
Abstract
Cognitive disturbances, mood disorders and fatigue are common in SLE patients with substantial adverse effects on function and quality of life. Attribution of these clinical findings to immune-mediated disturbances associated with SLE remains difficult and has compromised research efforts in these areas. Improved understanding of the role of the immune system in neurologic processes essential for cognition including synaptic plasticity, long term potentiation and adult neurogenesis suggests multiple potential mechanisms for altered central nervous system function associated with a chronic inflammatory illness such as SLE. This review will focus on the biology of cognition and neuroinflammation in normal circumstances and potential biologic mechanisms for cognitive impairment, depression and fatigue attributable to SLE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuropsychiatric illness has been associated with systemic lupus erythematosus (SLE) since the systemic manifestations of this disease were first described by Kaposi in 1872; yet, neuropsychiatric SLE (NPSLE) remains an enigma both clinically and biologically. NPSLE has been classified into 19 syndromes that encompass a wide spectrum of neurologic and psychiatric illness in the peripheral, central, and autonomic nervous systems (Table 1). Attribution of each of these syndromes to an immunologic process directly associated with immune dysregulation in SLE has been extremely difficult as neurologic effects from medications, hormonal and metabolic disturbances, comorbid disease, and infections can be clinically indistinguishable from each other and from SLE. Given the difficulties inherent in proposed attribution models where expert opinion remains the gold standard [1, 2], consideration of pathogenic mechanisms contributing to specific syndromes encompassed by NPSLE is critically important to identify unbiased biomarkers for attribution and the development of diagnostic and therapeutic approaches. This is particularly important for diffuse central nervous system (CNS) NPSLE syndromes where tissue biopsies are not an option, and biologic mechanisms are poorly understood as they are generally not the result of classic inflammatory processes characteristically found in other organs readily accessible to circulating immune components.
“Lupus brain fog” is an extremely common patient complaint that refers to periods of forgetfulness and confusion that are related to impaired cognition. Cognitive impairment is one of the central NPSLE syndromes and its prevalence, with associated deleterious effects on quality of life and individual productivity, ranges from 21 to 80 % [3–7]. This broad range is likely due to differences in assessment and attribution given the current inability of assessments to reliably distinguish causality of cognitive impairment. Debilitating depression and fatigue with substantial impact on cognition and quality of life are also very common in SLE with prevalences estimated between 17 and 75 % and 80 %, respectively [8–10]. Similar to cognitive impairment, attribution of depression and fatigue in SLE is notoriously difficult. To date, no biomarkers have been identified that provide sensitive and specific measures for cognitive and behavioral change attributable to SLE; yet, this remains essential for attribution, appropriate treatment, and for clinical trials of neuroprotection. This review will focus on the biology of cognition and potential biologic mechanisms for cognitive impairment, depression, and fatigue attributable to SLE.
Learning, memory consolidation, and synaptic plasticity
Cognition is the functional result of a process whereby learning and memory result from communication between neurons, astrocytes, glial cells, and immune cells through a variety of neurotransmitters, transcription factors, cytokines, and chemokines. The hippocampus is the primary brain structure involved in memory and cognition and is also one of two sites where adult neurogenesis occurs. Thus, the biologic basis for cognition relies on intact neural circuitry and adult neurogenesis within the hippocampus and neuroimmune signaling mechanisms.
Extensive research in brain biology has provided robust evidence for the role of the immune system in neurologic processes essential for cognition including synaptic plasticity, long-term potentiation (LTP), and adult neurogenesis (reviewed in [11–13]). The interplay between immune system and brain is comprised of several levels of communication: (1.) within the CNS between neurons and between neurons and microglia; (2.) within the vasculature in the meninges and choroid plexus between circulating T cells, endothelial cells, perivascular macrophages, and microglia; and (3.) within neuroendocrine pathways such as the hypothalamus–pituitary–adrenal pathway (HPA).
Microglia are resident macrophage-like cells that, in addition to surveillance and phagocytic activity, are important mediators of synaptic pruning and directly influence neuron synaptic activity through their immune properties [14]. Cytokines, chemokines and their receptors, complement receptors, Fc receptors, and MHC class I and class II molecules are expressed in microglia rendering them capable of interactions with immune cells and promoting damage in pathological states through the release of inflammatory cytokines and reactive oxygen and nitrogen species (reviewed in [15]). Neural circuitry is comprised of neurons, synapses, and a microglial support system. Neural circuits undergo modification throughout life through processes known as neural plasticity that include neuronal and glial cell death, removal of debris, and selective pruning of dendrites, axons, or terminals. Synaptic plasticity refers to the ability of a neural synapse to strengthen or weaken in response to increased or decreased signaling activity. Long-term potentiation (LTP) is a form of synaptic plasticity that is the physiologic process essential for laying down memory. It represents increased synaptic efficacy following high-frequency stimulation from afferent neurons [16], thereby resulting in increased sensitivity of neurons to repeat stimulation. Physiologically, the amplitude of the excitatory postsynaptic potential (EPSP) is greatly increased as a result of LTP. In response to the EPSP, glutamate is released into the synapse and binds to postsynaptic neuronal AMPA receptors. Glutamate binding to AMPA receptors permits influx of sodium ions resulting in depolarization of the postsynaptic neuron and activation of N-methyl-d-aspartate receptors (NMDAR) that allows calcium influx into the cell. Animal models measuring response to psychological stressors and learning paradigms have been used extensively to investigate the role of immune processes on neural plasticity, learning, memory, and LTP.
