
Abstract
Limited protective effects of commercially available vaccines necessitate the development of novel pneumococcal vaccines. We recently reported a pneumococcal systemic vaccine containing two proteins, Pneumococcal surface protein A (PspA of family 1 and 2) and a bacterium-like particle-based pneumococcal mucosal vaccine containing PspA2 and PspA4 fragments, both eliciting broad protective immune responses. We had previously reported that subcutaneous (s.c.+s.c.+s.c.) immunization with the systemic vaccine induced more pronounced humoral serum IgG responses, while intranasal (i.n.+i.n.+i.n.) immunization with the mucosal vaccine elicited a more pronounced mucosal secretory IgA (sIgA) response. We hypothesized that a combinatorial administration of the two vaccines might elicit more pronounced and broader protective immune responses. Therefore, this study aimed to determine the efficacy of combinatorial prime-boost immunization using both systemic and mucosal vaccines for a pneumococcal infection. Combinatorial prime-boost immunization (s.c.+i.n. and i.n.+s.c.) induced not only IgG, but also mucosal sIgA production at high levels. Systemic priming and mucosal boosting immunization (s.c.+i.n.) provided markedly better protection than homologous prime-boost immunization (s.c.+s.c.+s.c. and i.n.+i.n.+i.n.). Moreover, it induced more robust Th1 and Th17 cell-mediated immune responses than mucosal priming and systemic boosting immunization (i.n.+s.c.). These results indicate that combinatorial prime-boost immunization potentially induces a robust systemic and mucosal immune response, making it an optimal alternative for maximum protection against lethal pneumococcal infections.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Streptococcus pneumoniae is the primary pathogen causing invasive pneumococcal diseases, including pneumonia, meningitis, and otitis media [1]. Over 1 million individuals worldwide die from pneumococcal infections every year [2]. Moreover, pneumococcal infections account for approximately 20% of mortalities among children under 5 years of age [3]. Thus far, vaccination remains the best control measure to prevent pneumococcal infections. Current licensed vaccines including polysaccharide vaccines and conjugate vaccines effectively prevent invasive pneumococcal infections; however, they are not without their limitations. For example, the 23-valent polysaccharide vaccine (PPV23) was developed exclusively for a specific serotype capsule polysaccharide and is not effective in children under 5 years of age [4]. The 13-valent pneumococcal conjugate vaccines (PCV13) protect children by inducing a T cell response and potent immunogenicity; however, they show limited serotype coverage and cannot be predicted to replace serotypes not covered by vaccines [5,6,7]. Therefore, new vaccines with broader protection against various S. pneumoniae strains are required. Protein-based vaccines are promising candidates owing to a high degree of conservation [8, 9]. Many pneumococcal proteins, especially some cell-surface virulence factors, have been considered ideal antigens for pneumococcal protein vaccines in recent years [10,11,12].
Pneumococcal surface protein A (PspA) is a choline-binding protein that influences pneumococcal virulence and inhibits complement deposition on the bacterial cell surface [13, 14]. This protein contains three domains, the α-helical domain (αHD), the proline-rich domain (PRD), and the choline-binding domain that anchors the protein to the cell wall [15]. The PRD is a prominent structure of PspA, and the αHD is surface-exposed to antibodies [16, 17]. Based on sequence variations in the αHD, PspA is classified into three families encompassing six clades, family 1 including clades 1 and 2, family 2 including clades 3, 4, and 5, and family 3 including clade 6 [18, 19]. Although PspA is variable, anti-PspA antibodies are highly cross-reactive and cross-protective [20, 21]. As more than 96% of clinical isolates of S. pneumoniae express PspA from family 1 or 2, we selected PspA from the two major families as major antigens to develop a pneumococcal protein-based vaccine [18, 22, 23]. We had previously developed a pneumococcal systemic vaccine containing two PspA proteins of families 1 and 2 and a bacterium-like particle-based (BLP) pneumococcal mucosal vaccine displaying PspA2 and PspA4 fragments.
We previously evaluated the protective effects of systemic and mucosal vaccines [24]. The systemic vaccine comprised a mixture of fusion protein PsaA-PspA23 and a single protein PspA4. We found that subcutaneous immunization of mice with the systemic vaccine with an aluminum hydroxide adjuvant potentially protected against strains of different clades, including clades 1–5. The mucosal vaccine comprised a mixture of PspA2-PA-BLP and PspA4-PA-BLP (PA: protein anchor). BLPs are non-living shells of Lactococcus lactis with the original morphology and size, and are favorable for uptake by M cells on the mucosal surface and stimulation of innate immunity by TLR2 [25,26,27]. The PA domain comprises 3LysM motifs in the repeat region of AcmA and can be conjugated with antigens as a recombinant fusion protein [28,29,30]. Consequently, intranasal immunization of mice with the mucosal vaccine with a BLP adjuvant serving as a carrier induced broad protective immune responses [31]. These positive protective results indicated that two types of vaccines potentially serve as a promising substitute for capsular polysaccharide vaccines.
