
Abstract
Background
Spreading depolarization events following ischemic and traumatic brain injury are associated with poor patient outcome. Currently, monitoring these events is limited to patients in whom subdural electrodes can be placed at open craniotomy. This study examined whether these events can be detected using intra-cortical electrodes, opening the way for electrode insertion via burr hole.
Methods
Animal work was carried out on adult Sprague–Dawley rats in a laboratory setting to investigate the feasibility of recording depolarization events. Subsequently, 8 human patients requiring craniotomy for traumatic brain injury or aneurysmal subarachnoid hemorrhage were monitored for depolarization events in an intensive care setting with concurrent strip (subdural) and depth (intra-parenchymal) electrode recordings.
Results
(1) Depolarization events can be reliably detected from intra-cortically placed electrodes. (2) A reproducible slow potential change (SPC) waveform morphology was identified from intra-cortical electrodes on the depth array. (3) The depression of cortical activity known to follow depolarization events was identified consistently from both intra-cortical and sub-cortical electrodes on the depth array.
Conclusions
Intra-parenchymally sited electrodes can be used to consistently identify depolarization events in humans. This technique greatly extends the capability of monitoring for spreading depolarization events in injured patients, as electrodes can be sited without the need for craniotomy. The method provides a new investigative tool for the evaluation of the contribution of these events to secondary brain injury in human patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) [1, 2] and stroke (including sub-arachnoid hemorrhage (SAH), ischemic stroke, and intra-parenchymal brain hemorrhage) are important causes of mortality and morbidity worldwide [3]. An incomplete understanding of the pathophysiological responses to brain injuries such as these has slowed the development of effective management strategies in the neurocritical care setting. Brain injuries are no longer seen as single events but as dynamic, multifactorial processes involving a complex interplay of; structural damage [4], regional ischemia [5], inflammation [6], excitotoxicity [7], and metabolic crisis [8]. Management must reflect this dynamism and become individualized and focused on the various contributing causes of secondary brain injury [9].
An increasingly recognized link in this chain of injury mechanisms is the phenomenon of cortical spreading depolarization (SD)—a potentially modifiable therapeutic target. Spreading depolarizationsFootnote 1 are waves of sustained mass neuronal and astrocytic depolarization which originate spontaneously and propagate across the cerebral cortex in response to structural or ischemic brain injury. They are characterized on electrocorticography (ECoG) by a large negative slow potential change (SPC) followed by silencing of the electrical activity of the cortex (spreading depression). Spreading depolarizations occur frequently in patients undergoing craniotomy for aneurysmal SAH [10], TBI [11], malignant hemispheric stroke [12], and spontaneous intracerebral hematoma [13]. Repeated passage of SDs frequently results in vasoconstriction and oligemia [14], swelling and distortion of dendritic spines [15], and progressive cortical glucose depletion [16]. In patients with aneurysmal SAH, repetitive depolarizations with prolonged depression periods are associated with development of delayed ischemic strokes [14]. Spreading depolarization occurrence has been definitively linked to poor outcome in a recent study of the clinical significance of SD in TBI [17].
To our knowledge, we were the first group to reliably record SD in the context of human brain injury—by placement of subdural ECoG electrode strips directly onto the cortex at completion of emergency craniotomy [18]. Although very effective for this patient group, the requirement for craniotomy has restricted the number of patients who can be monitored for SD. In this collaborative study between two research centers, we investigated the detection of SD with a depth electrode array inserted into the brain parenchyma beside a subdural electrode strip (as “gold standard”) placed on the cortical surface at open craniotomy. These results were compared with those obtained from an animal model.
Detection with a depth electrode array would potentially allow for monitoring and recording of SD via burr hole access to the cortex. While this method is most immediately applicable to traumatic brain injury (where intraparenchymal monitoring via burr hole access is already standard of care), such a monitoring method would also allow screening for SD without the need for craniotomy in other pathologies such as (SAH) and stroke.
Materials and Methods
Human Studies of Depth Electrodes
Participants
Eight patients (age range 24–79 years) requiring craniotomy for traumatic brain injury (n = 6) or aneurysmal subarachnoid hemorrhage (n = 2) were enrolled in the Co-Operative Studies of Brain Injury Depolarizations (COSBID) at King’s College Hospital (London, UK). Inclusion criteria were the clinical need for neurosurgery for lesion evacuation, decompression, or aneurysm clipping, surgery within 7 days of injury, and age 18–80 years. Patients with Glasgow Coma Score (GCS) of three with fixed dilated pupils were excluded. As patients were comatose at time of inclusion, assent for study participation was obtained from legally authorized representatives. Consent was obtained from the patients themselves once capacity had been re-established at follow up. All study data were anonymized and securely stored. The study received ethical approval from the institutional review board and research was conducted in accordance with the Declaration of Helsinki.