Peripheral immune cells and cognition
Several murine studies have demonstrated the importance of CNS-reactive CD4+ T cells in normal cognition through their effects on neural plasticity [17, 18]. T cells have been shown to accumulate in cerebrospinal fluid (CSF) in specific areas in response to trauma or CNS signaling and analysis of the T cell repertoire has demonstrated enrichment for CNS-specific CD4+ T cells [19]. As direct interactions between T cells and brain parenchyma do not occur normally, the choroid plexus has been proposed as a potential site for indirect T cell communication [19]. Normal mice participating in learning tasks demonstrate adhesion molecule-mediated recruitment of CD4+ T cells to the meningeal space in the choroid plexus where they are activated and acquire a Th2 phenotype with high IL-4 expression [20]. Depletion of these T cells using SCID (no mature T or B cells), nude (no mature T cells), irradiated or IL-4-deficient mice resulted in impaired learning and memory [17, 18, 21], and repletion of normal T cells reversed the cognitive impairments [20]. Similarly, T cell trafficking to the choroid plexus is associated with episodes of stress in murine models [22, 23]. In humans, CNS-reactive T cells improve outcome in spinal cord trauma through recruitment of anti-inflammatory macrophages [24, 25]. Collectively, these data support a role for adaptive immunity in normal brain function, and disruption of CD4 T cell communication or choroid plexus signaling has potentially significant consequences on cognition as evidenced in studies of immune senescence and cognitive decline associated with aging [19]. Evidence for brain-reactive T cells has not been reported in SLE; however, the lupus-prone MRL/lpr mouse strain has been widely used as a model for NPSLE as these mice develop deficits in learning and memory and emotional reactivity coincident with the development of autoantibodies, circulating inflammatory mediators, and inflammation in peripheral organs. Histopathology reveals infiltration of mononuclear cells, predominantly T cells, into the choroid plexus and brain parenchyma that increases significantly with age and peripheral disease activity and is associated with the functional CNS disturbances [26].
Choroid plexus
The choroid plexus is a specialized interface between the brain and peripheral circulation that regulates the composition of CSF and allows neuroimmune communication [27]. It is comprised of a single epithelial layer surrounding an inner stroma that is vascularized with fenestrated blood vessels. The choroid plexus receives signals from both the brain parenchyma and peripheral circulation via chemokines, cytokines, and neurotropic factors. Toll-like receptors (TLR) are also found on the choroid plexus epithelium and mediate danger signals from the brain [28]. The choroid plexus up-regulates adhesion molecules in response to CNS injury allowing CD4+ memory T cells to transmigrate from the blood vessels to the stroma where they are activated if they encounter brain antigen presented in the context of resident antigen-presenting cells [29]. Activated CD4+ T cells produce IL-4 that modulates release of neurotropic factors that migrate to the brain parenchyma where they influence adult neurogenesis and microglial activation. Thus, the choroid plexus acts as a mediator for neuroimmune interactions allowing the brain and peripheral organs to communicate with each other and react appropriately to ongoing events. As evidenced in the MRL/lpr lupus model, an abundance of T cells in this area is associated with worsening CNS and peripheral disease manifestations [26].
Peripheral cytokine effects
Inflammatory cytokines (IL-1, IL-6, TNFα) and their receptors are expressed by microglia, astroglia, neurons, and brain endothelial cells [30, 31]. Increased levels of peripherally circulating inflammatory cytokines, as a result of administration or acute or chronic illness, have been associated with impaired cognitive performance in human subjects [32–36]. Peripherally circulating cytokines are thought to mediate changes in central cytokine expression by transport across the blood–brain barrier [37], microglia activation via the circumventricular organs [38], activated vascular endothelium via a prostaglandin-mediated mechanism [39], and peripheral vagal nerve stimulation resulting in central increase in IL-1 [40]. Murine studies suggest that microglia respond to increases in circulating inflammatory cytokines by switching to an inflammatory phenotype with intracerebral amplification of the peripheral inflammatory milieu and resultant impairments in memory function [41, 42]. In humans, elevated serum IL-6 levels in non-autoimmune community volunteers have been shown to inversely correlate with cognitive performance and hippocampal gray matter volumes [32, 43].