We previously reported that both types of vaccines elicited adequate cross-protection against both family 1 and family 2 pneumococcal strains; however, each vaccine induced its own antigen-specific immune responses [32]. The systemic vaccine induced more pronounced humoral serum IgG responses, while the mucosal vaccine elicited more pronounced mucosal secretory IgA responses. Therefore, we hypothesized that combinatorial administration of the two vaccines further enhances the protective effect against a pneumococcal infection. In this study, we investigated the efficacy of combinatorial prime-boost immunization with the systemic and mucosal vaccines for their protection against a pneumococcal infection.
Materials and methods
Bacterial strains and culture conditions
The S. pneumoniae strains used herein were ATCC10813 (PspA family 1 clade 2) and ATCC BAA-334 (PspA family 2 clade 3). They were cultured in Todd-Hewitt broth supplemented with 0.5% yeast extract (THY) at 37 °C without agitation and used for the bacterial challenge.
Mice
Four- to six-week-old female BALB/c mice, weighing 16–20 g, were purchased from the Changchun Institute of Biological Products. All animal experiments were approved by the Institutional Animal Care and Use Committee of Jilin University (Changchun, China).
Vaccine preparation
Antigens for the systemic vaccine comprised a mixture of the fusion protein PsaA-PspA23 and PspA4, which were cloned in Escherichia coli and purified via nickel affinity chromatography. Protein purity was determined using a sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) that was scanned via near-infrared imaging (Odyssey). The adjuvants comprised sterile phosphate-buffered saline (PBS) mixed with 10% Al(OH)3. Antigens were prepared as previously reported [33].
Antigens for the mucosal vaccine comprised recombinant proteins PspA2-PA and PspA4-PA, which were cloned in Escherichia coli BL21. BLPs were mixed with the antigens to interact with the PA anchor on the antigen for 30 min at 25 °C. The particles were then washed thrice with sterile PBS to eliminate unbound proteins, thus yielding PspA2-BLP and PspA4-BLP vaccines. BLPs were prepared as previously reported [34]. Proteins were quantified via SDS-PAGE and near-infrared imaging (Odyssey). The amount of BLPs was determined by comparing the absorbance at 600 nm with that of different known concentrations of BLP standards.
Immunizations
Systemic antigens were prepared from 5 μg of PsaA-PspA23 and 20 μg of PspA4 in 100 μl PBS for subcutaneous (s.c.) administration per dose. BALB/c mice were immunized subcutaneously with three doses at 14-week intervals with a 100-μl systemic vaccine or 100 μl buffer as a negative control. Mucosal antigens were prepared from 0.15 mg of PspA2-BLP and 0.3 mg of PspA4-BLP in 20 μl PBS for intranasal (i.n.) administration per dose. Mice were immunized intranasally with three doses at 14-day intervals with 20 μl mucosal vaccine or 20 μl BLP as a negative control. The commercial PPV23 and PCV13 were used as positive controls. Details of the immunization strategy are described in Table 1.
Determination of antibody titers
Serum, bronchoalveolar lavage fluid (BALF), and nasal wash (NW) samples were collected 2 weeks after the third immunization to evaluate antibody levels. Antigen-specific IgG, IgG1, and IgG2a levels in serum were measured via indirect enzyme-linked immunosorbent assay. The 96-well plates were coated with 0.5 μg antigens per well, and goat anti-mouse IgG, IgG1, or IgG2a (1:1000; Sigma) and HRP-conjugated rabbit anti-goat IgG (1:5000; Beijing Dingguo Changsheng Biotech) were used to detect corresponding antibody levels in serum samples. The optical density (OD) was measured at 450 nm, and the results are expressed as titers. The IgA titer in BALF and NW was similar to that of IgG in serum. The bound antibodies were detected with HRP-anti-mouse IgA (1:5000; Gene Tex) in 8 serial 2-fold dilutions.
S. pneumoniae challenge
Two weeks after the third immunization with antigens or PBS, mice were anesthetized with ether and intranasally challenged with an LD90 dose of S. pneumoniae strain ATCC 10813 (1 × 104 CFU/mouse) or ATCC BAA-334 (4 × 107 CFU/mouse) with 20 μl in each nostril. The mice were monitored for 14 days, and differences between survival rates in each group were analyzed.
Measurement of cytokine levels
Splenocytes (5 × 106) harvested from mice were cultured in 24-well plates for 12 h and then stimulated with 5 μl PspA (containing 1 μg PsaA-PspA23 and 4 μg PspA4), 5 μl ConA (containing 5 μg ConA) as a positive control, or 5 μl PBS as a negative control for 48 h. To detect TNF-α and IL-17A in the culture supernatants, the CBA Mouse Soluble Protein Master Buffer Kit (BD Biosciences) and the CBA Mouse Soluble Protein Flex Set (BD Biosciences) were used in accordance with the manufacturer’s protocols. TNF-α and IL-17A levels were analyzed using FCAP Array software.
Statistical analysis
Statistical analysis was performed using GraphPad Prism Software (La Jolla, CA, USA). Differences were determined using one-way analysis of variance with t tests for post hoc analysis. The results are presented as mean ± SEM values and P values < 0.05 were considered statistically significant.
