
Abstract
Systemic sclerosis (SSc) is an intractable multifaceted disease with high mortality. Although its pathogenesis is not fully understood, recent studies have advanced our knowledge on SSc. The cardinal pathological features of SSc are autoimmunity, vasculopathy, and fibrosis. The B cells in SSc are constitutively activated and lead to the production of a plethora of autoantibodies, such as anti-topoisomerase I and anti-centromere antibodies. In addition to these autoantibodies, which are valuable for diagnostic criteria or biomarkers, many other autoantibodies targeting endothelial cells, including endothelin type A receptor and angiotensin II type I receptor, are known to be functional and induce activation or apoptosis of endothelial cells. The autoantibody-mediated endothelial cell perturbation facilitates inflammatory cell infiltration, cytokine production, and myofibroblastic transformation of fibroblasts and endothelial cells. Profibrotic cytokines, such as transforming growth factor β, connective tissue growth factor, interleukin 4/interleukin 13, and interleukin 6, play a pivotal role in collagen production from myofibroblasts. Specific treatments targeting these causative molecules may improve the clinical outcomes of patients with SSc. In this review, we summarize recent topics on the pathogenesis (autoantibodies, vasculopathy, and fibrosis), animal models, and emerging treatments for SSc.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The prevalence of systemic sclerosis (SSc) ranges from 0.7/100,000 to 53/100,000 depending on the different ethnicities, and there are higher numbers in the USA than in Europe or Japan [1]. In a nationwide, cross-sectional hospital-based study in Japan, patients with SSc comprised 0.92% of dermatological patients [2]. SSc has a high mortality rate due to the development of SSc-associated interstitial lung disease and pulmonary arterial hypertension, which are the most frequent causes of disability and mortality in SSc [3,4,5].
Depending on the extent of skin involvement, SSc is classified into the following two main subtypes: limited cutaneous SSc and diffuse cutaneous SSc. Given the heterogeneity of clinical symptoms and signs, the American College of Rheumatology/the European League against Rheumatism developed new classification criteria in 2013 [6,7,8].
SSc is not an inherited disease, therefore, twins show a low disease concordance rate (4.7%) that is similar between monozygotic and dizygotic twin pairs [9]. However, genetic factors contribute to its susceptibility, as shown by a 60-fold higher occurrence of the disease in families compared to the general population [10,11,12]. Genetic linkage studies and genome-wide association studies have identified many susceptible genes, including the major histocompatibility complex (MHC) class II as well as non-MHC genes related to the metabolism of extracellular matrix molecules, innate immunity, macrophage activation, and T cell functions [10,11,12].
Systemic sclerosis (SSc) is a multisystem connective tissue disease characterized by three cardinal pathological features, including aberrant immune activation, vasculopathy, and tissue fibrosis with an unknown aetiology [3, 4, 13]. Although its pathogenesis is not fully understood, recent studies have advanced our knowledge on SSc. In this review, we summarize recent topics on the pathogenesis (autoantibodies, vasculopathy, and fibrosis), animal models, and emerging treatments for SSc.
Pathogenesis
Pathogenesis of SSc remains unknown, whereas it is commonly thought that autoimmunity and vasculopathy precede fibroblast activation and interstitial fibrosis [14, 15]. Autoantibodies to endothelial cells, ischemia–reperfusion injury following Raynaud’s phenomenon, generation of reactive oxygen species (ROS) with inflammatory cell infiltration, and subsequent cytokine production trigger myofibroblastic transformation of endothelial cells as well as fibroblasts and induce excessive production of collagens and other extracellular matrices [5, 8, 14, 15]. Various profibrotic cytokines, such as transforming growth factor β (TGFβ), interleukin (IL)-4, IL-13, IL-6, and IL-33 are likely to be involved in the development of fibrosis [13, 14, 16,17,18].
Autoantibody and vasculopathy
The immune system activation is exemplified by the activation of B cells and the production of autoantibodies [19,20,21,22,23]. B cells from patients with SSc overexpress the B cell stimulatory receptor CD19 by 54% in patients with early SSc and by 28% in those with long-standing disease compared to normal controls [22,23,24]. Notably, a small increase (15–29%) in the CD19 expression of B cells in transgenic mice induced autoantibody production [25]. In contrast, the function of CD22, an inhibitory B cell molecule, is inhibited by anti-CD22 autoantibodies present in patients with SSc [22, 23, 25]. An increased CD19/CD22 ratio may facilitate the sustained activation of B cells and consequent overproduction of various autoantibodies [22, 23, 25, 26].
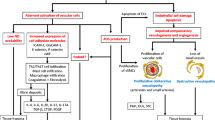
Anti-nuclear antibodies are found in the sera of the vast majority of SSc patients, and their antigenic specificity significantly correlates with the clinical characteristics of the disease [19,20,21]. Autoantibodies are currently the most reliable biomarkers for diagnosis, classification, and prediction of specific clinical features of SSc [19,20,21]. Certain autoantibodies, such as anti-topoisomerase I, anti-centromere, anti-RNAP III, anti-U3 RNP, anti-Th/To, and anti-U1 RNP antibodies, are closely associated with distinct clinical features and disease activities of SSc [19,20,21, 27, 28] (Fig. 1). Other autoantibodies targeting a variety of cytoplasmic, cell membrane, and extracellular autoantigens were also detected in SSc [21, 23]. Some of them, such as anti-endothelial cell, anti-intercellular adhesion molecule-1 (ICAM-1), anti-endothelin type A receptor (ETAR), and anti-angiotensin II type I receptor (AT1R) antibodies, may be functional and pathogenic [21, 23]. In addition to autoantibodies, a plethora of biomarkers related to immune reactions [29, 30], endothelial cell function [31, 32], and the extracellular matrix [33, 34] have been reported recently [19].

