
Abstract
Background
The pathogenesis of vasospasm (VS) post aneurysmal subarachnoid hemorrhage (SAH) is multifactorial and not completely understood. The authors hypothesize that circulating antiangiogenic factors play an important role in brain injury post SAH and that elevated levels predict the occurrence of symptomatic vasospasm.
Methods
In this study the authors measured the serum and cerebrospinal fluid (CSF) levels of soluble endoglin (sEng) and soluble fms-like tyrosine kinase 1 (sFlt1) in controls and SAH patients within 48 h of the bleed. Patients were prospectively followed and subcategorized into those with (sVS) and without symptomatic vasospasm (no-sVS).
Results
Compared to healthy controls, SAH patients had higher CSF levels of sEng (0.037 vs. 0.251 ng/ml; P = 0.02) and sFlt1 (0.068 vs. 0.679 ng/ml; P = 0.001). In the subgroup analysis, sVS patients had higher CSF levels of sEng and sFlt1 than no-sVS patients (sEng: 0.380 vs. 0.159 ng/ml, P = 0.02; sFlt1: 1.277 vs. 0.343 ng/ml, P = 0.01). The serum levels of sEng and sFlt1 were not statistically different among the different groups.
Conclusions
Based on these results the authors conclude that elevated CSF levels of sFlt1 and sEng herald the occurrence of symptomatic VS post SAH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebral vasospasm (VS) is a frequently encountered complication of subarachnoid hemorrhage (SAH) that is associated with increased morbidity and mortality [1]. Different mechanisms have been proposed to be involved in the pathogenesis of VS including synthesis of spasmogenic substances, endothelial dysfunction, oxidative stress, inflammation, disturbance in the equilibrium of endogenous vasoconstrictors (endothelin-1) and vasodilators (NO, nitric oxide), and vascular denervation [2]. Vascular mitogens, such as transforming growth factor (TGF)-β and vascular endothelial growth factor (VEGF), have been shown to increase in the cerebrospinal fluid (CSF) of patients with SAH and it has been proposed that they participate in cellular proliferation and vascular stiffening [3, 4]. The increase in these vascular growth factors may also be an adaptive response to vascular related brain injury as both mediators have neuroprotective properties. Studies performed in animal models showed that after cerebral ischemia VEGF induces endothelial proliferation and enhances neuronal survival and TGF-β reduces exitotoxitiy and caspase-mediated apoptosis [5–8]. VEGF and TGF-β are regulated by circulating antiangiogenic factors; soluble-fms-like tyrosine kinase 1 (sFlt1) neutralizes VEGF and soluble endoglin (sEng) sequesters, and thus inhibits, TGF-β [9, 10]. The role of sFlt1 and sEng in cerebrovascular disease has not been investigated; however, there is substantial evidence suggesting they may be detrimental as they antagonize the beneficial effects of VEGF and TGF-β. In addition, both antiangiogenic factors regulate the production of NO, a pivotal mediator of VS in SAH [11–13]. In this study the authors measured serum and CSF levels of sFlt1 and sEng in controls and SAH patients and investigated whether they are associated with the occurrence of symptomatic VS and outcome.
Methods
Subjects were recruited at the University of Illinois Medical Center at Chicago. Institutional review board approval was obtained prior to study initiation. Cases were eligible to participate in this study if they were 18 years of age or older and had non-traumatic aneurysmal SAH, aneurysm visualized by either digital subtraction catheter angiography or non-invasive cerebral angiography (either computed tomographic or magnetic resonance angiography), SAH Fisher grade 3, and the presence of external ventricular drain. The exclusion criteria were SAH due to causes other than a ruptured saccular aneurysm, VS noted on the admission angiography, pregnancy, previous neurological disability (defined as preadmission modified Rankin Score >2), and history of stroke, brain tumor, intracranial surgery, traumatic brain injury, previously treated cerebral aneurysm, or other vascular malformation. Controls were non-pregnant individuals ≥18 years of age with no previous neurological disorders undergoing CSF drainage for evaluation of headache or due to congenital hydrocephalus and shunt malfunctioning, and with normal CSF studies, including chemistry, cell count and differential, and cultures. Samples of CSF and serum were obtained within 48 h of symptoms onset, centrifuged at 270×g for 15 min at 5°C, and the supernatant stored at −80°C. sEng and sFlt1 levels were determined using commercially available ELISA tests (R&D systems). CSF sEng and sFlt1 indices were calculated as (sEng or sFlt1CSF × proteinserum)/(sEng or sFlt1serum × proteinCSF). SAH subjects were prospectively followed through their hospitalization and subcategorized into those with symptomatic vasospasm (sVS) and without symptomatic VS (no-sVS). Vasospasm was defined as >30% arterial narrowing on DSA not attributable to atherosclerosis, catheter-induced spasm, or vessel hypoplasia, as determined by an independent neuroradiologist. Symptomatic VS was defined as development of new focal neurological signs, deterioration in level of consciousness, or both, when the cause was felt to be ischemia attributable to VS after other possible causes of worsening have been excluded, or the appearance of new infarction on CT or MR when the cause was felt to be attributable to VS. Outcome at discharge was assessed by using the modified Ranking scale (mRS). SAH patients were dichotomized into those with good outcome (mRS ≤ 3) and bad outcome as (mRS ≥ 4). Differences between groups were studied using Student’s t-test for continuous variables or χ2 test for categorical variables. The sEng and sFlt1 levels in different groups were compared using the nonparametric Mann–Whitney U test. The CSF levels of sEng and sFlt1 in SAH patients were divided into quartiles (Q) and the rate of sVS calculated for each of them.
