centers. J Intensive Care Med. 2019. https://doi.org/10.1177/0885066619841603.
Whyte J, Hart T, Vaccaro M, et al. Effects of methylphenidate on attention deficits after traumatic brain injury: a multidimensional, randomized, controlled trial. Am J Phys Med Rehabil. 2004;83:401–20.
McNab JA, Edlow BL, Witzel T, et al. The human connectome project and beyond: initial applications of 300 mT/m gradients. Neuroimage. 2013;80:234–45.
Morales M, Margolis EB. Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci. 2017;18:73–85.
Norton L, Hutchison RM, Young GB, et al. Disruptions of functional connectivity in the default mode network of comatose patients. Neurology. 2012;78:175–81.
Bodien YG, Threlkeld ZD, Edlow BL. Default mode network dynamics in covert consciousness. Cortex. 2019;119:571.
Kondziella D, Fisher PM, Larsen VA, et al. Functional MRI for assessment of the default mode network in acute brain injury. Neurocrit Care. 2017;27:401–6.
Vanhaudenhuyse A, Noirhomme Q, Tshibanda LJ, et al. Default network connectivity reflects the level of consciousness in non-communicative brain-damaged patients. Brain. 2010;133:161–71.
Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 2012;76:33–50.
Gale AS. The effect of methylphenidate (ritalin) on thiopental recovery. Anesthesiology. 1958;19:521–31.
Janowsky DS, Leichner P, Clopton P, et al. Comparison of oral and intravenous methylphenidate. Psychopharmacology. 1978;59:75–8.
Joyce PR, Nicholls MG, Donald RA. Methylphenidate increases heart rate, blood pressure and plasma epinephrine in normal subjects. Life Sci. 1984;34:1707–11.
Wang GJ, Volkow ND, Hitzemann RJ, et al. Behavioral and cardiovascular effects of intravenous methylphenidate in normal subjects and cocaine abusers. Eur Addict Res. 1997;3:49–54.
Dodson ME, Fryer JM. Postoperative effects of methylphenidate. Br J Anaesth. 1980;52:1265–70.
Volkow ND, Wang GJ, Fowler JS, et al. Cardiovascular effects of methylphenidate in humans are associated with increases of dopamine in brain and of epinephrine in plasma. Psychopharmacology. 2003;166:264–70.
Carter CH, Maley MC. Parenteral use of methylphenidate (ritalin). Dis Nerv Syst. 1957;18:146–8.
Clark CR, Geffen GM, Geffen LB. Role of monoamine pathways in attention and effort: effects of clonidine and methylphenidate in normal adult humans. Psychopharmacology. 1986;90:35–9.
Chan YP, Swanson JM, Soldin SS, et al. Methylphenidate hydrochloride given with or before breakfast: II. Effects on plasma concentration of methylphenidate and ritalinic acid. Pediatrics. 1983;72:56–9.
Joyce PR, Donald RA, Nicholls MG, et al. Endocrine and behavioral responses to methylphenidate in normal subjects. Biol Psychiatry. 1986;21:1015–23.
Volkow ND, Wang GJ, Fowler JS, et al. Methylphenidate and cocaine have a similar in vivo potency to block dopamine transporters in the human brain. Life Sci. 1999;65:PL7–12.
Li CS, Morgan PT, Matuskey D, et al. Biological markers of the effects of intravenous methylphenidate on improving inhibitory control in cocaine-dependent patients. Proc Natl Acad Sci U S A. 2010;107:14455–9.
Christensen RO. A new agent for shortening recovery time in oral surgery. Oral Surg Oral Med Oral Pathol. 1958;11:999–1002.
Percheson PB, Carroll JJ, Screech G. Ritalin (methylphenidate): clinical experiences. Can Anaesth Soc J. 1959;6:277–82.
Gale AS. The comparative and additive effects of methylphenidate and bemegride. Anesthesiology. 1961;22:210–4.
Volkow ND, Wang GJ, Gatley SJ, et al. Temporal relationships between the pharmacokinetics of methylphenidate in the human brain and its behavioral and cardiovascular effects. Psychopharmacology. 1996;123:26–33.
Smith B, Adriani J. Studies on newer analeptics and the comparison of their action with pentylenetetrazole, nikethamide and picrotoxin. Anesthesiology. 1958;19:115.
Ticktin H, Epstein J, Shea JG, et al. Effect of methylphenidate hydrochloride in antagonizing barbiturate-induced depression. Neurology. 1958;8:267–71.
Volkow ND, Wang GJ, Fowler JS, et al. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry. 1998;155:1325–31.
Setsompop K, Gagoski BA, Polimeni JR, et al. Blipped-controlled aliasing in parallel imaging for simultaneous multislice echo planar imaging with reduced g-factor penalty. Magn Reson Med. 2012;67:1210–24.
Glover GH, Li TQ, Ress D. Image-based method for retrospective correction of physiological motion effects in fMRI: RETROICOR. Magn Reson Med. 2000;44:162–7.