Pathogenesis of systemic sclerosis. The cardinal pathological features of systemic sclerosis (SSc) are autoantibodies, vasculopathy, and fibrosis. B cells in SSc are constitutively activated with increased CD19 and decreased CD22 expression, which leads to the overproduction of a plethora of autoantibodies, such as anti-topoisomerase I, anti-RNAP III, anti-U3 RNP, anti-centromere, anti-Th/To, and anti-U1 RNP antibodies. These autoantibodies are associated with characteristic clinical features, such as diffuse SSc (dSSc), limited SSc (lSSc), interstitial lung disease (ILD), digital ulcer, renal crisis, malignancy, cardiomyopathy, myopathy, pulmonary artery hypertension (PAH), and overlap syndrome. Many other functional autoantibodies targeting endothelial cells, intercellular adhesion molecule 1 (ICAM-1), endothelin type A receptor (ETAR), and angiotensin II type I receptor (AT1R) as well as platelet-derived growth factor receptor (PDGFR) are known to induce activation or apoptosis of endothelial cell (EC). The stimulated ECs express reactive oxygen species (ROS), transforming growth factor β (TGFβ), interleukin-8 (IL-8), and vascular endothelial cell adhesion molecule 1 (VCAM-1). The autoantibody-mediated endothelial cell perturbation facilitates inflammatory cell (Inf) infiltration, cytokine production, and myofibroblastic transformation of fibroblasts (Fb) and ECs. Profibrotic cytokines, such as TGFβ, connective tissue growth factor (CTGF), IL-6, and IL-4/IL-13, play a pivotal role in collagen production from myofibroblasts (MFb). Perturbation of ECs and consequent profibrotic processes result in vasculopathy (including capillaroscopic abnormalities and intimal fibrosis) and tissue fibrosis (including skin fibrosis and ILD)
Raynaud’s phenomenon and abnormal nailfold capillary changes, which are the representative vascular manifestations of SSc, often appear before the onset of sclerosis [8, 15, 35]. Raynaud’s phenomenon is the abnormal thermal regulation of blood flow that is probably triggered by endothelial injury [15]. The skin fibrosis tends to occur on locations, such as fingers, distal extremities, and face, which are frequently exposed to cold temperatures [15]. Nailfold capillaroscopy reveals structural alterations in capillaries that include dilatation, distortion, and microhemorrhages that lead to progressive loss of the capillaries (Fig. 1) [36]. Microscopically, capillary damage of SSc is characterized by endothelial apoptosis, intimal and medial fibrous thickening, and adventitial fibrosis with perivascular infiltration of the macrophages, B cells, and T cells [15]. Precapillary arterioles then show endothelial proliferation and mononuclear inflammatory infiltrates followed by intimal proliferation and luminal narrowing [37]. The origin of the cells that populate the intima and participate in collagen production remains unknown. However, activated resident fibroblasts, circulating fibroblast precursors (fibrocytes), the endothelial cells, and pericytes have all been implicated [15]. In line with these findings, an endothelial cell to mesenchymal transition is shown in the lesional vascular systems in SSc [5].
The endothelial apoptosis or activation is likely mediated by functional autoantibodies [23, 38]. Anti-endothelial cell antibodies cause endothelial cell apoptosis [39]. Anti-ICAM-1 antibodies induce the production of ROS and expression of vascular cell adhesion molecule-1 (VCAM-1) which may facilitate the attachment of immune cells [40]. Anti-ETAR and anti-AT1R autoantibodies, which are detected in most SSc patients, are agonistic antibodies that upregulate the expression of TGF-β, IL-8, and VCAM-1 of endothelial cells and cause fibrosis, vasoconstriction, and recruitment of immune cells [41,42,43]. Both AT1R and ETAR are expressed on cells of both the vascular and immune system [38]. Angiotensin II increased the production of TGFβ and collagen by fibroblasts via AT1R [44]. Endothelin-1 also induced collagen production and vasoconstriction via ETAR [45]. The expressions of ETAR and AT1R are found to be highest in patients with early SSc [46]. The anti-ETAR and anti-AT1R antibodies from SSc patients induce obliterative vasculopathy when injected into mice [43]. The endothelial cell apoptosis has been demonstrated in the skin lesions of patients with SSc as well as in avian SSc models [47]. Moreover, many SSc patients have antibodies against a human cytomegalovirus-derived UL94 protein that shares homology with NAG-2 (tetraspan novel antigen-2), which is expressed on the surface of human endothelial cell and fibroblast [48, 49]. The anti-UL94 peptide antibodies bind to NAG-2 and induce “scleroderma-like” gene expression in endothelial cells and fibroblasts [48, 49]. Thus, the molecular mimicry mechanism links antibodies against the human-cytomegalovirus-derived protein UL94 to the pathogenesis of systemic sclerosis [48, 49].
