
Abstract
Rationale
The cardiovascular effects of psychostimulant drugs (methylphenidate, amphetamine, cocaine) have been mostly associated with their noradrenergic effects. However, there is some evidence that dopaminergic effects are involved in the cardiovascular actions of these drugs. Here, we evaluated this association in humans.
Methods
Positron emission tomography (PET) and [11C]raclopride, a dopamine (DA) D2 receptor radioligand that competes with endogenous DA for occupancy of the D2 receptors, were used to measure changes in brain DA after different doses of intravenous methylphenidate in 14 healthy subjects. Cardiovascular (heart rate and blood pressure) and catecholamine (plasma epinephrine and norepineprhine) responses were determined in parallel to assess their relationships to methylphenidate-induced changes in brain DA.
Results
Methylphenidate administration significantly increased heart rate, systolic and diastolic blood pressures and epinephrine concentration in plasma. The increases in blood pressure were significantly correlated with methylphenidate-induced increases of DA in striatum (r>0.78, P<0.001) and of plasma epinephrine levels (r>0.82, P<0.0005). In turn methylphenidate-induced DA increases in striatum were correlated with increases of epinephrine in plasma (r=0.85, P<0.0001). Subjects in whom methylphenidate did not increase DA had no change in blood pressure or in plasma epinephrine concentration.
Discussion
These results are consistent with the hypothesis that methylphenidate-induced increases in blood pressure are in part due to its central dopaminergic effects. They also suggest that methylphenidate's pressor effects may be in part mediated by DA-induced increases in peripheral epinephrine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Methylphenidate (MP) is the drug of choice for the treatment of attention deficit hyperactivity disorder (ADHD), which is the most common behavioral disorder of childhood (Swanson et al. 1991; Greenhill et al. 2002; Wilens et al. 2002). MP is also effective for the treatment of narcolepsy (Littner et al. 2001) and as an antidepressant in the treatment of medically ill elderly patients (Kaufman et al. 1984; Chiarello and Cole 1987; Rozans et al. 2001). Unfortunately MP also has reinforcing effects, particularly when taken intravenously or when snorted, which can lead to abuse and addiction (Parran and Jasinski 1991).
Among the side effects of MP are its cardiovascular effects; MP increases heart rate (HR) and blood pressure (BP; Ballard et al. 1976; Brown et al. 1984). Though cardiovascular effects are considered clinically insignificant at typical therapeutic doses (Findling et al. 2001), a recent study that did ambulatory pressure monitoring over 24 h reported significant increases in BP with therapeutic doses of MP in children with ADHD (Stowe et al. 2002). Also tachycardia and hypertension have been reported with unintentional overdoses, medication errors, and overdoses caused by abuse or by suicide attempts (Klein-Schwartz 2002). The brain mechanisms underlying the cardiovascular effects of MP have not been well defined. MP blocks the norepinephrine (NE) transporter (Kuczenski and Segal 1997), and the prevailing view is that its cardiovascular effects are mediated primarily through NE stimulation, but some lines of investigation suggest otherwise. First, the cardiovascular actions of cocaine, a drug pharmacologically similar to MP that also blocks dopamine (DA) and NE transporters, are mediated in part by DA (Tella and Goldberg 1998). Second, there is some evidence that DA contributes to cardiovascular regulation via central and peripheral mechanisms (van den Buuse 1998): stimulation of DA cells in the ventral tegmental area (VTA) increases BP, and this effect is antagonized by the DA D2 receptor blocker raclopride, suggesting that DA regulates cardiovascular function. Third, DA modulation of cardiovascular function appears to be in part mediated via its effects on peripheral catecholamines (Mannelli et al. 1997). Based on this literature, we developed the hypothesis that DA effects are also involved in the cardiovascular side effects of MP. Directed by this hypothesis, in this paper we address the relationships between MP-induced increases in brain DA and effects on cardiovascular function and peripheral catecholamine levels.