The demonstration that acute increases in serum levels of IL-6, IL-1, and TNFα in response to typhoid vaccination in healthy volunteers prompted selective decreases in spatial memory and parahippocampal glucose metabolism on PET scan within 4 hours [44] suggests a possible mechanism for transient alterations in cognitive processes. This may be applicable to chronic inflammatory states such as SLE where end organ damage related to immune dysregulation is mediated in part by peripherally circulating inflammatory cytokines including IFNα, TNFα, IL-1, IL-6, IL-10, BAFF/APRIL, IFNγ, IL-17, and IL-21 (reviewed in [45, 46]). These cytokines are produced by activated T helper cells, macrophages, and plasmacytoid dendritic cells at different points in disease progression, and levels may reflect disease activity. Some, such as IL-1 and IL-6 are also secreted by other cell types including neutrophils, epithelial cells, endothelial cells, and resident cells in specific tissues. They activate autoreactive lymphocytes in peripheral organ tissue to produce more inflammatory cytokines and chemokines, thus driving a local tissue inflammatory response. A relationship between systemic inflammation and cognitive impairment has been demonstrated in SLE where circulating IL-6 levels correlated inversely with performance on memory testing [47]. Similar cognitive deficits have been reported in SLE patients with high C-reactive protein levels indicative of circulating inflammatory molecules [48].
The IFNα amplification loop has been ascribed a central role in SLE pathogenesis, implicated in disease flares, and peripheral circulation of this cytokine is associated with numerous neuropsychiatric symptoms [49–51]. Treatment for Hepatitis C liver disease or malignancy with IFNα has given us a human model for neuropsychological effects of IFNα. Dose-dependent treatment effects include fatigue (80–90 %), seizures (1–4 %), depressive symptoms (30–50 %), and occasionally psychosis, confusion, and mania [52–54]. As IFNα does not easily cross the BBB, its central effects may be mediated by induction of other potent inflammatory cytokines such as IL-6, IL-1, and TNFα and decreased CNS neurotransmitter biosynthesis (serotonin, dopamine) [55–58]. Of note, idiopathic major depression is also associated with increased circulating levels of IFNα, IL-1, IL-6, and TNFα [59], and both major depression and brain responses to IFNα share common peripheral blood gene signatures of neuron survival and plasticity [60]. Functionally, IFNα therapy is associated with decreased glucose metabolism on PET imaging in the prefrontal cortex that correlates with depression scores [61] and hypermetabolism in the basal ganglia that correlates with fatigue [62]. Although peripherally circulating IFNα levels are difficult to assess in SLE patients, IFN-inducible genes in the peripheral blood have been identified in virtually all SLE patients, and the intensity of expression is related to disease activity [49]. Of note, increased SLE disease activity has been linked to increased vulnerability to depression in several studies [63–65]. IFNα has also been identified in NPSLE CSF and associated with diffuse central neurologic syndromes including psychosis, severe depression, seizures, and acute confusional state [66–68]. Similar to the decreased glucose metabolism reported in the prefrontal cortex of patients with IFNα treatment-associated depression, elevated CSF IFNα titers have also been associated with decreased glucose metabolism in the frontal lobes in 50 % of NPSLE subjects in one study [69]. Furthermore, immune complexes formed by CSF from NPSLE patients with diffuse syndromes (mood and anxiety disorders, seizures, psychosis, acute confusional states) combined with antigen stimulate expression of multiple cytokines in plasmacytoid dendritic cells including IFNα, IFNγ, IL-8, and MCP-1 [66]. These data implicate IFNα in the pathogenesis of depression and fatigue related to SLE as has been shown in idiopathic major depressive disorders and consequent to IFNα therapy. IFNα-mediated increases in microglial expression of TNFα, IL-1, and IL-6 may also impact hippocampal synaptic plasticity and cognition.
Numerous studies have focused on the association between chronic peripheral inflammatory processes, irrespective of etiology, and incapacitating fatigue (reviewed in [70]). As described previously, circulating inflammatory molecules gain access to the CNS where their pro-inflammatory effects are amplified by resident microglial cells, astrocytes, and neurons resulting in neuroinflammation [13]. It is therefore not surprising that extreme fatigue is highly prevalent in chronic neurodegenerative diseases characterized by neuroinflammation such as Parkinson’s disease and multiple sclerosis. Biologically, it has been proposed that chronic inflammatory states mediate a Toll-like receptor (TLR) radical cycle whereby peripherally circulating pro-inflammatory cytokines induce up-regulation of additional cytokines and reactive oxygen and nitrogen species via NF-kappaB signaling with resultant oxidative and nitrosative stress [71]. Oxidative and nitrosative stress mitigates damage to lipids, proteins, and DNA resulting in the formation of damage-associated molecular pattern molecules (DAMPS) that engage with pro-inflammatory TLRs and amplify the cycle [72]. Impaired clearance of nucleic acid-containing debris is one of the characteristic immune abnormalities identified in SLE [73]; this may contribute to increased availability of autoantigen for this TLR radical cycle, and excessive TLR activation is recognized as a potential target for therapy in SLE [74]. Pro-inflammatory cytokines implicated in chronic fatigue include IL-1, IL-6, and TNFα [75], all of which are present in the chronic inflammatory state found in SLE. Additionally, elevated levels of markers of oxidative stress associated with pro-inflammatory cytokines and reduced levels of omeg-3 fatty acids have been reported in SLE [76]. Of note, a recent placebo-controlled clinical trial of fish oil supplementation targeting oxidative stress in SLE demonstrated significant improvement in fatigue scores and some circulating inflammatory markers (ESR, IL-12) [77].