Protocol
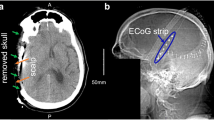
At the conclusion of surgery, a subdural electrode strip (TS06R-AP10X-0W6 Adtech, Racine, WI) was placed on the surface of the cortex. The strip consisted of a single row of 6 platinum contacts spaced at 10 mm intervals between centers. The contacts were embedded in a 0.7-mm-thick polyurethane strip which overlapped the edges of the contacts leaving a diameter of 2.3 mm exposed, recessed approximately 0.1 mm from the surface. In addition, a Spencer depth electrode array (SD08R-AP58X-000 Adtech, Racine, WI, USA) was inserted into the brain parenchyma at the apex of the gyrus adjacent to the subdural strip (Fig. 1A). The flexible depth array was 1.1 mm in diameter and consisted of eight cylindrical 1.3-mm-long platinum contacts spaced consecutively with 2.2 mm between electrode centers. Insertion was perpendicular or somewhat oblique to the cortical surface and was targeted for placement of at least two electrode contacts in the cortical mantle (Fig. 1B). In TBI patients, strip and depth electrode arrays were placed in cortical regions estimated at surgery to be pericontusional penumbra. In cases where no contused brain was seen at craniotomy or in the case of SAH, attempts were made to place strip electrodes along the course of a single gyrus in the affected territory.

A Intra-operative photograph, showing a Intraparenchymal placement of depth electrode array b Placement of subdural ECoG strip on surface of cortex. B Head CT following decompressive craniectomy showing a Intraparenchymal placement of depth electrode array b Cortical surface contacts of strip electrode array
Following surgery, probes were connected to monitors in the intensive care unit (ICU) and continuous data acquisition commenced. Electrocorticography data from subdural strip and depth electrodes were acquired through Octal bioamplifiers (ADInstruments, Sydney, Australia) with a lower frequency limit of 0.02 Hz and sampled at 200 Hz with two 16-channel Powerlab analog-to-digital converters (ADInstruments, Sydney, Australia). Subdural strip ECoG was acquired in a sequential bipolar chain configuration, an established montage for SD detection [18], and depth electrode ECoG was acquired in a common reference configuration. A Ag/AgCl scalp electroencephalographic (EEG) cup electrode placed over the contralateral mastoid process served as a far-field reference, and the same electrode type placed on the ipsilateral mastoid served as ground. These scalp electrodes were applied following skin preparation with abrasive gel (Bio-medical, NuPrep) and were secured using adhesive collodion (SLE). Conductive Electrolux gel (Unimed) was used to maintain electrical contact between the skin and the electrode and was re-applied daily to maintain a high signal-to-noise ratio. For retrospective data representation (including all figures in this manuscript) an average reference was derived from all functioning depth channels and subtracted from each raw data channel to give a pseudo-average reference array: this reduced reference noise and accentuated cortical depression.
Electrocorticography data were recorded with LabChart software version 7.2 (ADInstruments, Sydney, Australia). Data from a small head-mounted accelerometer (ACL300 Biometrics Ltd Newport, UK) used to monitor patient movement were also registered time-locked to the ECoG data using PowerLab/LabChart. These data assist in ECoG review since patient movements are a source of low-frequency artifact. All data were acquired continuously in the ICU for a maximum of 7 days and patients were treated according to standard brain injury protocols. After monitoring, both strip and depth electrodes were removed at the bedside using gentle traction. There were no complications associated with placement or removal of either electrode type.
SD Waveform Analysis
The characteristics of SDs in depth electrode recordings were quantified by various measures. For multiphasic SPCs occurring at frequencies <0.1 Hz, the duration of the initial negative phase was measured from the start of deviation from the baseline level (zero) until the return to baseline. Subsequent phases were measured similarly as the time between crossings of the baseline zero level (see section on SD waveform morphology in human patients). The spreading depression of high-frequency (0.5–30 Hz) spontaneous activity was measured by computing a leaky integral of the total power with a 60-s decay time constant [19]. This integral of a moving window of only recent activity allows the value to remain constant through time, rather than increase, when total cortical power remains constant, and to decrease or increase from this steady level when cortical activity changes. Depression durations were, therefore, measured from the start of decrease of the power integral to the start of its recovery. Speed of onset of high-frequency (HF) ECoG depression at a given contact was measured as the slope of the integral immediately following the onset of the depression. Slope was calculated as follows: the integral value 4 min after the start of the depression period was subtracted from the baseline integral value and the difference was normalized to the baseline level to determine the relative change, expressed in percentage decrease per min:
All integral slopes were negative indicating a consistent drop in high-frequency power associated with the SD wave.