Results
Preparation of antigens
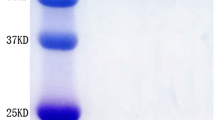
The fusion protein PsaA-PspA23 comprises full-length PsaA and the N-terminal clade-defining region of PspA2 and PspA3 [33]. Protein PspA4 contains the N-terminal and the proline-rich region of PspA4. These proteins were purified via nickel affinity chromatography and analyzed via SDS-PAGE and western blot analysis (using mouse anti-His tag monoclonal antibody as the primary antibody). As shown in Fig. 1a, the purities of PsaA-Pspa23 and PspA4 were > 85%, and their sizes were approximately 90 kDa and 65 kDa, respectively.

SDS-PAGE and western blot analysis of antigens. Lanes 1 and 3, systemic antigen PspaA-PspA23, 90 kDa; lanes 2 and 4, systemic antigen PspA4, 65 kDa; lanes 5 and 7, mucosal antigen PspA2-PA, 67 kDa; lanes 6 and 8, mucosal antigen PspA4-PA, 100 kDa
The fusion proteins PspA2-PA and PspA4-PA for binding BLPs were cloned in E. coli and mixed with BLPs in supersonic lysates. After binding with BLPs, PspA2-PA-BLP and PspA4-PA-BLP were analyzed via SDS-PAGE and western blot analysis (using mouse anti-PspA2 or anti-PspA4 monoclonal antibody as the primary antibody). As shown in Fig. 1b, the PspA2-PA-BLP and PspA4-PA-BLP displayed obvious bands in a background of degraded lactococcal components and their sizes were approximately 67 kDa and 100 kDa, respectively. Simultaneously, the binding efficiency of PspA2 or PspA4 with BLPs was determined via Odyssey near-infrared imaging; the binding capacities were 40 μg PspA2-PA/mg BLPs and 30 μg PspA4-PA/mg BLPs.
Heterologous prime-boost immunization induced a comparable level of systemic immune response
Systemic immunization (s.c.) with the systemic vaccine (PsaA-PspA23 and PspA4 with Al(OH)3 as the adjuvant) via subcutaneous administration resulted in higher antigen-specific IgG antibody levels in comparison with mucosal immunization with the intranasally (i.n.) administered mucosal vaccine (BLP-PspA2 and BLP-PspA4) [32]. To evaluate whether heterologous prime-boost immunization elicits a robust systemic immune response, antigen-specific IgG, IgG1, and IgG2a antibody levels were determined in serum 2 weeks after the final immunization (per the immunization scheme shown in Table 1).
As shown in Fig. 2a, b, the anti-PspA23 and anti-PspA4 IgG titers in all immunized groups were significantly greater than those in the negative control group (P < 0.001). Furthermore, IgG titers in the heterologous immunization group (s.c. + i.n. and i.n. + s.c.) were significantly lower than those in the systemic immunization group (s.c. + s.c. + s.c.) (P < 0.01), but comparable to those in the mucosal immunization group (i.n. + i.n. + i.n.). No significant difference was observed between the systemic priming-mucosal boosting group (s.c. + i.n.) and the mucosal priming-systemic boosting group (i.n. + s.c.). As shown in Fig. 2c, d, significant differences between IgG1 and IgG2a titers in anti-PspA23 and anti-PspA4 IgG titers were observed in all immunized groups (P < 0.001). These results indicate that the heterologous prime-boost immunization can induce a systemic immune response and elicit a Th2 immune response.

Induction of antigen-specific antibody responses in serum. a PspA23-specific and b PspA4-specific IgG titers. c PspA23-specific and d PspA4-specific IgG1 and IgG2a titers. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant
Heterologous prime-boost immunization induced a significant mucosal immune response
The intranasal administration of the mucosal vaccine (i.n.), but not the systemic immunization of the systemic vaccine (s.c.), induced the production of antigen-specific sIgA antibodies at high levels [32]. To determine whether heterologous prime-boost immunization induces an adequate mucosal immune response, the antigen-specific sIgA antibody levels were determined in BALF and NW samples 2 weeks after the final immunization. As shown in Fig. 3a, b, the heterologous immunization groups (s.c. + i.n. and i.n. + s.c.) displayed a significant increase in anti-PspA2 (P < 0.001 and P < 0.01) and anti-PspA4 sIgA (P < 0.001 and P < 0.05) antibody levels in BALF, although the titers were lower than those in the mucosal immunization group (i.n. + i.n. + i.n.) (P < 0.05). Furthermore, the systemic priming-mucosal boosting group (s.c. + i.n.) significantly induced the production of antigen-specific sIgA antibodies in comparison with the mucosal priming-systemic boosting group (i.n. + s.c.) (P < 0.01). As shown in Fig. 3c, d, the same tendency was observed with anti-PspA2 and anti-PspA4 sIgA antibodies in NW, although the difference between the systemic priming-mucosal boosting group (s.c. + i.n.) and the mucosal priming-systemic boosting group (i.n. + s.c.) was not significant.