The effects of intravenous MP on HR and BP and on peripheral NE and epinephrine (EP) concentrations were investigated and related to changes in extracellular DA in striatum (ST). Extracellular DA in ST was measured using positron emission tomography (PET) and [11C]raclopride, a radioligand which competes with DA for binding to DA D2 receptors (Seeman et al. 1989; Dewey et al. 1993). Since [11C]raclopride binding in the human brain is highly reproducible (Volkow et al. 1993), differences in binding between placebo and MP predominantly reflect MP-induced changes in synaptic DA, and consequent changes in availability of DA D2 receptors to the radioligand (Volkow et al. 1994). MP-induced changes in synaptic DA as assessed with PET and [11C]raclopride have been shown to be reproducible when subjects are tested and re-tested on separate occasions (Wang et al. 1999). The data on MP-induced effects on striatal DA were published in a prior paper that evaluated the role of DA on the reinforcing effects of MP (Volkow et al. 1999).
Methods
Subjects
The participants were 14 right-handed healthy subjects (8 males, 6 females, age 33±6 years, mean±SD). Subjects were excluded if they had a present or past history of drug or alcohol abuse or dependence (excluding nicotine/caffeine), as defined in Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV, or a current or past history of psychiatric, neurological, cardiovascular or endocrinological disease. None of the subjects was taking medications at the time of the study. Toxicological drug screens were performed prior to each PET scan. Studies were approved by the Institutional Review Board at Brookhaven National Laboratory and informed consent was obtained from all subjects after procedures were explained.
Scans
The design of the study called for four scans with [11C]raclopride (two scans per day over a 2-day period). However, because of scheduling problems, we completed the four scans only on 9 of the 14 subjects and, in the other 5 subjects, we only completed two scans. The first scan on a given day was after placebo and the second was 127 min later after an i.v. dose of MP established under single-blind conditions. Since the dose-related effects of MP on the measures of this study were not known, we performed a dose-ranging evaluation: subjects were randomly assigned to receive one of four i.v. doses of MP (0.025, 0.1, 0.25, 0.5 mg/kg). Table 1 shows the dose(s) of MP given to each one of the subjects. MP or placebo (3 cc saline) was injected 7 min prior to [11C]raclopride. Scans were done on a CTI-931 PET tomograph (6×6×6.5 mm full width half maximum) and were started immediately after injection of 3.8–10 mCi [11C]raclopride (specific activity 0.5–1.5 Ci/µM at end of bombardment; 2–24 µg injected dose) for a series of 20 emission scans obtained over 60 min as previously described (Volkow et al. 1993). Details on synthesis of [11C]raclopride, subject positioning, transmission and emission scans, arterial blood sampling for radiotracer quantification and metabolite analyses have been published (Volkow et al. 1993). Venous blood was drawn for quantification of plasma concentration of MP prior to and at 27 min and 47 min after MP using capillary GC/mass spectrometry (Srinivas et al. 1991). The plasma samples for MP concentration were lost for one of the subjects.
Cardiovascular and catecholamine measures
Recordings for HR and BP were obtained continuously throughout the placebo and MP scans. Arterial blood samples obtained for the determination of plasma catecholamines were transferred to a tube containing 20 ml of 9% ethylene diamine tetraacetic acid (EDTA) and of 6% glutathione, and the plasma was analyzed for EP and NE. 3,4-Dihydroxybenzylamine (DHBA) was added to plasma as an internal standard, and catecholamines absorbed onto alumina at pH 8.5 and eluted with perchloric acid (PCA; 0.1 M). The processed samples were quantified by high-performance liquid chromatography (HPLC) using a catecholamine column and an electrochemical detector (Bioanalytic Systems, West Lafayette, IN). Using this system, the detection limit for EP, NE and DHBA was 10 pg.
Image analysis and modeling
Regions of interest (ROI) were outlined for ST and cerebellum (CB), as previously described(Volkow et al. 1993). Briefly, ROI were initially outlined on the individual's summed baseline [11C]raclopride image (images obtained between 15 min and 54 min) and were then projected into the dynamic [11C]raclopride images to generate time activity curves for ST and CB. These time activity curves for tissue concentration along with the time activity curves for unchanged tracer in plasma were used to calculate [11C]raclopride's distribution volumes (DVs), which correspond to the equilibrium measurement of the ratio of tissue concentration to plasma concentration, in ST and CB using a graphical analysis technique for reversible systems (Logan et al. 1990). The ratio of DV in ST to that of DV in CB corresponds to (Bmax/Kd)+1 and is insensitive to changes in cerebral blood flow (Logan et al. 1994). The response to MP was quantified as the difference in Bmax/Kd between placebo and MP and expressed as percentage change from baseline [(Bmax/Kdplacebo−Bmax/KdMP)/Bmax/Kdplacebo]×100.