Animal Studies
Six male Sprague–Dawley rats weighing 325–400 g (Harlan Laboratories Inc.) were used in the experiments. Animals were housed in a 12-h light/dark cycle with food and water ad libitum according to standards of the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal procedures and care were approved by the University of Cincinnati Institutional Animal Care and Use Committee in accordance with the Animal Welfare Act and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council).
Anesthesia was induced with 5 % isoflurane delivered in 100 % oxygen and maintained at 2.5 % throughout surgery, and core body temperatures were maintained at 37 °C by a homeothermic heating blanket. Rats were mounted in a stereotaxic frame and the skull was exposed by midline scalp incision with retraction of the right temporal muscle. A large craniotomy was made (~10 × 5 mm), extending from midline to the lateral ridge and from lambda to bregma, for placement of electrodes. A second smaller craniotomy (2 mm2) was made 4 mm anterior and 2 mm lateral of bregma, for induction of SD. Small incisions were made in the dura at both craniotomies and the exposed brain was kept moist with physiologic saline. The femoral vein was cannulated for subsequent delivery of 20–30 mg/kg sodium pentobarbital during recordings, since pentobarbital does not affect SD waveforms [20].
A Spencer depth electrode array, identical to those used clinically (1.1 mm diameter, 1.3 mm length, and 0.88 mm inter-electrode distance), was inserted in the large craniotomy window so that two contacts resided in the parenchyma (n = 6). The superficial contact was positioned entirely in cortex while the deeper contact was subcortical (Fig. 2A). In three rats, a single platinum disk electrode, also identical to those used in clinical recordings (2.3 mm diameter exposed contact), was additionally placed on the dura in the large craniotomy. In all rats, a glass micropipette (2–3-μm tip diameter, 2–3 MΩ) was placed at a cortical depth of 300 μm adjacent to the clinical electrodes for comparison. A platinum sub-dermal needle (Grass Technologies, West Warwick, RI) served as reference and an Ag/AgCl sub-dermal wire electrode (Ives EEG Solutions, Inc., Newburyport, MA) served as ground.

Depth electrode recordings of SD in the rat. A Upper: Schematic of a Spencer depth electrode in the rat brain. One recording site of the depth electrode was positioned in cortex (black) while the other was sub-cortical (gray). Middle: Representative traces of DC recordings of an SD on cortical (black) and sub-cortical (gray) depth electrodes. Lower: Same recordings after applying a 0.01-Hz high-pass digital filter to simulate clinical amplifiers. * p < 0.05. B Upper: Mean (±SEM, n = 6) amplitude of the negative DC shift or SPC. Lower: Mean (±SEM, n = 6) duration of the negative DC shift or SPC
Single SDs were induced by 30-s application of 1 M KCl-soaked cotton ball to the frontal cortex, and applications were repeated every 20 min to record multiple SDs. Full-band signals were recorded with a direct current-coupled amplifier (g.USBamp, Guger Technologies, Graz, Austria) and exported to LabChart (ADInstruments, New South Wales, Australia) for analysis. Three SD from each rat were averaged for data analysis, and unpaired two-tailed t-tests were used for statistical comparisons. Animals were euthanized at the end of experiments for histologic processing and visualization of the electrode track.
Results
Animal Studies of Depth Electrodes
To determine the feasibility of depth electrode monitoring, we first characterized depth electrode recordings of SD in rat cerebral cortex in comparison to traditional recordings with glass micropipettes. Application of KCl to the frontal cortex (30 s) reliably produced a single SD wave that spread to the recording electrodes in parietal cortex and was recorded as a negative shift in the DC potential (Fig. 2A, middle) and spreading depression of spontaneous activity. DC shifts recorded by the platinum depth electrode located in cortex were 5.1 mV (±0.7) and 81.5 s (±6.4) in duration. These were considerably smaller in amplitude and longer in duration than recordings from micropipettes (20.2 ± 1.5 mV, 46.7 ± 2.9 s, p < 0.05, Fig. 2B). Each SD confirmed by the micropipette was also reliably measured on the cortical depth electrode.