Induction of antigen-specific antibody responses in bronchoalveolar lavage fluid (BALF) and nasal washes (NW). a PspA23-specific and b PspA4-specific sIgA titers in BALF. c PspA23-specific and d PspA4-specific secretory IgA titers in NW. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant
These results suggest that heterologous prime-boost immunization can induce a mucosal immune response and systemic priming, and that mucosal boost immunization is more effective at inducing an sIgA response.
Heterologous prime-boost immunization protected mice against a lethal pneumococcal challenge with different family strains
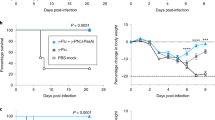
Heterologous prime-boost immunization generated marked IgG and sIgA antibody production, which is important for preventing pneumococcal infections. Hence, we evaluated the protective effect of the combinatorial prime-boost immunization with the vaccines through a pneumococcal challenge. Mice were immunized alone or alternately with a systemic or mucosal vaccine at 2-week intervals. Two weeks after the final immunization, mice were intranasally challenged with two pneumococcal strains of family 1 and family 2 and mortality was monitored for 14 days. The detailed results of the survival analysis are described in Fig. 4. As shown in Fig. 4a, when challenged with a lethal dose of ATCC 10813, all immunized groups displayed a significantly greater protective effect than their corresponding controls. The heterologous immunization groups (s.c. + i.n. and i.n. + s.c.) displayed a 100% survival rate, equivalent to that in the systemic vaccine group (s.c. + s.c. + s.c.), whereas the mucosal vaccine group (i.n. + i.n. + i.n.) only displayed an 80% survival rate. Furthermore, the positive controls of PPV23 and PCV13 displayed 80% and 100% protection, respectively.

Induction of protection against a lethal intranasal pneumococcal challenge. Two weeks after the final immunization, the mice were challenged (i.n.) with an LD90 dose of Streptococcus pneumoniae ATCC 10813 (a) and an LD90 dose of strain ATCC BAA-334 (b). Survival rates were monitored for 14 d (n = 10 per group). Log-rank test: *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant
As shown in Fig. 4b, on using strain ATCC BAA-334 for the pneumococcal challenge, the systemic priming-mucosal boosting group (s.c. + i.n.) displayed a 100% survival rate, which was greater than that of the mucosal vaccine group (i.n. + i.n. + i.n.); however, the mucosal priming-systemic boosting group (i.n. + s.c.) only displayed a 70% survival rate, being slightly lower than that of the mucosal vaccine group (i.n. + i.n. + i.n.), wherein the survival rate was 80%. Moreover, the negative control for BLP displayed a 60% survival rate, suggesting that mucosal application of BLP potentially inhibits S. pneumoniae infections. Furthermore, the commercial positive controls of PPV23 and PCV13 did not provide any protection, although this serotype 4 challenge strain exists in the protection spectrum of PPV23 and PCV13; this could be because we applied high 90% lethal challenge doses, which potentially decreased the performance of these vaccines. A similar discrepancy was observed in the systemic vaccine group (s.c. + s.c. + s.c.), which displayed only a 30% survival rate. All groups immunized with the mucosal vaccine or BLP displayed significantly higher protection than did the PBS negative control group.
These results indicate that the heterologous prime-boost immunization potentially provides effective protection against pneumococcal strains from both family 1 and 2. Furthermore, systemic priming and mucosal boost immunization induced more robust protection than systemic or mucosal immunization alone, and this protection was significant and exerted broad-spectrum effects against different clade strains.
Systemic priming and mucosal boost immunization elicit significant Th1 and Th17 cellular immune responses
To further investigate the type of cell-mediated response contributing to the protective effect of heterologous prime-boost immunization, TNF-α and IL-17A levels were determined in supernatants from splenocyte cultures re-stimulated with the PspA antigen for 48 h, 2 weeks after the final immunization. As shown in Fig. 5a, TNF-α levels in the systemic priming-mucosal boosting group (s.c. + i.n.) and systemic vaccine group (s.c. + s.c. + s.c.) were significantly higher than those in the negative control group (P < 0.01), but significantly lower than those in the mucosal vaccine group (i.n. + i.n. + i.n.) (P < 0.01). Notably, the TNF-α levels did not significantly increase in the mucosal priming-systemic boosting group (i.n. + s.c.).

Induction of Th1 and Th17 cell-mediated immune responses. On day 14 after the final immunization, splenocytes harvested from mice were cultured for 12 h and then stimulated with PBS, PsaA-PspA23, and PspA4. Two days later, TNF-α (a) and IL-17A (b) levels in the supernatant were detected via a CBA assay. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant
As shown in Fig. 5b, IL-17A levels were significantly higher in the prime-boost immunization groups than in the negative control group (P < 0.01 and P < 0.001) and significantly higher in the systemic priming-mucosal boosting group (s.c. + i.n.) than in the mucosal vaccine group (i.n. + i.n. + i.n.) (P < 0.05). The IL-17A levels were significantly lower in the mucosal priming-systemic boosting group (i.n. + s.c.) than in the mucosal vaccine group (i.n. + i.n. + i.n.) (P < 0.05), but comparable to those in the systemic vaccine group (s.c. + s.c. + s.c.). These results indicate that combinatorial systemic priming and mucosal boost immunization elicit an adequate Th1 and Th17 immune response and may increase the potential to induce a Th17 cell-mediated immune response in comparison with systemic or mucosal immunization alone.