Central mechanisms
Physiologic learning and memory: cytokines, chemokines, and neurotransmitters
Although pro-inflammatory cytokines IL-1β, TNFα, and IL-6 are most often associated with brain-destructive processes, murine studies support their critical importance in normal synaptic function and hippocampal memory consolidation [13, 78, 79]. Activated microglial cells express pro-inflammatory cytokines IL-1β, TNFα, IL-6; these have wide ranging effects including increased MHC class I expression on brain endothelial cells [80] and effects on hippocampal LTP [81]. Hippocampal microglial cell IL-1β mRNA expression increases following fear conditioning [82] or participation in a spatial recognition task [83]. Additionally, intracerebroventricular injection of low doses of IL-1β has been shown to facilitate spatial and contextual fear memories and memories of passive avoidance training [82, 84, 85]. Impaired learning in spatial memory and fear conditioning paradigms following treatment with IL-1 receptor antagonist and in mice with genetically altered IL-I signaling supports the role of IL-1 in normal, physiologic learning processes [82, 86]. However, high levels of IL-1β, administered centrally or peripherally, have detrimental effects on hippocampal-dependent learning tasks that included spatial learning, and long-term memory induced by fear conditioning paradigms through effects on LTP and reduced neurogenesis (reviewed in [13, 87]).
IL-6 and TNFα have variable effects on normal learning and memory that are dependent on timing and conditions including magnitude and duration of cytokine exposure (reviewed in [13]). TNFα has been shown to influence the expression of AMPA receptors on postsynaptic neurons, thereby impacting synaptic strength [88]. Synaptic scaling, a homeostatic plasticity mechanism that allows neurons to regulate their firing rate and provide stability to circuitry output, has also been shown to be mediated by glial cell TNFα [89, 90]. Neurons express IL-6 receptors, and in a physiologic state, low basal levels of IL-6 are increased during induction of hippocampal LTP [91, 92]. Studies with transgenic models suggest that high levels of both IL-6 and TNFα have detrimental effects on hippocampal-dependent learning in the context of inflammatory challenges through effects on synaptic transmission, LTP, and neurogenesis [93–96].
Fraktalkine, a chemokine secreted by neurons with cognate receptors on microglia, is also involved in the regulation of neuron development, neuronal plasticity including synaptic pruning by microglia, synaptic transmission, LTP, and adult neurogenesis [97]. Low levels of stress or certain types of emotional stimuli facilitate the formation of long-term memory and neural plasticity via input from the hypothalamic–pituitary axis (releasing endogenous glucocorticoids) and sympathetic nervous system (releasing norepinephrine, dopamine, serotonin). Hippocampal LTP is strengthened by low levels of these compounds, whereas high levels can be disruptive [98, 99].
Physiologic learning and memory: adult neurogenesis
Adult neurogenesis occurs in the dentate gyrus of the hippocampus (reviewed in [100, 101]). New neurons develop from progenitor stem cells in the subgranular zone in a process that takes approximately 7 weeks and is regulated by the local “neurogenic niche” comprised of glial cells, endothelial cells, microglial cells, macrophages, ependymal cells, and neurons [102]. These new neurons reside in the dentate gyrus, receive input from the entorhinal cortex and project axons to the pyramidal cells in the CA 3 region of the hippocampus where they form glutamatergic synapses. While the presence of these cells is not mandatory for basic function, they have been shown to contribute significantly to higher hippocampal function [100, 103], with considerable impact on learning, memory, anxiety, and mood states [104–106]. Approximately 9000 new progenitor cells are generated daily in the rodent hippocampus, and 80–90 % of these differentiate into neurons compared to approximately 700 new neurons generated daily in humans [107].
In addition to input from cell-to-cell contact, distant brain regions via neurotransmitters [dopamine, γ-aminobutyric acid (GABA), and serotonin], and signaling from transcription factors (reviewed in [11]), maintenance of adult neurogenesis is also dependent on the presence of an adaptive immune system as evidenced by reports that SCID and nude mice display impaired neurogenesis that is repaired by T cell replenishment [108]. Regulation of adult neurogenesis is known to be affected by environmental influences such as stress, inflammation, and physical activity with resulting effects on hippocampal function and behavior (reviewed in [11, 109]). Studies in murine models suggest that stressful experiences negatively impact adult neurogenesis through effects on progenitor cell proliferation, neuronal differentiation, and survival and microglial cells [11]. The functional impact is impaired performance on hippocampal cognitive tasks such as spatial navigation [110, 111]. Importantly, similar effects on adult neurogenesis have been reported following administration of exogenous glucocorticoids [112, 113]. Several neurodegenerative disease processes have been associated with impaired adult neurogenesis including Alzheimer’s disease, Parkinson’s disease, Huntinton’s disease, and HIV infection [114]. Conversely, numerous animal studies have demonstrated positive effects of physical exercise on adult neurogenesis including enhanced progenitor cell proliferation and differentiation [115–117]. While these results are understandably difficult to reproduce in humans, several studies have suggested the benefits of physical exercise on memory, attention, executive function, and the prevention of age-related cognitive decline, dementia, and neurodegenerative diseases [118, 119]. Exposure to an enriched environment and learning has also been shown to promote increased new neuron survival and incorporation into neural circuits, whereas inhibition of hippocampal adult neurogenesis results in impaired learning and memory responses [106, 120–123].