Data analysis
Differences in Bmax/Kd, the cardiovascular and the catecholamine measures between placebo and MP were separately tested for each one of the MP doses with paired t-tests. To assess the effects of MP on peripheral EP and NE concentrations, we obtained the area under the curve from time of injection up to 60 min. For the cardiovascular measures, we averaged the eight scores that were obtained between 4 min and 12 min, which were the time periods when peak effects for MP occurred and compared them against the baseline measures (eight scores obtained 1 h prior to MP injection). Pearson product moment correlation analyses were calculated between the changes in Bmax/Kd and the cardiovascular changes (% change from placebo). On those correlations that were found to be significant, we performed partial correlation analysis to determine whether the correlations remained significant after removing the contribution of dose and of concentration of MP in plasma (Kirk 1990).
Results
Due to scheduling and technical problems, not all subjects completed the four scans. Nine subjects were tested twice as planned, but five subjects were tested only once, so a total of 23 brain DA measures (PET raclopride studies) were performed (Table 1). Peripheral catecholamine measures were obtained for 17 of the 23 scans. The number of brain DA and peripheral catecholamine measures for each MP dose is shown in Table 2, and Table 3 shows the plasma MP concentrations achieved for the different doses.
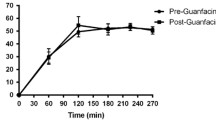
Overall, MP significantly increased HR and systolic and diastolic BPs, with maximum effects (in general) at the 0.25-mg/kg dose (Fig. 1). For HR, the increases were about the same magnitude and significant for the 0.5-mg/kg (55±35%, t=5, df 6, P<0.003) and 0.25-mg/kg (55±43%, t=3.5 df 7, P<0.01) doses, smaller (about half the magnitude) and not statistically significant for the 0.1-mg/kg (27±27%), and negligible for the 0.025-mg/kg (4±4%) doses (Fig. 1). The same pattern held for BP. Systolic BP increases were statistically significant for the 0.5-mg/kg (22±6%; t=9, df 6, P<0.0001) and 0.25-mg/kg (25±9%; t=9, df 7, P<0.0001) doses, but not for the 0.1-mg/kg (6±7%) and the 0.025-mg/kg (0±3%) doses. Also, diastolic BP increases were statistically significant for the 0.5-mg/kg (22±9%; t=7.5, df 6, P<0.0003) and the 0.25-mg/kg (20±4%; t=16, df 7, P<0.0001) doses but not for the 0.1-mg/kg (9±5%) and negligible for the 0.025-mg/kg (0±5%) doses.

Temporal course for the effects of the various doses of intravenous methylphenidate on heart rate and systolic and diastolic blood pressures. Increases were significant for the 0.5-mg/kg and 0.25-mg/kg doses
Increases in EP concentrations in plasma were statistically significant for the 0.5-mg/kg and the 0.25-mg/kg doses but not for the 0.1-mg/kg and 0.025-mg/kg doses. MP effects in NE were not statistically significant (Table 3).
MP decreased Bmax/Kd for the 0.5-mg/kg (19±9%, t=4.6, df 6, P<0.004) and the 0.25-mg/kg (15±11%, t=3.6, df 7, P<0.01) doses but not for the 0.1-mg/kg (12±7%, t=2.7, df 2, P<0.11) and 0.025-mg/kg (0±6%) doses. The correlations of the DA changes with the other dependent measures revealed some statistically significant effects. MP-induced changes in DA (% change in Bmax/Kd from placebo) were significantly correlated with systolic (r=0.68, df 22, P<0.0004) and diastolic (r=0.58, df 22, P<0.004) BPs but not with HR (r=0.27, df 1,22, P=0.21) (Fig. 2). MP-induced changes in DA were also correlated with MP-induced changes in EP (r=0.85, df 14, P<0.0001) but not with NE (r=0.36, df 15, P=0.18) (Fig. 3). After partialing out for the concentration of MP in plasma and for MP doses, the correlations remained significant for systolic (r=0.638; df 18, P<0.003) but not for diastolic (r=0.378; df 18, P<0.10) BPs and were also significant for EP (r=0.72, df 11, P<0.006).