Fibrosis
The degree of cell infiltration correlates with both the degree and progression of skin thickening, which results from excessive accumulation of type 1 collagen and extracellular matrix proteins [3, 8, 50, 51]. These infiltrated cells, especially endothelial cells and fibroblasts, are potential candidates for producing various cytokines, chemokines, and growth factors that induce fibrosis [3, 8, 50, 51]. Despite the differences in the extent and distribution of skin involvement, both limited and diffuse type SSc produce marked fibrosis followed by the preceded autoimmunity, vasculopathy, and perivascular inflammatory cell infiltration [8, 50, 51].
Representative profibrotic growth factors and cytokines encompass TGFβ, connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), IL-6, and IL-4/IL-13 (Fig. 1). After ligation with TGFβ receptor type II (TGFβRII) and TGFβRI, TGFβ induces gene transcription of type I collagen, α smooth muscle actin (αSMA), and CTGF via SMAD2/SMAD3 phosphorylation [3]. An increased TGFβRI/TGFβRII ratio was found in the SSc fibroblasts, which was implicated in the overactivation of TGFβ signaling [52]. The expression of αSMA is a hallmark of myofibroblastic transformation of activated fibroblasts, which is also frequently detected in SSc [53, 54]. CTGF is a cysteine-rich modular protein belonging to the CCN family of matricellular growth factors, all of which function as adaptor molecules connecting the cell surface to the extracellular matrix [3, 55]. The expression of CTGF is induced by TGF-β, ET-1, and hypoxia [3, 55]. The levels of CTGF are markedly elevated in the lesional skin of SSc patients [56] and in mouse models of scleroderma [57].
The expression of PDGF and its receptors is elevated in SSc fibroblasts and in the lesional skin [58]. Antibodies that stimulate the PDGF receptor (PDGFR) were frequently found in SSc patients [59]. The autoantibodies recognize native PDGFR and selectively activate Ha-Ras-ERK1/2 and ROS cascades to induce type I collagen gene expression and myofibroblast phenotype conversion in normal human fibroblasts [59]. The activated B cells secreted IL-6, which directly stimulates fibroblasts [60,61,62]. IL-6 induces the type 1 collagen expression via enhancing TGFβ-Smad3 signalling pathway [62]. Both IL-4 and IL-13 likely activate fibroblasts and induce type 1 collagen synthesis via a TGFβ-independent approach [63,64,65]. Serum levels of IL-4 and IL-13 were significantly higher in patients with SSc compared to normal controls [66]. It is likely that IL-13, rather than IL-4, plays a more dominant role in fibrosis [17, 67]. Notably, CD8+ T cells producing IL-13 have been shown to have skin-homing receptors in SSc skin and they upregulate type 1 collagen production when incubated with healthy dermal fibroblasts [68].
Animal models of systemic sclerosis
Inducible fibrotic models
Among inducible animal models, the most widely used and established ones are a bleomycin-induced SSc model and a sclerodermatous graft-versus-host disease model in mice [13, 69,70,71]. Recently, three new inducible murine models of SSc have been established by utilizing ROS [72], DNA topoisomerase I antigen with complete Freund’s adjuvant [73], and angiotensin II [74]. Basically, the focus of these inducible models was put on the induction of fibrosis tissue and, to a lesser extent, on the immunological aspects of SSc, instead of its vascular pathology [13].
Genetic animal models
The best-characterized genetic animal models of SSc are tight skin-1 (Tsk-1) mice, in which an in-frame tandem partial reduplication of the Fbn1 gene encoding fibrillin-1, which is a major component of microfibrils mediating elastic fiber assembly, is responsible for the phenotype [75]. Although the Tsk-1 mice exhibit TGFβ upregulation with the aberrant activation of B cells and autoantibody production, the fibrosis of Tsk-1 mice occurs in the hypodermis, which is not observed in human SSc [13]. Mice with constitutively active TGFβRI recapitulate clinical, histological, and biochemical features of human SSc [76]. In addition, mice expressing the Ctgf gene in fibroblasts [77] or Wnt10b in adipocytes [78] exhibit extensive fibrosis in the dermis and internal organs.
New genetic animal models
Fra-2 is one of the components of Fos (c-Fos, Fra-1, Fra-2, FosB) which dimerize with Jun (c-Jun, JunB, JunD) subunits to make AP-1 transcription factor [13]. The Fra2 transgenic mice developed dermal and pulmonary fibrosis following the apoptosis of endothelial cells [79, 80]. Urokinase-type plasminogen activator receptor (uPAR) is a glycosylphosphatidylinositol-anchored cell surface receptor expressed by several cell types, including lymphohaematopoietic cells, fibroblasts, and endothelial cells [13]. A major role of uPAR is to concentrate its ligand, uPA, at the cell-matrix interface. uPA has a serine protease activity that induces the conversion of plasminogen to plasmin and the activation of growth factors and pro-enzymes, such as matrix metalloproteinases [13]. Interestingly, the cleavage and/or inactivation of uPA/uPAR is associated with the transition of fibroblasts to myofibroblasts and subsequent fibrosis as well as with the functional and structural abnormalities of microvasculature in SSc [81,82,83,84]. In line with these findings, uPAR-deficient mice recapitulate the fibrotic and vascular features of SSc [85]. However, both Fra2 transgenic mice and uPAR-null mice totally lack immunological aspects, specifically autoantibody production, in SSc (Table 1) [13].