A second electrode on the depth array located subcortically also registered DC shifts, but with significantly attenuated amplitudes (2.0 ± 0.1 mV, Fig. 2A, middle, p < 0.05 compared to cortical depth electrode). However, the spreading depression of spontaneous activity induced by SD had similar durations between the deeper sub-cortical electrode (269.8 ± 34.6 s) and cortical depth electrode (279.4 ± 26.6 s).
We also compared depth electrode recordings of SD in the rat to those made with subdural surface disk electrodes, the gold standard for clinical ECoG monitoring. Morphologies of the SD waves were similar between the cortical surface (not shown) and depth electrodes, with no significant differences in the negative DC shift amplitude (surface: 6.8 ± 0.7; depth: 5.1 ± 0.6 mV) or duration (surface: 68.7 ± 9.0; depth: 81.5 ± 6.4 s). Thus, cylindrical depth electrodes located in cortex are capable of recording SD in the rat with waveform characteristics similar to those from cortical surface disk electrodes. Depth electrodes placed deep to the cortex experience reduced DC signal amplitude, but reliably detect spreading depression of spontaneous activity. To simulate the AC-coupled human data a 0.01-Hz high-pass digital filter was applied (Fig. 2A, lower). Peak-to-peak amplitudes of these waveforms did not significantly differ from negative DC shift amplitudes (Fig. 2B, upper; AC: 3.9 ± 0.7; DC: 5.1 ± 0.7 mV), and durations of the negative–positive phases generally approximated the negative DC shift durations (Fig. 2B, lower; AC: 96.5 ± 2.9; DC: 81.5 ± 6.4 s).
SD Waveform Morphology in Human Depth Recordings
A total of eight patients underwent ECoG monitoring with both subdural strip and intraparenchymal depth electrode arrays. Patients were monitored for at least 2 and up to 7 days, and a total of 405 h of data was obtained. To avoid data loss from DC drift over these long timescales the amplifiers were AC coupled with a 0.02-Hz high-pass cut-off frequency. In visual review of depth electrode recordings by two recognized experts in the field (JH and MF), multiple SDs were readily and easily identified by the same hallmark characteristics as described for subdural strip recordings (Fig. 3): (1) coincident onset of multiphasic shifts of the slow potential and depression of high-frequency (0.5–30 Hz) spontaneous activity; (2) rapid onset (<30 s) of depression followed by, after a variable period of 2–10 + min, gradual recovery of spontaneous activity marked by slow/sharp wave discharges of increasing frequency; and (3) spread of the SPC and depression between adjacent electrodes of the depth array (Fig. 3; also see below). Blinded assessment of these recordings by both expert and minimally trained non-expert reviewers revealed a superior positive predictive value (PPV) and sensitivity (Sens.) for the detection of SD events in depth electrode data. PPV = 1 versus 0.91, Sens. = 0.94 versus 0.83 for expert reviewers. PPV = 0.85 versus 0.73, Sens. = 0.76 versus 0.64 for non-expert reviewers. The negative predictive value (NPV) and specificity (Spec.) were effectively equal for depth and strip data in both expert and non-expert groups (see supplementary materials for a detailed discussion of analysis).

Concurrent recording from strip electrode and depth electrode arrays. The same 3 depolarization events are recorded. a Bipolar configuration of strip electrodes means that SPCs appear with opposite polarity in adjacent unfiltered ECoG channels. b Sequential decrease in cortical activity on the bandpass filtered (BPF) (0.5–30 Hz) data channels. c Monopolar configuration of depth electrodes means that SPCs appear with the same polarity in adjacent channels. d Narrowing of BPF (0.5–30 Hz) depth data bands confirms a reduction in cortical activity
A triphasic morphology was observed in the waveform of clinical depth electrode SPCs. This consisted of an initial negative deflection followed by a more rounded and prolonged positive deflection, and finally a second negativity (e.g., contacts 3–5, Fig. 4C). This SPC waveform reflects the negative DC shift of SD, with distortion due to the 0.02-Hz high-pass filter of the clinical amplifiers, and is the same as observed in monopolar recordings from subdural strip electrodes with the same amplifiers [21]. This waveform is the same as that simulated by applying a high-pass digital filter to DC recordings from the rat (Fig. 2A, lower).