Discussion
A pneumococcal protein-based systemic vaccine comprising PspA of families 1 and 2 and a BLP-based mucosal vaccine with PspA2 and PspA4 fragments can induce broad-spectrum protective immune responses against various pneumococcal infections [24, 31]. Although the two vaccines exerted significant protective effects, the immune response of each vaccine was unique owing to differences in adjuvants and optimal immunization routes [32]. Subcutaneous immunization with the systemic vaccine with Al(OH)3 as an adjuvant produced a high level of IgG antibodies, but no IgA antibodies. Intranasal immunization with the mucosal vaccine with BLP as an adjuvant induced the production of not only IgA, but also of IgG at high levels. However, the IgG levels were insufficient to provide complete protection in comparison with that provided by the systemic vaccine. Because mucosal IgA and systemic IgG antibodies constitute first-line defense against invading pathogens, they potentially prevent the colonization and infection of S. pneumoniae after host cell invasion. Therefore, we hypothesized that a combinatorial immunization with the two vaccine forms might markedly augment the protective effects against pneumococci. Therefore, this study evaluated the protective immune responses upon combinatorial immunization with systemic and mucosal vaccines. Herein, we evaluated the efficacy of 2-dose prime-boost immunization regimens using different forms of vaccines. In particular, we aimed to determine whether combinatorial prime-boost immunization provides better and broader protection against lethal pneumococcal infections.
We previously evaluated the immunogenicity and protective effects of systemic and mucosal vaccines, respectively, and reported that homologous prime-boost immunization with only three doses of systemic or mucosal vaccines elicited adequate production of specific IgG or IgA antibodies and provided optimal protection. The present results show that the combinatorial prime-boost immunization with one dose of both systemic and mucosal vaccines was sufficient to significantly increase IgG and IgA production in BALF and NW. In BALF, systemic priming and mucosal boosting immunization elicited significantly higher production of specific IgA than mucosal priming and systemic boosting immunization alone. This indicated that the combinatorial immunization, especially of systemic priming and mucosal boosting immunization, potentially induces a robust humoral immune response, inducing both systemic IgG antibody and mucosal IgA production. Furthermore, this immunization strategy reduces the number of doses and duration of immunization, thus reflecting its effectiveness. However, owing to differences in the pneumococcal strains and specificity of the antibodies, the potential protective effects of the highly specific IgG and IgA antibodies in the present model remain somewhat unclear. Some studies have reported that PspA-specific IgG can enhance complement deposition, thus potentially generating protection through complement-dependent phagocytosis [35, 36]. Furthermore, other studies have reported that PspA-specific IgA prevents bacterial colonization in the nasopharynx and protects against pneumococcal infections [37]. Therefore, it is necessary to determine the combinatorial protection efficiency of the PspA-specific IgG and IgA antibody titers against different pneumococcal strains.
Further studies have reported that combinatorial immunization potentially improves and augments the protective effects of the individual immunizations. The combinatorial prime-boost immunization groups herein displayed significantly increased survival rates after a challenge with different family strains in comparison with the negative control groups. However, the systemic priming-mucosal boosting group consistently displayed the highest survival rates against these strains in comparison with other groups. Homologous prime-boost immunization groups only displayed partial protection. In particular, the systemic vaccine group displayed a 100% survival rate upon a challenge with strain ATCC 10813, but only a 30% survival rate with strain ATCC BAA-334. Similarly, partial protection was observed in the positive controls of PPV23 and PCV13. The mucosal vaccine group displayed an 80% survival rate upon a challenge with the same two strains, but did not achieve complete protection. The BLP vaccine group displayed a 60% survival rate upon a challenge with strain ATCC BAA-334, probably owing to the inhibitory effect of BLP on bacterial colonization in the lungs, concurrent with previous reports on BLP-based vaccines [38, 39]. In general, these results suggest that different types of vaccines exert preferential protective effects on different strains; however, prime-boost immunization with systemic vaccines for priming followed by boosting with the mucosal vaccine displayed the most optimal protective effects among all vaccine groups. The mechanisms underlying the differential protective effects of various immunization regimens remain unclear. Previous studies reported that prime-boost immunization with different types of vaccines elicits unique immune responses for improved immunogenicity and protection [40, 41]. Thus, the aforementioned differences in protective effects not only depend on specific humoral antibodies, but also are potentially closely associated with different forms of cell-mediated immunity.