In contrast to these animal models, Klf5 +/−-; Fli1 +/− mice develop immune activation, vasculopathy, and fibrosis, which are the three cardinal features of SSc [13, 86]. Fli1 is a member of the Ets transcription factor family expressed in endothelial and hematopoietic cells under physiological conditions, and to a lesser extent in dermal fibroblasts [13]. Fli1 exerts a potent repressor of the COL1A1 and COL1A2 gene expression in dermal fibroblasts [87, 88]. The expression of Fli1 was decreased in dermal fibroblasts, dermal microvascular endothelial cells, and perivascular inflammatory cells in the lesional and non-lesional skin of SSc patients, especially in the lesional skin [88]. Fli1 haploinsufficiency is enough to reduce the production of type I collagen [87, 88]. However, Fli1 haploinsufficiency had no effect on CTGF expression [86]. Therefore, some additional factors are mandatory to upregulate the CTGF expression that is also the hallmark feature of dermal fibroblasts derived from SSc patients [13, 56]. Kruppel-like factor 5 (KLF5) is a potent repressor of the Ctgf gene [86]. Moreover, KLF5 was downregulated in the lesional skin of SSc [89]. Thus, Klf5 +/− ; Fli1 +/− double haploinsufficiency mice spontaneously develop remarkable dermal fibrosis, which is characterized by the increase in dermal thickness, the amount of collagen content, and the mRNA of the Col1a1, Col1a2, and Ctgf genes, at the age of 3 months [86]. In addition, the pathological cascade from vasculopathy (stenosis of arterioles and bushy capillaries) to tissue fibrosis was also present in these mice [86]. Furthermore, the Klf5 +/− ; Fli1 +/− mice mimicked the immunological aspects of human SSc, including increased CD19 expression in B cells, upregulated production of IL-6, and the production of autoantibodies (Table 1) [86].
Treatments and therapeutic perspectives
Although the treatment outcomes are not satisfactory, immunosuppressive agents, such as corticosteroids [8], methotrexate [90, 91], cyclophosphamide [92, 93], mycophenolate mofetil [94, 95], and intravenous immunoglobulin [96, 97], are currently being used [98]. ETAR antagonist and angiotensin-converting enzyme blockade have been used to treat clinical complications related to vasculopathies of patients with SSc [99, 100]. Injection of botulinum toxin is useful for preventing Raynaud’s phenomenon [101].
B cell depletion by rituximab (anti-CD20 antibody) improves lung function and skin thickening in patients with SSc [102,103,104]. Fresolimumab, a neutralizing antibody that targets all three isoforms of TGFβ, also decreases skin fibrosis with histological reduction of dermal myofibroblasts [105]. As for the anti-IL-6 receptor α antibody, a randomized controlled trial revealed that tocilizumab tended to reduce the skin thickening compared to placebo, but not significantly [106, 107]. Rapamycin binds to the FK-506 binding protein 12 and inhibits the function of mammalian target of rapamycin. Rapamycin prevents fibrosis of the skin and lungs as well as autoantibody production in a murine model of SSc [108]. In a phase I, single-blinded, randomized, parallel trial of rapamycin versus methotrexate, rapamycin was just as effective as methotrexate for skin sclerosis [109]. Pirfenidone, a pyridine with a simple chemical structure, is an antifibrotic agent, and it has been approved for the treatment of idiopathic pulmonary fibrosis worldwide [110, 111]. Although the molecular mechanisms underlying the antifibrotic effects of pirfenidone are not completely understood, it is thought to work predominantly by modulating TGFβ and TNFα signaling [8, 112]. Further clinical studies are needed to prove the effectiveness of pirfenidone on the skin and pulmonary fibrosis in SSc.
Conclusion
Pathogenesis of SSc is dominated by complex interactions between vascular, immunological, and fibrotic processes, but it is still poorly understood. SSc is an intractable disease with high mortality. Research efforts towards understanding the cellular and molecular basis of scleroderma have aimed to reveal novel molecular targets and diagnostic agents, which has led to early and accurate diagnosis as well as innovative therapies against this disease [10, 112]. The development of preclinical models, including animal models that accurately recapitulate human disease, will be essential tools for the ultimate goal of finding a cure for this disease. The list of potential molecular targets for the treatment of fibrosis is growing. Several of those studies to target pathogenetic molecules have direct translational implications for treating SSc in the very near future [6, 11, 111].
References
Distler O, Cozzio A. Systemic sclerosis and localized scleroderma—current concepts and novel targets for therapy. Semin Immunopathol. 2016;38(1):87–95.
Furue M, Yamazaki S, Jimbow K, Tsuchida T, Amagai M, Tanaka T, et al. Prevalence of dermatological disorders in Japan: a nationwide, cross-sectional, seasonal, multicenter, hospital-based study. J Dermatol. 2011;38(4):310–20.
Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117(3):557–67.
Desbois AC, Cacoub P. Systemic sclerosis: an update in 2016. Autoimmun Rev. 2016;15(5):417–26.
Jimenez SA, Piera-Velazquez S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of systemic sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. 2016;51:26–36.
van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–47.
Toki S, Motegi S, Yamada K, Uchiyama A, Kanai S, Yamanaka M, et al. Clinical and laboratory features of systemic sclerosis complicated with localized scleroderma. J Dermatol. 2015;42(3):283–7.
Yanaba K. Strategy for treatment of fibrosis in systemic sclerosis: present and future. J Dermatol. 2016;43(1):46–55.
Feghali-Bostwick C, Medsger TA Jr, Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 2003;48(7):1956–63.