In summary, neuroimmune interactions and adult neurogenesis are critical for the biologic interactions responsible for learning and memory. These interactions are complex, and disruption of the balance has potent impact on function and is implicated in several neurodegenerative and psychiatric illnesses. Impaired neurogenesis in the dentate gyrus has been demonstrated in the MRL/lpr murine model [124], and hippocampal atrophy has been associated with cognitive deficits in lupus patients [125, 126]. It is therefore not unreasonable to consider that decreased adult neurogenesis may be partially responsible for the impaired cognition and regional brain atrophy associated with SLE, although direct evidence for this has not been reported.
Central mechanisms in SLE
Vascular mechanisms, related to thrombosis from anti-phospholipid antibodies (APL) with resulting cerebrovascular disease and cognitive impairment [127, 128], have been recognized for some time; however, many lupus patients with cognitive dysfunction do not have APL, and others have APL but no evidence of impaired cognition [129, 130]. The most common postmortem findings in patients with active CNS disease are widespread small vessel vasculopathy, brain infarction, hemorrhage or normal brain tissue [131–134]. Nonetheless, cerebrospinal fluid (CSF) from lupus patients with diffuse CNS disease (acute confusional state, psychosis, mood disorders, severe cognitive impairment, seizures) has been informative, and murine models provide additional mechanistic information. In the context of immune modulation of neural plasticity and adult neurogenesis, the chronic inflammatory state in SLE suggests possible mechanisms for altered neuropsychological conditions such as cognitive impairment, depression, and fatigue.
CSF cytokines and chemokines in SLE
High titers of a number of pro-inflammatory cytokines and chemokines have been identified in the CSF of NPSLE patients including IL-1, IL-6, IL-10, IFNα, IFNγ, TNFα, BAFF, APRIL, fraktalkine [135], IL-8/CXCL8, MCP-1/CCL2, IP-10/CXCL10, RANTES/CCL5, MIG/CXCL9) [135–141], and follow-up CSF assessment after resolution of NPSLE symptoms has demonstrated significant decreases in all pro-inflammatory molecules back to levels similar to the non-NPSLE lupus controls [141]. Of note, all of these clinical studies of CSF and NPSLE have included SLE subjects severely ill with multiple CNS manifestations; none have focused exclusively on cognitive impairment, depression, or fatigue. CSF levels of IL-6 and IL-8 have been shown to correlate with intrathecal levels of neurofilament triplet protein (NFL) and glial fibrillary acidic protein (GFAP), proteins indicative of neuronal damage and repair, in SLE subjects with NPSLE compared to SLE subjects without CNS involvement and healthy controls [142]. Of note, NFL and GFAP levels were also significantly elevated in the SLE subjects with no overt CNS manifestations compared to healthy controls suggesting both sub-clinical and clinically evident cytokine-mediated CNS damage. Elevated CSF metalloproteinase-9 (MMP-9) levels correlate with CSF IL-6, IL-8, NFL, and GFAP levels in NPSLE CSF compared to SLE patients without CNS involvement and healthy controls [143]. This association suggests MMP-9-mediated brain parenchymal toxicity that is induced by inflammatory cytokines; this is corroborated by a study demonstrating that systemic treatment of NZB/W lupus mice with a MMP blocker, cystamine, results in decreased neuronal apoptosis [144]. Evidence of an association between CSF IL-6 and the IgG index [139] suggests the possibility that increased intrathecal IL-6 may increase B cell activation in the CNS. Recently, CSF levels of BAFF and APRIL were demonstrated to be significantly increased in patients with diffuse NPSLE syndromes compared to SLE patients without CNS symptoms [140]. BAFF and APRIL are known to promote B cell survival, isotype-switching, and CD-40L-independent antibody production. Significantly elevated intrathecal titers of IL-1, IL-6, and IL-10 have also been demonstrated in older MRL/lpr mice, correlating with onset of impaired cognitive processes [145]. In the context of cytokine effects on physiologic learning and memory, the elevated titers of intrathecal pro-inflammatory cytokines and chemokines demonstrated in human SLE and murine models are likely to disrupt physiologic synaptic plasticity in the hippocampus in addition to effects on MMP-9 expression and neural toxicity.