Lindquist MA, Waugh C, Wager TD. Modeling state-related fMRI activity using change-point theory. Neuroimage. 2007;35:1125–41.
Cribben I, Wager TD, Lindquist MA. Detecting functional connectivity change points for single-subject fMRI data. Front Comput Neurosci. 2013;7:143.
Hutchison RM, Womelsdorf T, Allen EA, et al. Dynamic functional connectivity: promise, issues, and interpretations. Neuroimage. 2013;80:360–78.
Killick R, Eckley I. Changepoint: an R package for changepoint analysis. J Stat Softw. 2014;58:1–19.
Piarulli A, Bergamasco M, Thibaut A, et al. EEG ultradian rhythmicity differences in disorders of consciousness during wakefulness. J Neurol. 2016;263:1746–60.
Engemann DA, Raimondo F, King JR, et al. Robust EEG-based cross-site and cross-protocol classification of states of consciousness. Brain. 2018;141:3179–92.
Cimenser A, Purdon PL, Pierce ET, et al. Tracking brain states under general anesthesia by using global coherence analysis. Proc Natl Acad Sci USA. 2011;108:8832–7.
Giacino JT, Kalmar K, Whyte J. The JFK Coma Recovery Scale-Revised: measurement characteristics and diagnostic utility. Arch Phys Med Rehabil. 2004;85:2020–9.
Edlow BL, McNab JA, Witzel T, et al. The structural connectome of the human central homeostatic network. Brain Connect. 2016;6:187–200.
Acknowledgements
We thank the members of the Patient and Family Advisory Board of the Massachusetts General Hospital Laboratory for NeuroImaging of Coma and Consciousness for their feedback and insights regarding the ethical conduct of this clinical trial. We acknowledge Maryam Masood and Zora DiPucchio for their contributions to the regulatory oversight of the trial. We also thank Dr. David A. Schoenfeld for helpful statistical consultation.
Funding
The study was funded by the NIH Director’s Office (DP2HD101400), National Institute of Neurological Disorders and Stroke (K23NS094538, R21NS109627, RF1NS115268), American Academy of Neurology/American Brain Foundation, James S. McDonnell Foundation, Rappaport Foundation, and Tiny Blue Dot Foundation. We also acknowledge support from the National Institute of Neurological Disorders and Stroke Clinical Trial Methodology Course (R25NS088248). This work was conducted with support from Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL 1TR002541) and financial contributions from Harvard University and its affiliated academic healthcare centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic healthcare centers, or the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
BLE: study conception and design; data acquisition, processing, and analysis; data interpretation; drafting the article; final approval of the version to be published. MEB: design of treatment compounding and operating procedures and pharmacokinetic monitoring, critical review of manuscript, final approval of the version to be published. DWZ: design of EEG pharmacodynamic biomarker; data acquisition, processing, and analysis; critical review of manuscript, final approval of the version to be published. ASF: design of statistical plan; critical review of manuscript, final approval of the version to be published. SBS: design of structural MRI predictive biomarker; data acquisition, processing, and analysis; critical review of manuscript, final approval of the version to be published. ZDT: design of functional MRI pharmacodynamic biomarker; data acquisition, processing, and analysis; critical review of manuscript, final approval of the version to be published. SC: design of dynamic functional connectivity-based pharmacodynamic analysis; critical review of manuscript, final approval of the version to be published. JEK: design of functional MRI sequence; data acquisition, processing, and analysis; critical review of manuscript, final approval of the version to be published. SC: design of functional MRI physiologic monitoring; data acquisition, processing, and analysis; critical review of manuscript, final approval of the version to be published. SLM: design of functional MRI physiologic monitoring; data acquisition, processing, and analysis; critical review of manuscript, final approval of the version to be published. TPB: study design; critical review of manuscript, final approval of the version to be published. JJF: study design, ethical guidance, final approval of the version to be published. JTG: study design; critical review of manuscript, final approval of the version to be published. LRH: study design; critical review of manuscript, final approval of the version to be published. KS: development of investigational therapy; study design; critical review of manuscript, final approval of the version to be published. ENB: development of investigational therapy; study design; critical review of manuscript, final approval of the version to be published. YGB: study conception and design; data acquisition, processing, and analysis; data interpretation; drafting the article; final approval of the version to be published.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant conflicts of interest to disclose.
Ethical approval/Informed consent
All biomarker data reported here were obtained with written informed consent provided by healthy control subjects or by surrogate decision-makers for patients with altered consciousness, as part of a separate Institutional Review Board-approved protocol.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article refers to the commentary by https://doi.org/10.1007/s12028-020-01065-4.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Edlow, B.L., Barra, M.E., Zhou, D.W. et al. Personalized Connectome Mapping to Guide Targeted Therapy and Promote Recovery of Consciousness in the Intensive Care Unit. Neurocrit Care 33, 364–375 (2020). https://doi.org/10.1007/s12028-020-01062-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-020-01062-7