Fuschiotti P. Current perspectives on the immunopathogenesis of systemic sclerosis. Immunotargets Ther. 2016;5:21–35.
Makino T, Jinnin M. Genetic and epigenetic abnormalities in systemic sclerosis. J Dermatol. 2016;43(1):10–8.
Murdaca G, Contatore M, Gulli R, Mandich P, Puppo F. Genetic factors and systemic sclerosis. Autoimmun Rev. 2016;15(5):427–32.
Asano Y. Recent advances in animal models of systemic sclerosis. J Dermatol. 2016;43(1):19–28.
Vettori S, Cuomo G, Iudici M, D’Abrosca V, Giacco V, Barra G, et al. Early systemic sclerosis: serum profiling of factors involved in endothelial, T-cell, and fibroblast interplay is marked by elevated interleukin-33 levels. J Clin Immunol. 2014;34(6):663–8.
Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013;65(8):1953–62.
Denton CP. Advances in pathogenesis and treatment of systemic sclerosis. Clin Med (Lond). 2016;16(1):55–60.
O’Reilly S. Role of interleukin-13 in fibrosis, particularly systemic sclerosis. Biofactors. 2013;39(6):593–6.
Bergmann C, Distler JH. Canonical Wnt signaling in systemic sclerosis. Lab Investig. 2016;96(2):151–5.
Hasegawa M. Biomarkers in systemic sclerosis: their potential to predict clinical courses. J Dermatol. 2016;43(1):29–38.
Perosa F, Prete M, Di Lernia G, Ostuni C, Favoino E, Valentini G. Anti-centromere protein a antibodies in systemic sclerosis: significance and origin. Autoimmun Rev. 2016;15(1):102–9.
Günther J, Rademacher J, van Laar JM, Siegert E, Riemekasten G. Functional autoantibodies in systemic sclerosis. Semin Immunopathol. 2015;37(5):529–42.
Yoshizaki A. B lymphocytes in systemic sclerosis: abnormalities and therapeutic targets. J Dermatol. 2016;43(1):39–45.
Sakkas LI, Bogdanos DP. Systemic sclerosis: new evidence re-enforces the role of B cells. Autoimmun Rev. 2016;15(2):155–61.
Mavropoulos A, Simopoulou T, Varna A, Liaskos C, Katsiari CG, Bogdanos DP, et al. Breg cells are numerically decreased and functionally impaired in patients with systemic sclerosis. Arthritis Rheumatol. 2016;68(2):494–504.
Odaka M, Hasegawa M, Hamaguchi Y, Ishiura N, Kumada S, Matsushita T, et al. Autoantibody-mediated regulation of B cell responses by functional anti-CD22 autoantibodies in patients with systemic sclerosis. Clin Exp Immunol. 2010;159(2):176–84.
Sato S, Hasegawa M, Fujimoto M, Tedder TF, Takehara K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J Immunol. 2000;165(11):6635–43.
Saigusa R, Asano Y, Nakamura K, Yamashita T, Ichimura Y, Takahashi T, et al. Association of anti-RNA polymerase III antibody and silicone breast implants in patients with systemic sclerosis. J Dermatol. 2016;43(7):808–10.
Saigusa R, Asano Y, Nakamura K, Miura S, Ichimura Y, Takahashi T, et al. Association of anti-RNA polymerase III antibody and malignancy in Japanese patients with systemic sclerosis. J Dermatol. 2015;42(5):524–7.
Yanaba K, Hayashi M, Yoshihara Y, Nakagawa H. Serum levels of soluble programmed death-1 and programmed death ligand-1 in systemic sclerosis: association with extent of skin sclerosis. J Dermatol. 2016;43(8):954–7.
Nakamura K, Asano Y, Taniguchi T, Minatsuki S, Inaba T, Maki H, et al. Serum levels of interleukin-18-binding protein isoform a: clinical association with inflammation and pulmonary hypertension in systemic sclerosis. J Dermatol. 2016;43(8):912–8.
Miura S, Asano Y, Saigusa R, Yamashita T, Taniguchi T, Takahashi T, et al. Serum omentin levels: a possible contribution to vascular involvement in patients with systemic sclerosis. J Dermatol. 2015;42(5):461–6.
Miura S, Asano Y, Saigusa R, Yamashita T, Taniguchi T, Takahashi T, et al. Serum vaspin levels: a possible correlation with digital ulcers in patients with systemic sclerosis. J Dermatol. 2015;42(5):528–31.
Wu CY, Asano Y, Taniguchi T, Sato S, Yu HS. Serum level of circulating syndecan-1: a possible association with proliferative vasculopathy in systemic sclerosis. J Dermatol. 2016;43(1):63–6.
Wu CY, Asano Y, Taniguchi T, Sato S, Yu HS. Serum heparanase levels: a protective marker against digital ulcers in patients with systemic sclerosis. J Dermatol. 2015;42(6):625–8.
Campbell PM, LeRoy EC. Pathogenesis of systemic sclerosis: a vascular hypothesis. Semin Arthritis Rheum. 1975;4(4):351–68.
Fleischmajer R, Perlish JS. Capillary alterations in scleroderma. J Am Acad Dermatol. 1980;2:161–70.
Haustein UF, Herrmann K, Böhme HJ. Pathogenesis of progressive systemic sclerosis. Int J Dermatol. 1986;25(5):286–93.
Cabral-Marques O, Riemekasten G. Vascular hypothesis revisited: role of stimulating antibodies against angiotensin and endothelin receptors in the pathogenesis of systemic sclerosis. Autoimmun Rev. 2016;15(7):690–4.