Autoantibodies and cognition, mood disturbances
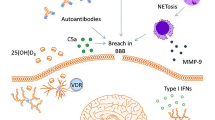
SLE is an autoimmune disease set apart from others by the plethora of autoantibodies; it is therefore not surprising that autoantibodies are implicated in NPSLE. The conundrum is that autoantibodies do not cross an intact BBB, and serum levels of brain-reactive antibodies in SLE have not correlated consistently with neuropsychiatric symptoms. However, CSF autoantibody levels have been shown to correlate significantly with neuropsychiatric manifestations, and evidence for BBB disruption in SLE is supported by reports of an elevated albumin concentration gradient between CSF and plasma [146–148], an elevated IgG index [149] and elevated serum levels of proteins whose origins are exclusive to the brain parenchyma such as S100B [150]. Of note, an elevated IgG index may also reflect intrathecal production of autoantibody. Evidence for this has been reported in one study of human NPSLE [151], and MRL/lpr lupus-prone mice exhibit increased brain atrophy that correlates with increased cellular infiltrate including CD19+ B cells and CD138+ plasma cells, suggesting the ability for intrathecal antibody production following BBB disruption [26, 152]. Proposed mechanisms for BBB disruption in SLE mediated by inflammatory cytokines, chemokines, complement C5a, anti-endothelial cell antibodies, anti-NMDAR antibodies and TWEAK (TNF-like weak inducer of apoptosis) are reviewed elsewhere [153–155]. Brain-reactive autoantibodies implicated in SLE include antibodies to N-methyl d-aspartate receptors (anti-NMDAR), ribosomal P (anti-P), MAP-2 (microtubule-associated protein 2), MMP-9, Ro, RNP, and phospholipid (APL) (reviewed in [156–158]). Although antibody-mediated toxicity can involve direct binding effects, complement activation leading to an inflammatory cascade, or antibody-dependent cellular cytotoxicity, paucity of brain inflammation reported in NPSLE neuropathology suggests that neurotoxic effects or alterations of neuroinflammatory pathways by autoantibodies or other soluble substances penetrating the BBB may be more important. The following are descriptions of two brain-reactive autoantibodies that have been identified in CSF from NPSLE patients and proven to have functional consequences.
Anti-NMDAR antibodies comprise a subset of anti-dsDNA antibodies that cross react with a peptide sequence found on the NR2A and NR2B subunits of NMDAR on neurons [159]. These antibodies preferentially bind activated NMDAR to amplify calcium influx, and effects on cell function are dependent on antibody concentration; low antibody levels increase NMDAR-mediated synaptic responses, affecting synaptic plasticity, whereas high levels result in excitotoxic cell death [160]. These experimental observations may provide a basis for understanding both transient and long-lasting cognitive and behavioral changes observed in SLE patients. Non-autoimmune mice immunized to generate anti-NMDAR antibodies demonstrate impaired memory and responses to behavioral training that correlate pathologically with neuronal damage in the CA1 region of the hippocampus and the amygdala, respectively [161, 162]. Phenotypical changes attributable to antibody-mediated neuronal dysfunction requires breach of the BBB, and the regional location of damage is a consequence of methods used to breach the BBB such that administration of epinephrine results in anti-NMDAR antibodies targeting the amygdala and administration of LPS results in hippocampal damage [161, 162]. More specifically, anti-NMDAR antibodies target place cells in the CA1 hippocampal region with resulting impaired spatial memory [163]. Accordingly, serum anti-NMDAR antibody levels have not reliably correlated with traditional neuropsychological testing, but they have been shown to correlate with spatial memory [163] and mood disturbances [164]. Anti-NMDAR antibodies have been eluted from SLE brain tissue postmortem, and elevated levels in CSF have been shown to correlate with severe manifestations of NPSLE including seizures, acute confusional state, mood and anxiety disorders, psychosis, and cognitive impairment [165–168]. Imaging studies have demonstrated correlations between serum anti-NMDAR ab levels and reduced hippocampal volume [125, 126] and between cognitive performance, serum antibody level, and increased hippocampal glucose metabolism on PET studies [169]. To date, these studies demonstrate anti-NMDAR antibody-mediated damage that involves synaptic plasticity and neuronal excitotoxic apoptosis associated with functional consequences in mice and human subjects.
Anti-P ab were first described in SLE 30 years ago [170]. They have been identified in serum and CSF of SLE patients with lupus psychosis, depression and cognitive disturbance, and their prevalence is estimated between 15 and 21 % [168, 171–175]. These antibodies were originally described as recognizing a P epitope in the highly conserved C-terminal regions of three ribosomal P proteins, but the functional consequences of antibody binding remained elusive [170]. It was subsequently determined that anti-P antibodies cross react with neuronal surface P antigen (NSPA) resulting in induction of calcium influx and neuronal apoptosis [176]. More recent studies demonstrate that NSPA is expressed in postsynaptic regions on neurons in the CA1 and ventral CA3 regions of the hippocampus, and the dentate gyrus and access of circulating anti-P antibodies to brain following BBB disruption impair memory in the absence of cell death [177, 178]. Anti-P binding to NSPA enhances postsynaptic transmission in CA1 neurons, thereby abrogating LTP mediated by increased AMPAR and NMDAR activity [177, 178].
Interestingly, functional consequences of both the anti-NMDAR and anti-P antibodies depend on location and antibody concentration. Both antibodies are neurotoxic at high concentrations and adversely impact synaptic plasticity at low concentrations. Both anti-NMDAR and anti-P antibodies mediate cognitive disturbances in the hippocampus. Anti-NMDAR antibodies also mediate behavioral disturbances in the amygdala, and anti-P antibodies are reported to affect depression and hyposmia through effects in the cingulated and olfactory piriform cortices [179]. Given their separate mechanisms and effects on NMDAR, AMPAR activity, and LTP, it has been suggested that anti-P antibodies may also potentiate the effects of anti-NMDAR antibodies [177].