Data from a depth array located within cortex. A graphical representation of inferred depth electrode position (based on CT imaging and pattern of SPC changes at specific contacts) on (A) representative MRI head image. B diagram indicating tissue regions a CSF space. b cortical gray matter. c subcortical white matter. C Upper Clearly defined SPC waves recorded from depth contacts estimated to lie within cortex (numbers 3 ,4 ,5). These displayed a typical negative deflection followed by a positive deflection. An attenuated SPC morphology was seen at contacts estimated to lie at the gray/white matter interface (numbers 1 and 2). Lower BPF (0.5–30 Hz) data indicate a reduction of cortical activity following the passage of depolarization waves. D Quantification of the morphology of an SPC event recorded with a depth electrode in a human patient. a duration of negative deflection, b duration of positive deflection, c total SPC duration d peak-to-peak amplitude
The typical negative–positive–negative SPC morphology in clinical recordings was best observed on electrode contacts judged to be located in the cortex (contacts 3, 4, and 5 in Fig. 4C) and was significantly attenuated in contacts estimated to be lying at the margins of cortical tissue. Exact localization of depth array contacts was not possible but a consistent increase in noise artifact was seen in contacts estimated to lie immediately above the cortex in the sub-arachnoid space (contacts 6, 7, and 8 in Fig. 4C). A variety of SPC morphologies was seen at contacts estimated to lie at the gray/white matter interface or deeper (contacts 1 and 2 in Fig. 4C). For analysis of waveform morphology, therefore, only events displaying typical morphology were used. This included 62 depth electrode SD events recorded on individual channels in five patients.
The SPC morphologies were paramaterized according to Fig. 4D. Peak-to-peak amplitudes (marked as “d”) of human SPCs ranged 0.78–5.263 mV and the mean was 1.72 mV. These values are consistent with those previously reported from humans using subdural electrodes and the same amplifier/filter settings [19], and slightly smaller than those in the rat (Fig. 2). This is likely attributable to the precise placement of depth electrodes in upper layers of cortex under experimental conditions. Comparison of the duration of depression of high-frequency spontaneous signal (0.5–30 Hz) with SPC wave parameters revealed no correlation with negative deflection time in seconds (Fig. 4D: duration “a,” Pearson r = −0.07, p = 0.29), a moderate correlation with positive deflection (Fig. 4D duration b, Pearsons r = 0.45, p = 0.0001) and a weak correlation with total SPC duration (Fig. 4D duration “c,” Pearsons r = 0.36 p = 0.02).
Recording Characteristics in Gray versus White Matter in Patients
In four of the eight patients monitored, serial CT scanning showed depth electrodes lying within cortical tissue. In the remaining four, a number of the depth contacts were shown to be located deep to the cortical mantle in the underlying white matter. In these patients, SPCs at the cortical contacts (Fig. 5C unfiltered contact 8) as well as concurrent recordings with strip electrodes confirmed the occurrence of SDs. However, SPCs were not recorded by the depth contacts located in the white matter (Fig. 5C, unfiltered contacts 6–1). In two patients, deep-lying probes initially showed no evidence of SPCs on the majority of electrode contacts. When retracted to place contacts within cortical tissue (confirmed with CT imaging) high-amplitude SPCs were found, clearly identifying SDs (Fig. 5D).

Data from a depth array located deep to cortex subsequently retracted to lie in cortex. A graphical representation of inferred depth electrode position (shown here on a non-specific MRI head image), based on CT imaging and pattern of SPC and ECoG changes at specific contacts. B graphical representation of cortical field potential (d) and depth electrode with contacts lying in white matter tracts (c) underlying cortex (b). One contact (number 8, red) is inferred to lie at the gray/white matter interface. C Upper Unfiltered data from seven of the eight depth electrode contacts (contact 7—orange on 6A and 6B—did not record meaningful data). An SD wave was confirmed by concurrent recording from a strip electrode (data not shown). The most superficial electrode contact (red) displays an SPC wave with aberrant morphology, while deeper lying contacts show no SPCs. Lower The band pass-filtered data from these electrode contacts display a distinct decrease in cortical activity in the most superficial contact and a decay of this effect in progressively deeper contacts. The leaky integral of the 0.5–30 Hz band is superimposed on contacts 8, 5, 3, and 1 to show decreasing slope at contacts with increased distance from cortex. D Upper Unfiltered data from the same electrode array after retraction by 1 cm. Typical SPC waveforms are recorded from contacts 2 and 3. Lower Band pass-filtered data show a clear decrease in cortical activity most marked in cortically placed contacts. The leaky integral of the 0.5–30 Hz band is again superimposed to more clearly demonstrate this reduction
In contrast to SPCs, the spreading depression of activity induced by SD (quantified using a leaky integral) was readily observed on electrodes located in white matter. In Fig. 4, for example, a clear amplitude depression was found at all electrodes. We also found that the distance from cortical tissue had a substantial effect on the extent of amplitude depression (Fig. 5C). This relationship was quantified by measuring the slope (rate of decrease) of the leaky integral plots as a function of distance from cortex for each contact in four separate depth array positions. Figure 6 shows the results for ten different SDs. Electrodes closest to cortex (e.g., 7 and 8) showed the greatest decrease in spontaneous activity (a steeper integral slope) while deeper electrodes showed smaller depressions. The Pearson’s correlation coefficient of r = −0.761, p < 0.001(n = 70) indicates a strong correlation between slope of the integral decay and the proximity of the contact to the cortex. The consistency of this relationship supports the hypothesis of a cortical field potential projected into the white matter.