Bordron A, Dueymes M, Levy Y, Jamin C, Leroy JP, Piette JC, et al. The binding of some human antiendothelial cell antibodies induces endothelial cell apoptosis. J Clin Invest. 1998;101(10):2029–35.
Wolf SI, Howat S, Abraham DJ, Pearson JD, Lawson C. Agonistic anti-ICAM-1 antibodies in scleroderma: activation of endothelial pro-inflammatory cascades. Vasc Pharmacol. 2013;59(1–2):19–26.
Kill A, Tabeling C, Undeutsch R, Kühl AA, Günther J, Radic M, et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res Ther. 2014;16(1):R29. doi:10.1186/ar4457.
Xia Y, Kellems RE. Angiotensin receptor agonistic autoantibodies and hypertension: preeclampsia and beyond. Circ Res. 2013;113(1):78–87.
Becker MO, Kill A, Kutsche M, Guenther J, Rose A, Tabeling C, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med. 2014;190(7):808–17.
Kawaguchi Y, Takagi K, Hara M, Fukasawa C, Sugiura T, Nishimagi E, et al. Angiotensin II in the lesional skin of systemic sclerosis patients contributes to tissue fibrosis via angiotensin II type 1 receptors. Arthritis Rheum. 2004 Jan;50(1):216–26.
Akamata K, Asano Y, Aozasa N, Noda S, Taniguchi T, Takahashi T, et al. Bosentan reverses the pro-fibrotic phenotype of systemic sclerosis dermal fibroblasts via increasing DNA binding ability of transcription factor Fli1. Arthritis Res Ther. 2014;16(2):R86. doi:10.1186/ar4529.
Günther J, Kill A, Becker MO, Heidecke H, Rademacher J, Siegert E, et al. Angiotensin receptor type 1 and endothelin receptor type a on immune cells mediate migration and the expression of IL-8 and CCL18 when stimulated by autoantibodies from systemic sclerosis patients. Arthritis Res Ther. 2014;16(2):R65. doi:10.1186/ar4503.
Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98(3):785–92.
Lunardi C, Bason C, Navone R, Millo E, Damonte G, Corrocher R, et al. Systemic sclerosis immunoglobulin G autoantibodies bind the human cytomegalovirus late protein UL94 and induce apoptosis in human endothelial cells. Nat Med. 2000;6(10):1183–6.
Lunardi C, Dolcino M, Peterlana D, Bason C, Navone R, Tamassia N, et al. Antibodies against human cytomegalovirus in the pathogenesis of systemic sclerosis: a gene array approach. PLoS Med. 2006;3(1):e2.
Roumm AD, Whiteside TL, Medsger TA Jr, Rodnan GP. Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification, subtyping, and clinical correlations. Arthritis Rheum. 1984;27(6):645–53.
Gilbane AJ, Denton CP, Holmes AM. Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis Res Ther. 2013;15(3):215.
Pannu J, Gore-Hyer E, Yamanaka M, Smith EA, Rubinchik S, Dong JY, et al. An increased transforming growth factor beta receptor type I:type II ratio contributes to elevated collagen protein synthesis that is resistant to inhibition via a kinase-deficient transforming growth factor beta receptor type II in scleroderma. Arthritis Rheum. 2004;50(5):1566–77.
Serratì S, Chillà A, Laurenzana A, Margheri F, Giannoni E, Magnelli L, et al. Systemic sclerosis endothelial cells recruit and activate dermal fibroblasts by induction of a connective tissue growth factor (CCN2)/transforming growth factor β-dependent mesenchymal-to-mesenchymal transition. Arthritis Rheum. 2013;65(1):258–69.
Leask A. Towards an anti-fibrotic therapy for scleroderma: targeting myofibroblast differentiation and recruitment. Fibrogenesis Tissue Repair. 2010;3:8. doi:10.1186/1755-1536-3-8.
Leask A. Getting out of a sticky situation: targeting the myofibroblast in scleroderma. Open Rheumatol J. 2012;6:163–9.
Igarashi A, Nashiro K, Kikuchi K, Sato S, Ihn H, Fujimoto M, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol. 1996;106(4):729–33.
Lakos G, Takagawa S, Chen SJ, Ferreira AM, Han G, Masuda K, et al. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol. 2004;165(1):203–17.
Gay S, Jones RE Jr, Huang GQ, Gay RE. Immunohistologic demonstration of platelet-derived growth factor (PDGF) and sis-oncogene expression in scleroderma. J Invest Dermatol. 1989;92(2):301–3.
Baroni SS, Santillo M, Bevilacqua F, Luchetti M, Spadoni T, Mancini M, et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med. 2006;354(25):2667–76.
Hasegawa M, Hamaguchi Y, Yanaba K, Bouaziz JD, Uchida J, Fujimoto M, et al. B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am J Pathol. 2006;169(3):954–66.
Castelino FV, Bain G, Pace VA, Black KE, George L, Probst CK, et al. An Autotaxin-LPA-IL-6 amplification loop drives scleroderma fibrosis. Arthritis Rheumatol. 2001;68(12):2964–74.
O’Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via gremlin protein. J Biol Chem. 2014;289(14):9952–60.
Postlethwaite AE, Holness MA, Katai H, Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J Clin Invest. 1992;90(4):1479–85.