IgG isotype analysis revealed that both systemic and mucosal vaccines induced high titers of IgG1, indicating a potent induction of the Th2-biased cell-mediated immune response. Similarly, combinatorial immunization with systemic and mucosal vaccines elicited a strong Th2 bias. By measuring antigen-induced cytokine production by the splenocytes, several studies have shown that heterologous prime-boost immunization can induce strong Th1 and Th17 responses [42, 43]. Therefore, we detected the Th1 and Th17 cytokine products secreted upon antigenic stimulation of spleen cells to determine whether the combined prime-boost immunity of our vaccine could also elicit strong cellular immune responses. Cytokine analysis revealed that the mucosal vaccine induced higher levels of TNF-α and IL-17A than the systemic vaccine, suggesting a higher potential of the mucosal vaccine to induce Th1- and Th17-type immune responses. These findings are concurrent with those of previous studies on vaccine adjuvants, wherein an Alu-based vaccine elicited only a Th2 antibody response, while the BLP-based vaccine elicited Th1 and Th17 cell-mediated responses (by interacting with TLR2) [27, 44]. However, it is rather unexpected that systemic priming and mucosal boosting immunization not only induced peak IL-17A production, but also provided optimal protection. These results confirm the importance of IL-17 in protecting against S. pneumoniae [45]. Other studies have reported that the IL-17-mediated protective mechanism promotes pneumococcal clearance through recruitment and activation of neutrophils and macrophages [46]. Moreover, the protective efficacy of Th-17 mediated protein-specific immune responses has become increasingly apparent and has been reported for numerous pneumococcal protein-based vaccines [47, 48]. Therefore, further studies are required to investigate the specific association between the Th17 cell-mediated immune response and protective ability of the present combinatorial vaccines. Furthermore, although the present results indicate combinatorial involvement of the Th1, Th2, and Th17 components of the immune system in response to immunization with systemic or mucosal vaccines, the Th17 immune response potentially plays a more prominent role in the combinatorial immune-elicited protection reported herein.
An ideal vaccination regimen is expected to induce adequate humoral and cellular immune responses to provide protection. In this study, the systemic and mucosal immunization induced the highest levels of IgG or IgA antibodies, but did not induce dominant IL-17A production or provide complete protection, consistent with their antibody levels. In contrast, systemic priming and mucosal boosting immunization not only induced peak IL-17A production, but also provided optimal protection, although its IgG or IgA antibody levels were not the highest. This inconsistency was also reported in our previous study, wherein humoral antibodies were found to be effective in promoting the clearance of pneumococci, but protection against lethal pneumococcal infection required both humoral and cellular immune responses [32, 44]. Other studies have also confirmed the important role of cellular and humoral immunity in jointly providing protection against pneumococcal infection [49, 50]. In addition, we acknowledge that our understanding of the protective mechanism induced by this vaccination regimen is only partial. Therefore, future investigation on the combined effects of both humoral and cellular immune responses triggered by the combinatorial immunization in our models will be required.
In conclusion, this study shows the potential benefits of combinatorial prime-boost immunization with a systemic and mucosal vaccine to induce heterologous protection against pneumococcal strains. The present results suggest that the combinatorial systemic priming and mucosal boosting immunization strategy in mice yields optimal protection against a lethal challenge of pneumococcal strains of different families. Further studies are required to illustrate the mechanism underlying the role of combinatorial immunization in the protection against pneumococcal infections. The present results provide insights into the development and optimization of an effective pneumococcal vaccine.
References
Mitchell TJ. Streptococcus pneumoniae: infection, inflammation and disease. Adv Exp Med Biol. 2006;582:111–24.
Zhanel GG, Wolter KD, Karlowsky JA. Clinical cure rates in subjects treated with azithromycin for community-acquired respiratory tract infections caused by azithromycin-susceptible or azithromycin-resistant Streptococcus pneumoniae: analysis of phase 3 clinical trial data-authors' response. J Antimicrob Chemother. 2015;70:3170–1.
Bryce J, Boschi-Pinto C, Shibuya K, Black RE, Group WHOCHER. WHO estimates of the causes of death in children. Lancet. 2005;365:1147–52.
O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet. 2009;374:893–902.
Mera R, Miller LA, Fritsche TR, Jones RN. Serotype replacement and multiple resistance in Streptococcus pneumoniae after the introduction of the conjugate pneumococcal vaccine. Microb Drug Resist. 2008;14:101–7.
Dagan R, Poolman J, Siegrist CA. Glycoconjugate vaccines and immune interference: a review. Vaccine. 2010;28:5513–23.
Deng X, Church D, Vanderkooi OG, Low DE, Pillai DR. Streptococcus pneumoniae infection: a Canadian perspective. Expert Rev Anti-Infect Ther. 2013;11:781–91.
Tai SS. Streptococcus pneumoniae protein vaccine candidates: properties, activities and animal studies. Crit Rev Microbiol. 2006;32:139–53.
Moffitt KL, Malley R. Next generation pneumococcal vaccines. Curr Opin Immunol. 2011;23:407–13.
Ochs MM, Williams K, Sheung A, Lheritier P, Visan L, Rouleau N, et al. A bivalent pneumococcal histidine triad protein D-choline-binding protein A vaccine elicits functional antibodies that passively protect mice from Streptococcus pneumoniae challenge. Hum Vaccin Immunother. 2016;12:2946–52.
Xu Q, Pryharski K, Pichichero ME. Trivalent pneumococcal protein vaccine protects against experimental acute otitis media caused by Streptococcus pneumoniae in an infant murine model. Vaccine. 2017;35:337–44.