Results
Between March 2010 and December 2010, 19 SAH subjects and 6 controls were recruited into this study. Five control cases (83%) underwent lumbar puncture for evaluation of headache of sudden onset and only one of them had history of migraine headaches. The sixth control case was an individual with congenital communicating hydrocephalus who underwent lumbar drainage placement due to ventriculoperitoneal shunt malfunctioning. Compared to SAH subjects, controls were more likely to be younger and male, and equally likely to be hypertensive (Table 1). Within the SAH group, the mean time to obtain the CSF and serum samples was (36 ± 11) h. Angiographic evidence of VS occurred in 11 (58%) SAH patients and six of these were symptomatic (sVS). Compared to sVS, patients who did not develop symptomatic VS (no-sVS) had a higher GCS at presentation (11.3 ± 3.6 vs. 7.1 ± 3.5).
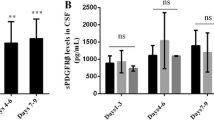
The serum levels of sEng and sFlt1 in the control group were approximately 100-fold and 10-fold higher than in the CSF, respectively (Table 2). Compared to controls, SAH patients had similar serum levels and approximately 7-fold increase in the CSF levels of sEng and sFlt1 (Table 2). The sEng and sFlt1 CSF indices were >1 suggesting that these mediators are, at least in part, intrathecally synthesized. Within the SAH group, the CSF levels of sEng were 2.5-times higher in patient with sVS than in those with no-sVS. Similarly, the CSF levels of sFtl1 were 3.7-times higher in the sVS group compared to no-sVS group. The rate of sVS according to sEng and sFlt1 quartiles, as measured in the CSF, is shown in Fig. 1. Patients were also dichotomized based on the mRS into those with good (mRS ≤ 3) and bad (mRS ≥ 4) outcome. Patients with poor outcome had higher levels of sEng in the CSF (P = 0.018) and a trend toward having higher levels of sFlt1 (Fig. 2); this, however, did not reach statistical significance (P > 0.05).

Rate of sVS per quartile of sEng and sFlt1 as measured in the CSF

CSF levels of sEng and sFlt1 in patients with good (mRS ≤ 3) and bad outcome (mRS ≥ 4)
Discussion
The results show that an early elevation of sEng and sFlt1 occurs in the CSF of SAH patients, particularly in those who will develop sVS in the subsequent days, and that the rate of occurrence of this complication is directly related to the levels of these antiangiogenic factors. Although there are no reliable reference values for sEng and sFlt1 in healthy individuals or stroke patients, the serum levels obtained in this study for both antiangiogenic factors were within the limits of what has been previously reported by others [14–17]. The systemic concentration of both mediators is significantly higher than in CSF; therefore, the increased levels of sEng and sFlt1 in this compartment is likely to be related, at least in part, to the blood extravasated into the subarachnoid space. However, the significantly elevated CSF indices observed in this study suggest that a fraction of these antiangiogenic factors is produced intrathecally.
The role of antiangiogenic factors in the occurrence of VS has not been investigated but there is substantial indirect evidence suggesting that sFtl1 and sEng may participate in the pathogenesis of brain injury after stroke, particularly in SAH. VEGF signals mainly through two tyrosine-kinase receptors: VEGF-R1 and VEGF-R2 [10]. During cerebral ischemia there is an upregulation of VEGF leading to AKT and ERK 1/2-mediated endothelial proliferation and neuronal survival [5]. In addition, VEGF-receptor type 2 regulates endothelial NO synthase which is a key enzyme in the pathogenesis of VS after SAH [18]. The production of TGF-β also increases during ischemia and it has been proposed that this represents an adaptive response against brain injury as TGF-β has neuroprotective properties [19]. In vitro studies performed in ischemic conditions showed that TGF-β1 and TGF-β3 protect neurons against NMDA-mediated excitotoxicity and caspase-mediated apoptosis, and intranasal delivery of TGF-β1 in a mouse model of cerebral ischemia improved cell survival, neurogenesis, and outcome [6, 7, 20].