Regression lines for the correlation between methylphenidate-induced changes in dopamine D2 receptor availability in striatum and methylphenidate-induced increases in heart rate (r=0.27, n.s.), systolic pressure (r=0.68, P<0.0005) and diastolic pressure (r=0.58, P<0.005)

Regression lines for the correlation between methylphenidate-induced changes in dopamine D2 receptor availability in striatum and methylphenidate-induced increases in epinephrine (r=0.85, P<0.0001) and norepinephrine (r=0.36, n.s.)
Because the 23 DA measures were not independent since 9 subjects were tested twice (5 subjects only once), we tested separate correlations that included only the first brain DA measure for each subject (total of 14 data points). The same results were obtained and the correlations with DA changes were significant for systolic (r=0.79, df 14, P<0.001) and diastolic (r=0.76, df 14, P<0.002) BPs and for EP (r=0.88, df 9, P<0.001).
The correlation between MP-induced changes in EP and changes in the cardiovascular measures were significant for systolic (r=0.82, df 14, P<0.0002) and diastolic (r=0.86, df 15, P<0.0001) BPs but not for HR (r=0.4, df 14, P=15) (Fig. 3). Similar results were obtained when the correlations were done using only one data point per subject and corresponded for the correlation between changes in EP and systolic BP to r=0.86 (df 9, P<0.002) and diastolic BP to r=0.91 (df 9, P<0.0005).
Discussion
The positive correlation between MP-induced increases in DA and increases in systolic BP is consistent with the hypothesis that this cardiovascular effect of MP might be partially mediated by central DA effects. The fact that this correlation remained significant after partialing out for dose effects and the concentration of MP in plasma suggests that this is not a spurious correlation reflecting an association between higher doses and greater increases in BP. Instead, after adjusting for dose, the covariation between DA and systolic BP should be due to individual differences in responsivity to MP. If an individual failed to show an increase in DA to even a high dose of MP, then that individual tended under the same conditions to lack response on the physiological response of systolic BP.
Evidence for the relevance of DA to the pressor effects of other stimulant drugs has been previously documented. Cocaine, which is a drug pharmacologically similar to MP (Volkow et al. 1995; Rush and Baker 2001), increases BP and HR when administered to rodents. These effects have been shown to be antagonized by the DA D2 receptor blocker eticlopride (Tella 1996). Similarly, the DA-selective reuptake inhibitor, GBR12909, also increased BP and HR and eticlopride also blocked these effects indicating that DA mediates the cardiovascular effects of these drugs through DA D2 receptors. This is likely to be a central effect of DA since the peripheral DA D2 antagonist drug domperidone did not block the cardiovascular effects of these DA enhancing drugs. Similarly, human studies have indicated that increases in BP induced by amphetamine have a DA component since they are attenuated by the DA D2 receptor antagonist haloperidol (Angrist et al. 1974, 2001). Also stimulation of the VTA has been shown to increase BP, which is an effect antagonized by the DA D2 receptor antagonist raclopride (Cornish et al. 1997; van den Buuse 1997). Though most studies have investigated the effects of DA D2 receptor agonists and antagonist drugs on stimulant's cardiovascular effects there is also evidence that D1 receptors are involved (Schindler et al. 2002). Overall, these findings point to an involvement of DA in the pressor responses to stimulant drugs and are consistent with studies documenting an involvement of DA in cardiovascular regulation (van den Buuse 1998). Though preclinical studies have provided evidence of the involvement of central DA in regulating stimulant induced increases in HR (Tella 1996), we did not observe a correlation between MP-induced increases in DA and HR. The reason(s) for this discrepancy are unclear but could reflect a differential involvement of DA-regulated brain regions or mechanism(s) regulating HR and BP. For example, the mesolimbic DA system via the VTA potentiates the effects of vasopressin on mean arterial pressure but not in HR (van Den Buuse and Catanzariti 2000). Since the PET raclopride method can only measure MP-induced changes in ST, we cannot assess the role of non-striatal DA regulated regions in MP-induced cardiovascular effects. It is also possible that the failure to observe a correlation between central DA and HR could reflect peripheral adaptation responses that counteract the central effects on cardiovascular function. For the case of amphetamine, it has been suggested that peripheral baroreceptor responses interfere with its effects in HR but not in BP (Angrist et al. 2001).