Variation of leaky integral slope as a function of contact depth. This graph shows the change in leaky integral value per minute measured over a 4-min period expressed as a percentage of the baseline value before the SD event. This is shown for ten SD events, recorded at four different electrode positions at varying depths below the cortical mantle. Recordings were taken from three patients. Insufficient retraction in one patient provided two sets of white matter recordings. The line of best fit predicts a change in the leaky integral slope of 1.4 %/min as the cortex is approached by one electrode array contact (≈2.2 mm)
Discussion
We have shown that SD waves can be reliably and sensitively detected in the injured human brain with a depth electrode array inserted into the brain parenchyma. The potential to measure this activity via an electrode array introduced through a burr hole can enable the reliable identification of SDs in patients who do not require a craniotomy, and, therefore, considerably broadens the patient population in which SD can be monitored.
While SDs have been clearly associated with poor outcome following TBI [17], their clinical significance in all types of acute brain injury is still being defined. The phenomenon has been implicated in the expansion of (a) ischemic lesions [22] and (b) brain contusions following TBI [11]. SD is likely to be a mediating mechanism by which systemic insults such as hyperthermia and hypotension lead to progression of secondary brain injury, although the majority of SDs occur spontaneously, in the absence of such insults [13]. Thus, there is a pressing need for more investigation into the role of SD but to date the phenomenon has been reliably recorded in humans only from cortical electrode strips placed at craniotomy.
Waveform Analysis of SPC
The DC shift of SD occurs at frequencies <0.02 Hz and is, therefore, distorted by the AC-coupled amplifiers used in these clinical studies. In depth electrode recordings, we identified the typical morphology for SPCs that has been identified previously for subdural electrode recordings made with the same AC-coupled amplifiers (0.02-Hz high-pass filter). This consisted of an initial negative deflection followed by a slower, more rounded positive deflection, and is a hallmark signature of SD. This waveform generally, though not strictly, reflects the derivative of the underlying negative DC potential shift indicating mass tissue depolarization [21].
This was further confirmed here in our animal studies. The SPC waveforms of SD had prominent negative and positive phases when similar 0.01 high-pass filtering was applied (Fig. 2). The negative phase corresponded to the declining slope, or depolarization phase, of the negative DC shift, while the positive phase corresponded to the recovery (increasing slope), or repolarization phase, of the negative DC shift (compare Fig. 2A, middle and lower). Thus, the SPCs waveforms observed in clinical recordings are explained by, and directly linked to, the underlying biophysical processes of SD.
This link between AC-coupled SPCs and negative DC shifts may further explain the correlation we observed between SPC durations and depression durations. We found that depression durations had the strongest correlation with the positive phase of the SPC. This positive phase, corresponding to tissue repolarization, is likely the most accurate measure of depolarization duration, since the time to maximal depolarization is relatively fixed and the total duration of depolarization is determined by the speed of repolarization. We have shown previously that the duration of depolarization, as measured by the negative DC shift, correlates to the depression duration [23, 24]. Thus, the present data extend this finding to measures of the AC-coupled SPC, notably the duration of the positive phase. We caution, however, that conclusions about depolarization duration cannot be drawn from AC-coupled recordings, and that the SPCs observed should be used only to identify SD [21, 23].
Signals Dependent on Location of Depth Contacts
Our data clearly indicate that depth electrode contacts must lie within cortical tissue in order to detect the SPC of SD. It is also clear that it is possible to measure changes in high-frequency ECoG amplitude in electrode contacts lying in white matter tracts; however, the sensitivity of this measurement declines with increasing distance of contacts from the cortex. This is most likely a result of increasing distance from a cortically generated electrical field potential (see Fig. 5B).