Odutola A, Ota MOC, Antonio M, Ogundare EO, Saidu Y, Foster-Nyarko E, et al. Efficacy of a novel, protein-based pneumococcal vaccine against nasopharyngeal carriage of Streptococcus pneumoniae in infants: a phase 2, randomized, controlled, observer-blind study. Vaccine. 2017;35:2531–42.
Orihuela CJ, Radin JN, Sublett JE, Gao G, Kaushal D, Tuomanen EI. Microarray analysis of pneumococcal gene expression during invasive disease. Infect Immun. 2004;72:5582–96.
Ren B, Szalai AJ, Hollingshead SK, Briles DE. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect Immun. 2004;72:114–22.
Hollingshead SK, Becker R, Briles DE. Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect Immun. 2000;68:5889–900.
Daniels CC, Coan P, King J, Hale J, Benton KA, Briles DE, et al. The proline-rich region of pneumococcal surface proteins A and C contains surface-accessible epitopes common to all pneumococci and elicits antibody-mediated protection against sepsis. Infect Immun. 2010;78:2163–72.
Vadesilho CF, Ferreira DM, Gordon SB, Briles DE, Moreno AT, Oliveira ML, et al. Mapping of epitopes recognized by antibodies induced by immunization of mice with PspA and PspC. Clin Vaccine Immunol. 2014;21:940–8.
Hollingshead SK, Baril L, Ferro S, King J, Coan P, Briles DE, et al. Pneumococcal surface protein A (PspA) family distribution among clinical isolates from adults over 50 years of age collected in seven countries. J Med Microbiol. 2006;55:215–21.
Croney CM, Coats MT, Nahm MH, Briles DE, Crain MJ. PspA family distribution, unlike capsular serotype, remains unaltered following introduction of the heptavalent pneumococcal conjugate vaccine. Clin Vaccine Immunol. 2012;19:891–6.
Moreno AT, Oliveira ML, Ferreira DM, Ho PL, Darrieux M, Leite LC, et al. Immunization of mice with single PspA fragments induces antibodies capable of mediating complement deposition on different pneumococcal strains and cross-protection. Clin Vaccine Immunol. 2010;17:439–46.
Briles DE, Hollingshead SK, King J, Swift A, Braun PA, Park MK, et al. Immunization of humans with recombinant pneumococcal surface protein A (rPspA) elicits antibodies that passively protect mice from fatal infection with Streptococcus pneumoniae bearing heterologous PspA. J Infect Dis. 2000;182:1694–701.
Qian J, Yao K, Xue L, Xie G, Zheng Y, Wang C, et al. Diversity of pneumococcal surface protein A (PspA) and relation to sequence typing in Streptococcus pneumoniae causing invasive disease in Chinese children. Eur J Clin Microbiol Infect Dis. 2012;31:217–23.
Brandileone MC, Andrade AL, Teles EM, Zanella RC, Yara TI, Di Fabio JL, et al. Typing of pneumococcal surface protein A (PspA) in Streptococcus pneumoniae isolated during epidemiological surveillance in Brazil: towards novel pneumococcal protein vaccines. Vaccine. 2004;22:3890–6.
Yu J, Chen X, Li B, Gu T, Meng X, Kong W, et al. A pneumococcal vaccine combination with two proteins containing PspA families 1 and 2 can potentially protect against a wide range of Streptococcus pneumoniae strains. Immunol Res. 2018;66:528–36.
Ramirez K, Ditamo Y, Rodriguez L, Picking WL, van Roosmalen ML, Leenhouts K, et al. Neonatal mucosal immunization with a non-living, non-genetically modified Lactococcus lactis vaccine carrier induces systemic and local Th1-type immunity and protects against lethal bacterial infection. Mucosal Immunol. 2010;3:159–71.
Van Braeckel-Budimir N, Haijema BJ, Leenhouts K. Bacterium-like particles for efficient immune stimulation of existing vaccines and new subunit vaccines in mucosal applications. Front Immunol. 2013;4:282.
Keijzer C, Haijema BJ, Meijerhof T, Voorn P, de Haan A, Leenhouts K, et al. Inactivated influenza vaccine adjuvanted with bacterium-like particles induce systemic and mucosal influenza A virus specific T-cell and B-cell responses after nasal administration in a TLR2 dependent fashion. Vaccine. 2014;32:2904–10.
Joris B, Englebert S, Chu CP, Kariyama R, Daneo-Moore L, Shockman GD, et al. Modular design of the Enterococcus hirae muramidase-2 and Streptococcus faecalis autolysin. FEMS Microbiol Lett. 1992;70:257–64.
Bateman A, Bycroft M. The structure of a LysM domain from E. coli membrane-bound lytic murein transglycosylase D (MltD). J Mol Biol. 2000;299:1113–9.
Steen A, Palumbo E, Deghorain M, Cocconcelli PS, Delcour J, Kuipers OP, et al. Autolysis of Lactococcus lactis is increased upon D-alanine depletion of peptidoglycan and lipoteichoic acids. J Bacteriol. 2005;187:114–24.