After SAH there is an elevation in the CSF levels of VEGF and TGF-β [4, 21]. It’s been hypothesized that VEGF is deleterious for the brain as it induces vascular remodeling and persistent wall stiffening [3]. Alternatively, it has been proposed that VEGF is neuroprotective against cerebrovascular injury as it induces vascular sprouting and angiogenesis, increases brain plasticity, and enhances vascular relaxation probably by boosting endothelial NO levels [22–24]. TGF-β in the CSF originates from the extravasated blood and from the choroids plexus. TGF-β is a vasodilator that has been shown to stimulate endothelial NO synthase, to inhibit the deleterious inducible NO synthase, and to impair ET-1-mediated blood vessel contraction [21, 25, 26]. VEGF and TGF-β are regulated by endogenous antiangiogenic factors. During hypoxia VEGF-R1 undergoes proteolytic cleavage and its extracellular portion (called sFlt1) is released to the circulation [27]. By binding to VEGF, sFlt1 decreases the bioavailability of this vascular factor and inhibits VEGF-mediated angiogenesis and NO-mediated vasorelaxation [10, 28]. In addition, under pathological conditions a soluble form the TGF-β co-receptor endoglin (a.k.a. sEng) is formed; sEng is an antiangiogenic factor that interferes TGF-β signaling [9]. The role of sFlt1 and sEng in vascular disease has been recently highlighted by studies showing that these two mediators interfere with VEGF and TGF-β in placental endothelial cells and attenuate endothelial NO synthase activation [28, 29].
The evidence aforementioned suggests that sEng and sFlt1 participate in the pathogenesis of brain injury post stroke, and particularly SAH, as they sequester the TGF-β and VEGF from the circulation. Based on this information and the results obtained in this study, it was proposed a novel pathogenic mechanism for VS and poor outcome after SAH. The authors hypothesize that the rapid increase in intracranial pressure (ICP) and the consequent global hypoperfusion and hypoxia that occurs after SAH induces an early surge in the production of sEng and sFlt1. By decreasing the bioavailability of TGF-β and VEGF, these two antiangiogenic factors impair NO homeostasis and cause neuronal and endothelial cell death leading ultimately to vasospasm and poor outcome (Fig. 3).

Possible role of sEng and sFlt1 in the pathogenesis of vasospasm and outcome after SAH (see text)
This study is a pilot project and, therefore, the main limitation is the small sample. The distribution of the clinical characteristics of the cohort studied, however, is representative of what has been historically described to occur in SAH patients [30, 31]. The results are highly encouraging as they suggest novel mechanisms that may participate in the occurrence of VS post SAH. As elevations of sEng and sFlt1 occur early in the disease and prior to the occurrence of sVS, both mediators have the potential to be used as biomarkers of this complication and as targets of novel therapeutic treatments. In the next phase the authors will attempt to validate the observations in a larger sample.
References
Johnston SC, Selvin S, Gress DR. The burden, trends, and demographics of mortality from subarachnoid hemorrhage. Neurology. 1998;50:1413–8.
Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3:256–63.
Borel CO, McKee A, Parra A, Haglund MM, Solan A, Prabhakar V, Sheng H, Warner DS, Niklason L. Possible role for vascular cell proliferation in cerebral vasospasm after subarachnoid hemorrhage. Stroke. 2003;34:427–33.
Miller CA, Lombard FW, Wu CT, Hubbard CJ, Silbajoris L, Borel CO, Niklason LE. Role of vascular mitogens in subarachnoid hemorrhage-associated cerebral vasculopathy. Neurocrit Care. 2006;5:215–21.
Shimotake J, Derugin N, Wendland M, Vexler ZS, Ferriero DM. Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke. 2010;41:343–9.
Prehn JH, Backhauss C, Krieglstein J. Transforming growth factor-beta 1 prevents glutamate neurotoxicity in rat neocortical cultures and protects mouse neocortex from ischemic injury in vivo. J Cereb Blood Flow Metab. 1993;13:521–5.
Zhu Y, Culmsee C, Klumpp S, Krieglstein J. Neuroprotection by transforming growth factor-beta1 involves activation of nuclear factor-kappaB through phosphatidylinositol-3-OH kinase/Akt and mitogen-activated protein kinase-extracellular-signal regulated kinase1, 2 signaling pathways. Neuroscience. 2004;123:897–906.
Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C, Klumpp S, Krieglstein J. Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–909.
Bernabeu C, Conley BA, Vary CP. Novel biochemical pathways of endoglin in vascular cell physiology. J Cell Biochem. 2007;102:1375–88.
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling—in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–71.