Clinical Utility
The use of depth electrode ECoG recordings is most readily applicable in the context of TBI, where monitoring probes are frequently inserted at the bedside via an access bolt as standard of care. Two studies have shown that an intraparenchymal electrode array can detect subclinical seizures and predict worsening of acquired brain injury [25, 26] but neither study was designed to detect SD.
We are now beginning a study using depth electrode arrays together with ICP, Licox, and microdialysis probes placed at the bedside in the neurocritical care unit via a multilumen cranial access bolt. The question arises whether intra-parenchymal probes will continually stimulate SD waves in surrounding cortex. The COSBID group has monitored many patients with penetrating probes (Hemedex, Licox, depth electrode, and microdialysis) that do not have SDs, which makes it unlikely that they are inducing them.
The data presented in this current clinical study come necessarily from patients with both subdural strip electrodes and depth electrode arrays placed at craniotomy. The electrodes were placed under direct vision in pericontusional cortical areas and as far as possible along the course of a single gyrus in the affected territory. This situation differs considerably from standard placement of monitoring probes through a bolt device in the conventional right frontal lobe location. Interpretation of data obtained from burr hole sited depth electrodes needs to take account of:
-
(a)
The electrode placement may be remote from damaged tissue, and might not detect SD waves occurring in perilesional cortex. Our practice is to place bolts as far as possible within 2 cm of lesioned cortex as judged from admission CT scans but our ability to target lesions is limited by the need to avoid eloquent cortex. Woitzik et al. [27] have used laser speckle imaging of cortical blood flow at craniectomy for malignant hemispheric stroke, which clearly demonstrates propagation of SD waves at least 7 cm from initiation point but the true extent of SD propagation is unknown.
-
(b)
The electrode might fail to target the gyral apex and become either a subdural electrode or enter a cortical sulcus. In either case we predict that both the SPC and HF components of the SD signature will be of reduced amplitude, and the temporal delay between adjacent contacts be increased.
-
(c)
The electrode contacts might be placed deep to the cortical mantle. The present study (Fig. 5) shows the differential effect this has on the SPC and HF parts of the SD wave signature.
A final issue to be considered is the difference in clinical management between patient groups. The most severely injured patients require surgical management by craniotomy and/or decompressive craniectomy, with associated resection of non-viable tissue and removal of hematoma. These are factors that might increase the frequency of SD events. Bolt-monitored patients will be managed medically in the neurocritical care unit. They are more likely to require more intensive management for raised intracranial pressure with sedative drugs, cooling, or hyperventilation. These are factors that may decrease the frequency of SD events.
The optimal management of severe brain injury is still a matter of debate. Some treatment decisions, such as the evacuation of expanding mass lesions, follow established, logical pathways [28]. Other decisions, such as the correct level and duration of sedation to apply and optimum temperature management, are less clearly defined. Clinicians may be obliged to make treatment decisions in the neurocritical care unit without recourse to real time, direct indices of cortical physiology. This lack of information applies both to the immediate clinical context and to the understanding of the pathophysiological processes underlying secondary brain injury [29].
The use of multi-modal monitoring offers a more detailed picture of secondary brain injury and may help to explain the heterogeneous response to this pathology, i.e., why some patients have a particularly poor outcome [17]. This technique offers a more complete understanding of secondary injury and the role of SD in its progression. It can contribute to the development and patient-specific targeting of therapies designed to minimize secondary injury. The use of a specific therapy, such as NMDA receptor antagonists or therapeutic hypothermia [30], may be much more effectively employed when in the confirmed presence of a target pathophysiology.
Notes
For the purposes of this study no distinction was made between events of short and long duration [10] and all are referred to as “SD.”
References
Faul M, Xu L, Wald MM, Coronado V, Dellinger AM. Traumatic brain injury in the United States: National estimates of prevalence and incidence, 2002–2006. Inj Prev. 2010;16:A268.
Coronado VG, Xu L, Basavaraju SV, et al. Surveillance for traumatic brain injury-related deaths–United States, 1997–2007. MMWR Surveill Summ. 2011;60:1–32.
Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–220.
Kurland D, Hong C, Aarabi B, Gerzanich V, Simard JM. Hemorrhagic progression of a contusion after traumatic brain injury: a review. J Neurotrauma. 2012;29:19–31.
von Oettingen G, Bergholt B, Gyldensted C, Astrup J. Blood flow and ischemia within traumatic cerebral contusions. Neurosurgery. 2002;50:781–8 discussion 788–790.