Jinnin M, Ihn H, Yamane K, Tamaki K. Interleukin-13 stimulates the transcription of the human alpha2(I) collagen gene in human dermal fibroblasts. J Biol Chem. 2004;279(40):41783–91.
Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol. 2004;173(6):4020–9.
Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J Rheumatol. 1997;24(2):328–32.
Fallon PG, Richardson EJ, McKenzie GJ, McKenzie AN. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J Immunol. 2000;164(5):2585–91.
Fuschiotti P, Larregina AT, Ho J, Feghali-Bostwick C, Medsger TA Jr. Interleukin-13-producing CD8+ T cells mediate dermal fibrosis in patients with systemic sclerosis. Arthritis Rheum. 2013;65(1):236–46.
Takagawa S, Lakos G, Mori Y, Yamamoto T, Nishioka K, Varga J. Sustained activation of fibroblast transforming growth factor-beta/Smad signaling in a murine model of scleroderma. J Invest Dermatol. 2003;121(1):41–50.
Jaffee BD, Claman HN. Chronic graft-versus-host disease (GVHD) as a model for scleroderma. I. Description of model systems. Cell Immunol. 1983;77(1):1–12.
Ruzek MC, Jha S, Ledbetter S, Richards SM, Garman RD. A modified model of graft-versus-host-induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease. Arthritis Rheum. 2004;50(4):1319–31.
Servettaz A, Goulvestre C, Kavian N, Nicco C, Guilpain P, Chéreau C, et al. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J Immunol. 2009;182(9):5855–64.
Yoshizaki A, Yanaba K, Ogawa A, Asano Y, Kadono T, Sato S. Immunization with DNA topoisomerase I and Freund’s complete adjuvant induces skin and lung fibrosis and autoimmunity via interleukin-6 signaling. Arthritis Rheum. 2011;63(11):3575–85.
Stawski L, Han R, Bujor AM, Trojanowska M. Angiotensin II induces skin fibrosis: a novel mouse model of dermal fibrosis. Arthritis Res Ther. 2012;14(4):R194. doi:10.1186/ar4028.
Siracusa LD, McGrath R, Ma Q, Moskow JJ, Manne J, Christner PJ, et al. A tandem duplication within the fibrillin 1 gene is associated with the mouse tight skin mutation. Genome Res. 1996;6(4):300–13.
Sonnylal S, Denton CP, Zheng B, Keene DR, He R, Adams HP, et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007;56(1):334–44.
Sonnylal S, Shi-Wen X, Leoni P, Naff K, Van Pelt CS, Nakamura H, et al. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 2010;62(5):1523–32.
Wei J, Melichian D, Komura K, Hinchcliff M, Lam AP, Lafyatis R, et al. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: a novel mouse model for scleroderma? Arthritis Rheum. 2011;63(6):1707–17.
Eferl R, Hasselblatt P, Rath M, Popper H, Zenz R, Komnenovic V, et al. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc Natl Acad Sci U S A. 2008;105(30):10525–30.
Maurer B, Busch N, Jüngel A, Pileckyte M, Gay RE, Michel BA, et al. Transcription factor fos-related antigen-2 induces progressive peripheral vasculopathy in mice closely resembling human systemic sclerosis. Circulation. 2009;120(23):2367–76.
Kanno Y, Kaneiwa A, Minamida M, Kanno M, Tomogane K, Takeuchi K, et al. The absence of uPAR is associated with the progression of dermal fibrosis. J Invest Dermatol. 2008;128(12):2792–7.
Bernstein AM, Twining SS, Warejcka DJ, Tall E, Masur SK. Urokinase receptor cleavage: a crucial step in fibroblast-to-myofibroblast differentiation. Mol Biol Cell. 2007;18(7):2716–27.
D’Alessio S, Fibbi G, Cinelli M, Guiducci S, Del Rosso A, Margheri F, et al. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004;50(10):3275–85.
Margheri F, Manetti M, Serratì S, Nosi D, Pucci M, Matucci-Cerinic M, et al. Domain 1 of the urokinase-type plasminogen activator receptor is required for its morphologic and functional, beta2 integrin-mediated connection with actin cytoskeleton in human microvascular endothelial cells: failure of association in systemic sclerosis endothelial cells. Arthritis Rheum. 2006;54(12):3926–38.
Manetti M, Rosa I, Milia AF, Guiducci S, Carmeliet P, Ibba-Manneschi L, et al. Inactivation of urokinase-type plasminogen activator receptor (uPAR) gene induces dermal and pulmonary fibrosis and peripheral microvasculopathy in mice: a new model of experimental scleroderma? Ann Rheum Dis. 2014;73(9):1700–9.
Noda S, Asano Y, Nishimura S, Taniguchi T, Fujiu K, Manabe I, et al. Simultaneous downregulation of KLF5 and Fli1 is a key feature underlying systemic sclerosis. Nat Commun. 2014;5:5797. doi:10.1038/ncomms6797.
Czuwara-Ladykowska J, Shirasaki F, Jackers P, Watson DK, Trojanowska M. Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. J Biol Chem. 2001;276(24):20839–48.
Kubo M, Czuwara-Ladykowska J, Moussa O, Markiewicz M, Smith E, Silver RM, et al. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in fibrotic scleroderma skin. Am J Pathol. 2003;163(2):571–81.
Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A. 2003;100(21):12319–24.
van den Hoogen FH, Boerbooms AM, Swaak AJ, Rasker JJ, van Lier HJ, van de Putte LB. Comparison of methotrexate with placebo in the treatment of systemic sclerosis: a 24 week randomized double-blind trial, followed by a 24 week observational trial. Br J Rheumatol. 1996;35(4):364–72.
Pope JE, Bellamy N, Seibold JR, Baron M, Ellman M, Carette S, et al. A randomized, controlled trial of methotrexate versus placebo in early diffuse scleroderma. Arthritis Rheum. 2001;44(6):1351–8.
Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–66.
Hoyles RK, Ellis RW, Wellsbury J, Lees B, Newlands P, Goh NS, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54(12):3962–70.
Koutroumpas A, Ziogas A, Alexiou I, Barouta G, Sakkas LI. Mycophenolate mofetil in systemic sclerosis-associated interstitial lung disease. Clin Rheumatol. 2010;29(10):1167–8.
Gerbino AJ, Goss CH, Molitor JA. Effect of mycophenolate mofetil on pulmonary function in scleroderma-associated interstitial lung disease. Chest. 2008;133(2):455–60.
Poelman CL, Hummers LK, Wigley FM, Anderson C, Boin F, Shah AA. Intravenous immunoglobulin may be an effective therapy for refractory, active diffuse cutaneous systemic sclerosis. J Rheumatol. 2015;42(2):236–42.
Takehara K, Ihn H, Sato S. A randomized, double-blind, placebo-controlled trial: intravenous immunoglobulin treatment in patients with diffuse cutaneous systemic sclerosis. Clin Exp Rheumatol. 2013;31(2 Suppl 76):151–6.
Kowal-Bielecka O, Landewé R, Avouac J, Chwiesko S, Miniati I, Czirjak L, et al. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR scleroderma trials and research group (EUSTAR). Ann Rheum Dis. 2009;68(5):620–8.
Denton CP, Pope JE, Peter HH, Gabrielli A, Boonstra A, van den Hoogen FH, et al. Long-term effects of bosentan on quality of life, survival, safety and tolerability in pulmonary arterial hypertension related to connective tissue diseases. Ann Rheum Dis. 2008;67(9):1222–8.
Lopez-Ovejero JA, Saal SD, D’Angelo WA, Cheigh JS, Stenzel KH, Laragh JH. Reversal of vascular and renal crises of scleroderma by oral angiotensin-converting-enzyme blockade. N Engl J Med. 1979;300(25):1417–9.
Motegi S, Yamada K, Toki S, Uchiyama A, Kubota Y, Nakamura T, et al. Beneficial effect of botulinum toxin a on Raynaud’s phenomenon in Japanese patients with systemic sclerosis: a prospective, case series study. J Dermatol. 2016;43(1):56–62.
Daoussis D, Melissaropoulos K, Sakellaropoulos G, Antonopoulos I, Markatseli TE, Simopoulou T, et al. A multicenter, open-label, comparative study of B-cell depletion therapy with rituximab for systemic sclerosis-associated interstitial lung disease. Semin Arthritis Rheum. 2016; doi:10.1016/j.semarthrit.2016.10.003.
Smith V, Pizzorni C, Riccieri V, Decuman S, Brusselle G, DE Pauw M, et al. Stabilization of microcirculation in patients with early systemic sclerosis with diffuse skin involvement following rituximab treatment: an open-label study. J Rheumatol. 2016;43(5):995–6.
Bosello S, De Santis M, Lama G, Spanò C, Angelucci C, Tolusso B, et al. B cell depletion in diffuse progressive systemic sclerosis: safety, skin score modification and IL-6 modulation in an up to thirty-six months follow-up open-label trial. Arthritis Res Ther. 2010;12(2):R54. doi:10.1186/ar2965.
Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. 2015;125(7):2795–807.
Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. 2016;387(10038):2630–40.
Shima Y, Hosen N, Hirano T, Arimitsu J, Nishida S, Hagihara K, et al. Expansion of range of joint motion following treatment of systemic sclerosis with tocilizumab. Mod Rheumatol. 2015;25(1):134–7.
Yoshizaki A, Yanaba K, Yoshizaki A, Iwata Y, Komura K, Ogawa F, et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 2010;62(8):2476–87.
Su TI, Khanna D, Furst DE, Danovitch G, Burger C, Maranian P, et al. Rapamycin versus methotrexate in early diffuse systemic sclerosis: results from a randomized, single-blind pilot study. Arthritis Rheum. 2009;60(12):3821–30.
Albera C, Costabel U, Fagan EA, Glassberg MK, Gorina E, Lancaster L, et al. Efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis with more preserved lung function. Eur Respir J. 2016;48(3):843–51.
Lopez-de la Mora DA, Sanchez-Roque C, Montoya-Buelna M, Sanchez-Enriquez S, Lucano-Landeros S, Macias-Barragan J, et al. Role and new insights of Pirfenidone in fibrotic diseases. Int J Med Sci. 2015;12(11):840–7.
Distler JH, Feghali-Bostwick C, Soare A, Asano Y, Distler O, Abraham DJ. Frontiers of antifibrotic therapy in systemic sclerosis. Arthritis Rheumatol. 2017;69(2):257–67.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Furue, M., Mitoma, C., Mitoma, H. et al. Pathogenesis of systemic sclerosis—current concept and emerging treatments. Immunol Res 65, 790–797 (2017). https://doi.org/10.1007/s12026-017-8926-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-017-8926-y