Conclusion
Neuroinflammation is the proverbial double-edged sword as it is clear that immune cells and inflammatory molecules are integral components of physiologic processes that regulate learning and memory, but unrestrained acute and chronic inflammation has broad deleterious effects on cognition, mood, and fatigue. The chronic inflammatory state in SLE with recurrent episodes of acute escalation provides the necessary ingredients for altered CNS processes that govern cognition and mood. We have reviewed known mechanisms for neuroimmune communication governing cognitive processes and cited evidence for abnormal neuroimmune communication in animal models and human SLE studies. Clinically, central NPSLE syndromes such as cognitive impairment, mood disturbances, and fatigue are both transient and chronic, and neuronal loss alone from direct antibody or MMP-mediated toxicity or ischemia related to thrombosis, embolic events, or atherosclerotic disease would not account for transient phenomena. However, aberrant cytokine- and chemokine-mediated signaling via peripheral or central mechanisms may temporarily impact synaptic plasticity and LTP, thus providing a mechanism for transient cognitive impairment. Both anti-NMDAR and anti-P antibodies have been shown to mediate hippocampal neuron synaptic plasticity in addition to their neurotoxic effects, providing additional mechanisms for transient cognitive or mood disturbances. Through different metabolic pathways, the same pro-inflammatory cytokines have been shown to promote oxidative and nitrosative stress with resulting production of DAMPS and TLR engagement leading to symptoms of extreme fatigue. IFNα deserves particular attention because of its importance in disease pathogenesis and dose-dependent associations of therapeutic IFNα with neuropsychological symptoms of fatigue, depression, and seizures.
Cognitive impairment, fatigue, and depression are among the most commonly patient-reported symptoms in SLE with debilitating effects on quality of life, and the ultimate goal is to develop appropriate therapeutic strategies. Consideration of known and emerging mechanisms of the biologic processes underlying these syndromes, if attributable to SLE, points to therapeutic targets and potential biomarkers, although much additional research needs to be done to substantiate the role of neuroinflammation in NPSLE. Interestingly, not all therapies need be immunosuppressive as evidenced by the success of fish oil for antioxidant properties in fatigue [77]. Conversely, anti-cytokine therapies, such as those that target IFNα and its receptors, may provide benefit through centrally mediated processes that are unrelated to effects on peripheral organs.
References
Bortoluzzi A, et al. Development and validation of a new algorithm for attribution of neuropsychiatric events in systemic lupus erythematosus. Rheumatology (Oxford). 2015;54(5):891–8.
Hanly JG, et al. Neuropsychiatric events at the time of diagnosis of systemic lupus erythematosus: an international inception cohort study. Arthritis Rheum. 2007;56(1):265–73.
Peretti CS, et al. Cognitive impairment in systemic lupus erythematosus women with elevated autoantibodies and normal single photon emission computerized tomography. Psychother Psychosom. 2012;81(5):276–85.
Kozora E, et al. Immune function and brain abnormalities in patients with systemic lupus erythematosus without overt neuropsychiatric manifestations. Lupus. 2012;21(4):402–11.
Nowicka-Sauer K, et al. Neuropsychological assessment in systemic lupus erythematosus patients: clinical usefulness of first-choice diagnostic tests in detecting cognitive impairment and preliminary diagnosis of neuropsychiatric lupus. Clin Exp Rheumatol. 2011;29(2):299–306.
Petri M, et al. Depression and cognitive impairment in newly diagnosed systemic lupus erythematosus. J Rheumatol. 2010;37(10):2032–8.
Ainiala H, et al. The prevalence of neuropsychiatric syndromes in systemic lupus erythematosus. Neurology. 2001;57(3):496–500.
Krupp LB, et al. A study of fatigue in systemic lupus erythematosus. J Rheumatol. 1990;17(11):1450–2.
Palagini L, et al. Depression and systemic lupus erythematosus: a systematic review. Lupus. 2013;22(5):409–16.
Schmeding A, Schneider M. Fatigue, health-related quality of life and other patient-reported outcomes in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2013;27(3):363–75.
Crowther AJ, Song J. Activity-dependent signaling mechanisms regulating adult hippocampal neural stem cells and their progeny. Neurosci Bull. 2014;30(4):542–56.
Marin I, Kipnis J. Learning and memory… and the immune system. Learn Mem. 2013;20(10):601–6.
Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25(2):181–213.
Tremblay ME, et al. The role of microglia in the healthy brain. J Neurosci. 2011;31(45):16064–9.
Ransohoff RM, El Khoury J. Microglia in health and disease. Cold Spring Harb Perspect Biol. 2015. doi:10.1101/cshperspect.a020560.
Shors TJ, Matzel LD. Long-term potentiation: what’s learning got to do with it? Behav Brain Sci. 1997;20(4):597–614.
Kipnis J, et al. Dual effect of CD4+ CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci USA. 2004;101(Suppl 2):14663–9.
Ron-Harel N, et al. Age-dependent spatial memory loss can be partially restored by immune activation. Rejuvenation Res. 2008;11(5):903–13.