Holmin S, Soderlund J, Biberfeld P, Mathiesen T. Intracerebral inflammation after human brain contusion. Neurosurgery. 1998;42:291–8 discussion 298–299.
Obrenovitch TP, Urenjak J. Is high extracellular glutamate the key to excitotoxicity in traumatic brain injury? J Neurotrauma. 1997;14:677–98.
Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25:763–74.
Menon DK. Procrustes, the traumatic penumbra, and perfusion pressure targets in closed head injury. Anesthesiology. 2003;98:805–7.
Woitzik J, Dreier JP, Hecht N, et al. Delayed cerebral ischemia and spreading depolarization in absence of angiographic vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2012;32:203–12.
Strong AJ, Boutelle MG, Vespa PM, Bullock MR, Bhatia R, Hashemi P. Treatment of critical care patients with substantial acute ischemic or traumatic brain injury. Crit Care Med. 2005;33:2147–9 author reply 2149.
Dohmen C, Sakowitz OW, Fabricius M, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63:720–8.
Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:17–35.
Dreier JP, Major S, Manning A, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132:1866–81.
Takano T, Tian GF, Peng W, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–62.
Feuerstein D, Manning A, Hashemi P, et al. Dynamic metabolic response to multiple spreading depolarizations in patients with acute brain injury: an online microdialysis study. J Cereb Blood Flow Metab. 2010;30:1343–55.
Hartings JA, Bullock MR, Okonkwo DO, et al. Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. Lancet Neurol. 2011;10:1058–64.
Strong AJ, Fabricius M, Boutelle MG, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738–43.
Fabricius M, Fuhr S, Bhatia R, et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129:778–90.
Kudo C, Nozari A, Moskowitz MA, Ayata C. The impact of anesthetics and hyperoxia on cortical spreading depression. Exp Neurol. 2008;212:201–6.
Hartings JA, Watanabe T, Dreier JP, Major S, Vendelbo L, Fabricius M. Recovery of slow potentials in AC-coupled electrocorticography: application to spreading depolarizations in rat and human cerebral cortex. J Neurophysiol. 2009;102:2563–75.
Nakamura H, Strong AJ, Dohmen C, et al. Spreading depolarizations cycle around and enlarge focal ischaemic brain lesions. Brain. 2010;133:1994–2006.
Hartings JA, Watanabe T, Bullock MR, et al. Spreading depolarizations have prolonged direct current shifts and are associated with poor outcome in brain trauma. Brain. 2011;134:1529–40.
Dreier JP, Major S, Manning A, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132:1866–81.
Stuart RM, Waziri A, Weintraub D, et al. Intracortical EEG for the detection of vasospasm in patients with poor-grade subarachnoid hemorrhage. Neurocrit Care. 2010;13:355–8.
Waziri A, Claassen J, Stuart RM, et al. Intracortical electroencephalography in acute brain injury. Ann Neurol. 2009;66:366–77.
Woitzik J, Hecht N, Pinczolits A, et al. Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology. 2013;80:1095–102.
Valadka AB, Robertson CS. Surgery of cerebral trauma and associated critical care. Neurosurgery. 2007;61:203–20 discussion 220–201.
Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9.
Hertle DN, Dreier JP, Woitzik J, et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain. 2012;135:2390–8.
Acknowledgments
This work was funded in part by a Wellcome Trust/Department of Health Healthcare Innovation Challenge Fund (Grant HICF—1010-080) and in part by the U.S. Army CDMRP PH/TBI Research Program (contract number W81XWH-08-2-0016) and the Mayfield Education and Research Foundation. We thank Chi Leng Leong for help with the figures. All animal work was carried out at the University of Cincinnati. All human clinical investigations were carried out at Kings College Hospital, London, UK.
Conflict of interest
Toby Jeffcote, Jason M Hinzman, Sharon L Jewell, Robert M Learney, Clemens Pahl, Christos Tolias, Daniel C Walsh, Agnieszka Zakrzewska, Martin E Fabricius, Anthony J Strong, Jed A Hartings, and Martyn G Boutelle declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Jed A. Hartings and Martyn G. Boutelle contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Jeffcote, T., Hinzman, J.M., Jewell, S.L. et al. Detection of Spreading Depolarization with Intraparenchymal Electrodes in the Injured Human Brain. Neurocrit Care 20, 21–31 (2014). https://doi.org/10.1007/s12028-013-9938-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-013-9938-7