The results from the present study are in agreement with previous studies documenting increases in EP but not in NE after intravenous MP (Joyce et al. 1984). Here we show that MP-induced increases in EP were associated with DA changes. Evidence that increases in EP induced by drugs that block the DAT were in part mediated by a centrally mediated effect of DA had previously been shown for cocaine and for GBR12909 for which the EP increases were blocked by eticlopride but not by domperidone (Tella 1996). However, there is also evidence that DA modulates EP release from the adrenals in part through peripheral DA D2 receptors (Kujacic et al. 1995). Because the increases in EP were also correlated with MP's pressor effects, we speculate that MP-induced increases in BP are indirectly mediated by DA-induced EP release. However, because MP also blocks the NE transporter, we cannot rule out the possibility that MP-induced increases in EP are due to its central NE effects (Kuczenski and Segal 1997). Furthermore while the changes in DA induced by MP accounted for a significant proportion of the variability in the pressor responses (r2=46%), this does not account for it completely.
The association of DA changes was significant for MP-induced changes in EP but not NE. The sources of these two peripheral measures of catecholamines differ, EP mostly reflects release from the adrenals while NE in plasma is considered a spillover from sympathetic nervous system activation at the tissue level (Ganong 2001). Thus, we speculate that the exclusive relationship of DA with the MP-induced rise in EP levels and BP is a reflection of DA-induced sympatho-adrenal stimulation, which in turn mediates the pressor response.
Although this study evaluated the effects of MP, the possibility that similar mechanisms underlie the cardiovascular effects elicited by other stimulant drugs such as amphetamine and cocaine, which also increase DA and NE in brain should not be excluded.
There are several limitations that should be considered for this study. First, there was considerable missing data due to scheduling and technical problems. This may be inherent to studies that require multiple PET scans and administration of psychoactive drugs. Also the expense of the PET scans poses a limitation to the total number of subjects that can be studied. Second, the PET DA D2 radioligand competition method provides only a relative estimate of changes in DA concentration that underestimates the actual magnitude of the DA changes (Laruelle 2000). In fact it has been estimated that a 1% decrease in [11C]raclopride binding corresponds to at least an eightfold increase in extracellular DA (Breier et al. 1997). Third, we used correlations to try to understand the underlying relationships between MP-induced changes in brain DA and changes in HR, BP and catecholamine levels. Significant correlations do not identify causative factors, but do provide candidates that can be evaluated in light of the literature and additional factors. However, the conclusions based on correlations are always tentative and provide a piece in the puzzle not a definitive answer about complex relationships. Fourth, this study was initially designed to assess the role of DA on the reinforcing effects of MP (Volkow et al. 1999) and thus the design was not optimal to evaluate its role in its cardiovascular effects.
In summary, this study provides the first documentation of a significant relationship between increases in brain DA and the increases in BP in response to MP in human subjects. These results are consistent with the hypothesis that DA plays a role in the cardiovascular responses to MP and suggest that DA, a neurotransmitter that regulates locomotor activity, cognition and reward (Le Moal and Simon 1991), may also be involved in maintaining cardiovascular homeostasis in humans.