Lu J, Guo J, Wang D, Yu J, Gu T, Jiang C, et al. Broad protective immune responses elicited by bacterium-like particle-based intranasal pneumococcal particle vaccine displaying PspA2 and PspA4 fragments. Hum Vaccin Immunother. 2019;15:371–80.
Yu J, Li B, Chen X, Lu J, Wang D, Gu T, et al. Comparison of immunogenicity and protection of two pneumococcal protein vaccines based on PsaA and PspA. Infect Immun. 2018;86.
Lu J, Sun T, Wang D, Dong Y, Xu M, Hou H, et al. Protective immune responses elicited by fusion protein containing PsaA and PspA fragments. Immunol Investig. 2015;44:482–96.
Audouy SA, van Roosmalen ML, Neef J, Kanninga R, Post E, van Deemter M, et al. Lactococcus lactis GEM particles displaying pneumococcal antigens induce local and systemic immune responses following intranasal immunization. Vaccine. 2006;24:5434–41.
Mukerji R, Mirza S, Roche AM, Widener RW, Croney CM, Rhee DK, et al. Pneumococcal surface protein A inhibits complement deposition on the pneumococcal surface by competing with the binding of C-reactive protein to cell-surface phosphocholine. J Immunol. 2012;189:5327–35.
Ren B, Li J, Genschmer K, Hollingshead SK, Briles DE. The absence of PspA or presence of antibody to PspA facilitates the complement-dependent phagocytosis of pneumococci in vitro. Clin Vaccine Immunol. 2012;19:1574–82.
Fukuyama Y, King JD, Kataoka K, Kobayashi R, Gilbert RS, Oishi K, et al. Secretory-IgA antibodies play an important role in the immunity to Streptococcus pneumoniae. J Immunol. 2010;185:1755–62.
Bruna-Romero O, Rocha CD, Tsuji M, Gazzinelli RT. Enhanced protective immunity against malaria by vaccination with a recombinant adenovirus encoding the circumsporozoite protein of Plasmodium lacking the GPI-anchoring motif. Vaccine. 2004;22:3575–84.
Nganou-Makamdop K, van Roosmalen ML, Audouy SA, van Gemert GJ, Leenhouts K, Hermsen CC, et al. Bacterium-like particles as multi-epitope delivery platform for Plasmodium berghei circumsporozoite protein induce complete protection against malaria in mice. Malar J. 2012;11:50.
Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol. 2009;21:346–51.
Lu S. Two is better than one. Lancet Infect Dis. 2011;11:889–91.
Qiu Y, Zhang X, Wang H, Zhang X, Mo Y, Sun X, et al. Heterologous prime-boost immunization with live SPY1 and DnaJ protein of Streptococcus pneumoniae induces strong Th1 and Th17 cellular immune responses in mice. J Microbiol. 2017;55:823–9.
Feunou PF, Kammoun H, Debrie AS, Locht C. Heterologous prime-boost immunization with live attenuated B. pertussis BPZE1 followed by acellular pertussis vaccine in mice. Vaccine. 2014;32:4281–8.
Chen X, Li B, Yu J, Zhang Y, Mo Z, Gu T, et al. Comparison of four adjuvants revealed the strongest protection against lethal pneumococcal challenge following immunization with PsaA-PspA fusion protein and AS02 as adjuvant. Med Microbiol Immunol. 2019;208:215–26.
Lu YJ, Gross J, Bogaert D, Finn A, Bagrade L, Zhang Q, et al. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog. 2008;4:e1000159.
Zhang Z, Clarke TB, Weiser JN. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest. 2009;119:1899–909.
Xu Q, Surendran N, Verhoeven D, Klapa J, Ochs M, Pichichero ME. Trivalent pneumococcal protein recombinant vaccine protects against lethal Streptococcus pneumoniae pneumonia and correlates with phagocytosis by neutrophils during early pathogenesis. Vaccine. 2015;33:993–1000.
Elhaik Goldman S, Dotan S, Talias A, Lilo A, Azriel S, Malka I, et al. Streptococcus pneumoniae fructose-1,6-bisphosphate aldolase, a protein vaccine candidate, elicits Th1/Th2/Th17-type cytokine responses in mice. Int J Mol Med. 2016;37:1127–38.
Cohen JM, Khandavilli S, Camberlein E, Hyams C, Baxendale HE, Brown JS. Protective contributions against invasive Streptococcus pneumoniae pneumonia of antibody and Th17-cell responses to nasopharyngeal colonisation. PLoS One. 2011;6:e25558.
Wilson R, Cohen JM, Jose RJ, de Vogel C, Baxendale H, Brown JS. Protection against Streptococcus pneumoniae lung infection after nasopharyngeal colonization requires both humoral and cellular immune responses. Mucosal Immunol. 2015;8:627–39.
Acknowledgments
We gratefully acknowledge Editage (www.editage.cn) for the editorial support in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
All mouse experiments in this paper were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, Y., Guo, X., Guo, M. et al. Combined prime-boost immunization with systemic and mucosal pneumococcal vaccines based on Pneumococcal surface protein A to enhance protection against lethal pneumococcal infections. Immunol Res 67, 398–407 (2019). https://doi.org/10.1007/s12026-019-09107-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-019-09107-6