Wu HM, Huang Q, Yuan Y, Granger HJ. VEGF induces NO-dependent hyperpermeability in coronary venules. Am J Physiol. 1996;271:H2735–9.
Jerkic M, Rivas-Elena JV, Prieto M, Carron R, Sanz-Rodriguez F, Perez-Barriocanal F, Rodriguez-Barbero A, Bernabeu C, Lopez-Novoa JM. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004;18:609–11.
Sabri M, Ai J, Macdonald RL. Nitric oxide related pathophysiological changes following subarachnoid haemorrhage. Acta Neurochir Suppl. 2011;110:105–9.
Masuyama H, Nakatsukasa H, Takamoto N, Hiramatsu Y. Correlation between soluble endoglin, vascular endothelial growth factor receptor-1, and adipocytokines in preeclampsia. J Clin Endocrinol Metab. 2007;92:2672–9.
Molvarec A, Szarka A, Walentin S, Szucs E, Nagy B, Rigo J Jr. Circulating angiogenic factors determined by electrochemiluminescence immunoassay in relation to the clinical features and laboratory parameters in women with pre-eclampsia. Hypertens Res. 2010;33:892–8.
Quantakine. Human Endoglin/CD 15 Immunoassay. R&D Systems, Inc. 2009. http://www.rndsystems.com/pdf/dndg00.pdf. Accessed 04 July 2011.
Quantakine. Human Soluble VEGF R1/FLT-1 Immunoasssay. R&D Systems, Inc. 2009. http://www.rndsystems.com/pdf/DVR100B.pdf. Accessed 04 July 2011.
Cui X, Chopp M, Zacharek A, Zhang C, Roberts C, Chen J. Role of endothelial nitric oxide synthetase in arteriogenesis after stroke in mice. Neuroscience. 2009;159:744–50.
Ata KA, Lennmyr F, Funa K, Olsson Y, Terent A. Expression of transforming growth factor-beta1,2,3 isoforms and type I and II receptors in acute focal cerebral ischemia: an immunohistochemical study in rat after transient and permanent occlusion of middle cerebral artery. Acta Neuropathol. 1999;97:447–55.
Ma X, Labinaz M, Goldstein J, Miller H, Keon WJ, Letarte M, O’Brien E. Endoglin is overexpressed after arterial injury and is required for transforming growth factor-beta-induced inhibition of smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2000;20:2546–52.
Flood C, Akinwunmi J, Lagord C, Daniel M, Berry M, Jackowski A, Logan A. Transforming growth factor-beta1 in the cerebrospinal fluid of patients with subarachnoid hemorrhage: titers derived from exogenous and endogenous sources. J Cereb Blood Flow Metab. 2001;21:157–62.
Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–38.
Yang JP, Liu HJ, Liu XF. VEGF promotes angiogenesis and functional recovery in stroke rats. J Invest Surg. 2010;23:149–55.
Yang JP, Liu HJ, Wang ZL, Cheng SM, Cheng X, Xu GL, Liu XF. The dose-effectiveness of intranasal VEGF in treatment of experimental stroke. Neurosci Lett. 2009;461:212–6.
Tong XK, Hamel E. Transforming growth factor-beta 1 impairs endothelin-1-mediated contraction of brain vessels by inducing mitogen-activated protein (MAP) kinase phosphatase-1 and inhibiting p38 MAP kinase. Mol Pharmacol. 2007;72:1476–83.
Vodovotz Y, Bogdan C. Control of nitric oxide synthase expression by transforming growth factor-beta: implications for homeostasis. Prog Growth Factor Res. 1994;5:341–51.
Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, Risau W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–76.
Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, Epstein FH, Romero R, Thadhani R, Karumanchi SA. CPEP Study Group soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992–1005.
Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D’Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M, Karumanchi SA. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–9.
Crowley RW, Medel R, Dumont AS, Ilodigwe D, Kassell NF, Mayer SA, Ruefenacht D, Schmiedek P, Weidauer S, Pasqualin A, Macdonald RL. Angiographic vasospasm is strongly correlated with cerebral infarction after subarachnoid hemorrhage. Stroke. 2011;42:919–23.
Bederson JB, Connolly ES Jr, Batjer HH, Dacey RG, Dion JE, Diringer MN, Duldner JE Jr, Harbaugh RE, Patel AB, Rosenwasser RH. American Heart Association Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 2009;40:994–1025.
Disclosures
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Testai, F.D., Aiyagari, V., Hillmann, M. et al. Proof of Concept: Endogenous Antiangiogenic Factors Predict the Occurrence of Symptomatic Vasospasm Post Subarachnoid Hemorrhage. Neurocrit Care 15, 416–420 (2011). https://doi.org/10.1007/s12028-011-9559-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-011-9559-y