References
Angrist B, Lee HK, Gershon S (1974) The antagonism of amphetamine-induced symptomatology by a neuroleptic. Am J Psychiatry 131:817–819
Angrist B, Sanfilipo M, Wolkin A (2001) Cardiovascular effects of 0.5 milligrams per kilogram oral d-amphetamine and possible attenuation by haloperidol. Clin Neuropharmacol 24:139–144
Ballard JE, Boileau RA, Sleator EK, Massey BH, Sprague RL (1976) Cardiovascular responses of hyperactive children to methylphenidate. JAMA 236:2870–2874
Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D (1997) Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci USA 94:2569–2574
Brown RT, Wynne ME, Slimmer LW (1984) Attention deficit disorder and the effect of methylphenidate on attention, behavioral, and cardiovascular functioning. J Clin Psychiatry 45:473–476
Chiarello RJ, Cole JO (1987) The use of psychostimulants in general psychiatry. A reconsideration. Arch Gen Psychiatry 44:266–295
Cornish JL, Wilks DP, Van den Buuse M (1997) A functional interaction between the mesolimbic dopamine system and vasopressin release in the regulation of blood pressure in conscious rats. Neuroscience 81:69–78
Dewey SL, Smith GS, Logan J, Brodie JD, Fowler JS, Wolf AP (1993) Striatal binding of the PET ligand 11C-raclopride is altered by drugs that modify synaptic dopamine levels. Synapse 13:350–356
Findling RL, Short EJ, Manos MJ (2001) Short-term cardiovascular effects of methylphenidate and adderall. J Am Acad Child Adolesc Psychiatry 40:525–529
Ganong WF (2001) Review of medical physiology, 12th edn. McGraw-Hill, New York
Greenhill LL, Pliszka S, Dulcan MK, Bernet W, Arnold V, Beitchman J, Benson RS, Bukstein O, Kinlan J, McClellan J, Rue D, Shaw JA, Stock S (2002) American academy of child and adolescent psychiatry. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 41:26S–49S
Joyce PR, Nicholls MG, Donald RA (1984) Methylphenidate increases heart rate, blood pressure and plasma epinephrine in normal subjects. Life Sci 34:1707–1711
Kaufman MW, Cassem NH, Murray GB, Jenike M (1984) Use of psychostimulants in medically ill patients with neurological disease and major depression. Can J Psychiatry 29:46–49
Kirk RE (1990) Statistics: an introduction. Holt, Rinehart, and Winston, Fort Worth
Klein-Schwartz W (2002) Abuse and toxicity of methylphenidate. Curr Opin Pediatr 14:219–223
Kuczenski R, Segal DS (1997) Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. Neurochemistry 68:2032–2037
Kujacic M, Hansson LO, Carlsson A (1995) Acute dopaminergic influence on plasma adrenaline levels in the rat. Eur J Pharmacol 273:247–257
Laruelle M (2000) Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab 20:423–451
Le Moal M, Simon H (1991) Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev 71:155–234
Littner M, Johnson SF, McCall WV, Anderson WM, Davila D, Hartse SK, Kushida CA, Wise MS, Hirshkowitz M, Woodson BT (2001) Standards of practice committee. Practice parameters for the treatment of narcolepsy: an update for 2000. Sleep 24:451–466
Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schlyer D, MacGregor RR, Hitzemann R, Bendriem B, Gatley SJ, Christman DR (1990) Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(1)-cocaine PET studies in human subjects. J Cereb Blood Flow Metab 10:740-747
Logan J, Volkow ND, Fowler JS, Wang G-J, Dewey SL, MacGregor R, Schlyer D, Gatley SJ, Pappas N, King P, Hitzemann R, Vitkun S (1994) Effects of blood flow on [11C] raclopride binding in the brain: model simulations and kinetic analysis of PET data. J Cereb Blood Flow Metab 14:995–1010
Mannelli M, Lazzeri C, Ianni L, La Villa G, Pupilli C, Bellini F, Serio M, Franchi F (1997) Dopamine and sympathoadrenal activity in man. Clin Exp Hypertens 19:163–179
Parran TV, Jasinski DR (1991) Intravenous methylphenidate abuse: prototype for prescription drug abuse. Arch Intern Med 151:781–783
Rozans M, Dreisbach A, Lertora JJ, Kahn MJ (2002) Palliative uses of methylphenidate in patients with cancer: a review. J Clin Oncol 20:335–339
Rush CR, Baker RW (2001) Behavioral pharmacological similarities betwen methylphenidate and cocaine in cocaine abusers. Exp Clin Psychopharmacol 9:59–73
Schindler CW, Gilman JP, Bergman J, Mello NK, Woosley RL, Goldberg SR (2002) Interactions between cocaine and dopamine agonists on cardiovascular function in squirrel monkeys. J Pharmacol Exp Ther 300:180–187
Seeman P, Guan HC, Niznik HB (1989) Endogenous dopamine lowers the dopamine D2 receptor density as measured by 3H raclopride: implications for positron emission tomography of the human brain. Synapse 3:96–97
Srinivas NR, Hubbard JW, Quinn D, Korchinski ED, Midha K (1991) Extensive and enantioselective presystemic metabolism of dl-threo-methylphenidate in humans. Prog Neuropsychopharmacol Biol Psychiatry15:213–220
Stowe CD, Gardner SF, Gist CC, Schulz EG, Wells TG (2002) 24-hour ambulatory blood pressure monitoring in male children receiving stimulant therapy. Ann Pharmacother 36:1142–1149
Swanson JM, Cantwell D, Lerner M, McBurnett K, Hanna G (1991) Effects of stimulant medication on learning in children with ADHD. J Learn Disabil 24:219–230
Tella SR (1996) Possible novel pharmacodynamic action of cocaine: cardiovascular and behavioral evidence. Pharmacol Biochem Behav 54:343–354
Tella SR, Goldberg SR (1998) Monoamine transporter and sodium channel mechanisms in the rapid pressor response to cocaine. Pharmacol Biochem Behav 59:305–312
van den Buuse M (1997) Pressor responses to brain dopaminergic stimulation. Clin Exp Pharmacol Physiol 24:764–769
van den Buuse M (1998) Role of the mesolimbic dopamine system in cardiovascular homeostasis. Stimulation of the ventral tegmental area modulates the effect of vasopressin on blood pressure in conscious rats. Clin Exp Pharmacol Physiol 25:661–668
van den Buuse M, Catanzariti R (2000) Stimulation of the ventral tegmental area enhances the effect of vasopressin on blood pressure in conscious rats. Br J Pharmacol 129:29–36
Volkow ND, Fowler JS, Wang G-J, Dewey SL, Schlyer D, MacGregor R, Logan J, Alexoff D, Shea C, Hitzemann R, Angrist B, Wolf AP (1993) Reproducibility of repeated measures of 11C raclopride binding in the human brain. J Nucl Med 34:609–613
Volkow ND, Wang G-J, Fowler JS, Logan J, Schlyer D, Hitzemann R, Lieberman J, Angrist B, Pappas N, Mac Gregor R, Burr G, Cooper T, Wolf AP (1994) Imaging endogenous dopamine competition with [11C]raclopride in the human brain. Synapse 16:255–262
Volkow ND, Ding Y-S, Fowler JS, Wang GJ, Logan J, Gatley SJ, Dewey SL, Ashby C, Lieberman J, Hitzemann R, Wolf AP (1995) Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in human brain. Arch Gen Psychiatry 52:456–463
Volkow ND, Wang G-J, Fowler JS, Logan J, Gatley SJ, Wong C, Hitzemann RJ, Pappas N (1999) Reinforcing effects of psychostimulants in humans are associated with increases in brain dopamine and occupancy of D2 receptors. J Pharmacol Exp Ther 291:409–415
Wang G-J, Volkow ND, Fowler JS, Logan J, Pappas NR, Natusil N, Wong CT, Hitzemann RJ (1999) Reproducibility of repeated measures of endogenous dopamine competition with [C-11]raclopride in the human brain. J Nucl Med 40:1285–1291
Wilens TE, Spencer TJ, Biederman J (2002) A review of the pharmacotherapy of adults with attention-deficit/hyperactivity disorder. J Atten Disord 5:189–202
Acknowledgements
This research was supported in part by the U.S. Department of Energy under contract DE-ACO2–76CH00016 and NIDA grants DA09490 and DA06278. We wish to thank David Schlyer, for Cyclotron operations; Donald Warner for PET operations; Colleen Shea, Robert MacGregor, Victor Garza, Richard Ferrieri and Payton King for radiotracer preparation and analysis; Thomas P Cooper for methylphenidate plasma analysis and Noelwah Netusil and Pauline Carter for patient care.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Volkow, N.D., Wang, GJ., Fowler, J.S. et al. Cardiovascular effects of methylphenidate in humans are associated with increases of dopamine in brain and of epinephrine in plasma. Psychopharmacology 166, 264–270 (2003). https://doi.org/10.1007/s00213-002-1340-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-002-1340-7