Baruch K, Schwartz M. CNS-specific T cells shape brain function via the choroid plexus. Brain Behav Immun. 2013;34:11–6.
Derecki NC, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207(5):1067–80.
Wolf SA, et al. CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol. 2009;182(7):3979–84.
Lewitus GM, Cohen H, Schwartz M. Reducing post-traumatic anxiety by immunization. Brain Behav Immun. 2008;22(7):1108–14.
Lewitus GM, et al. Vaccination as a novel approach for treating depressive behavior. Biol Psychiatry. 2009;65(4):283–8.
Hauben E, et al. Vaccination with a Nogo-A-derived peptide after incomplete spinal-cord injury promotes recovery via a T-cell-mediated neuroprotective response: comparison with other myelin antigens. Proc Natl Acad Sci USA. 2001;98(26):15173–8.
Schwartz M, Baruch K. Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: boosting autoimmunity to fight-off chronic neuroinflammation. J Autoimmun. 2014;54:8–14.
Ma X, Foster J, Sakic B. Distribution and prevalence of leukocyte phenotypes in brains of lupus-prone mice. J Neuroimmunol. 2006;179(1–2):26–36.
Marques F, Sousa JC. The choroid plexus is modulated by various peripheral stimuli: implications to diseases of the central nervous system. Front Cell Neurosci. 2015;9:136.
Butchi NB, et al. TLR7 and TLR9 trigger distinct neuroinflammatory responses in the CNS. Am J Pathol. 2011;179(2):783–94.
Szmydynger-Chodobska J, et al. Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J Cereb Blood Flow Metab. 2012;32(1):93–104.
Schobitz B, De Kloet ER, Holsboer F. Gene expression and function of interleukin 1, interleukin 6 and tumor necrosis factor in the brain. Prog Neurobiol. 1994;44(4):397–432.
Vitkovic L, et al. Cytokine signals propagate through the brain. Mol Psychiatry. 2000;5(6):604–15.
Marsland AL, et al. Brain morphology links systemic inflammation to cognitive function in midlife adults. Brain Behav Immun. 2015;48:195–204.
Bucks RS, et al. Selective effects of upper respiratory tract infection on cognition, mood and emotion processing: a prospective study. Brain Behav Immun. 2008;22(3):399–407.
Krabbe KS, et al. Low-dose endotoxemia and human neuropsychological functions. Brain Behav Immun. 2005;19(5):453–60.
Reichenberg A, et al. Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry. 2001;58(5):445–52.
Gibertini M. IL1 beta impairs relational but not procedural rodent learning in a water maze task. Adv Exp Med Biol. 1996;402:207–17.
Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37(1):26–32.
Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9(6):429–39.
Ek M, et al. Inflammatory response: pathway across the blood-brain barrier. Nature. 2001;410(6827):430–1.
Hosoi T, Okuma Y, Nomura Y. Electrical stimulation of afferent vagus nerve induces IL-1beta expression in the brain and activates HPA axis. Am J Physiol Regul Integr Comp Physiol. 2000;279(1):R141–7.
Perry VH. Stress primes microglia to the presence of systemic inflammation: implications for environmental influences on the brain. Brain Behav Immun. 2007;21(1):45–6.
Chen J, et al. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22(3):301–11.
Marsland AL, et al. Interleukin-6 covaries inversely with cognitive performance among middle-aged community volunteers. Psychosom Med. 2006;68(6):895–903.
Harrison NA, et al. Peripheral inflammation acutely impairs human spatial memory via actions on medial temporal lobe glucose metabolism. Biol Psychiatry. 2014;76(7):585–93.
Davis LS, Hutcheson J, Mohan C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31(10):781–9.
Azevedo PC, Murphy G, Isenberg DA. Pathology of systemic lupus erythematosus: the challenges ahead. Methods Mol Biol. 2014;1134:1–16.
Kozora E, et al. Inflammatory and hormonal measures predict neuropsychological functioning in systemic lupus erythematosus and rheumatoid arthritis patients. J Int Neuropsychol Soc. 2001;7(6):745–54.
Shucard JL, et al. C-reactive protein and cognitive deficits in systemic lupus erythematosus. Cogn Behav Neurol. 2007;20(1):31–7.
Chiche L, et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014;66(6):1583–95.
Crow MK. Interferon pathway activation in systemic lupus erythematosus. Curr Rheumatol Rep. 2005;7(6):463–8.
Ronnblom L. The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups J Med Sci. 2011;116(4):227–37.
Raison CL, et al. Neuropsychiatric adverse effects of interferon-alpha: recognition and management. CNS Drugs. 2005;19(2):105–23.
Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry. 2004;56(11):819–24.
Nashan D, et al. Understanding and managing interferon-alpha-related fatigue in patients with melanoma. Melanoma Res. 2012;22(6):415–23.
Wichers MC, et al. Interferon-alpha-induced depressive symptoms are related to changes in the cytokine network but not to cortisol. J Psychosom Res. 2007;62(2):207–14.
Kamata M, et al. Effect of single intracerebroventricular injection of alpha-interferon on monoamine concentrations in the rat brain. Eur Neuropsychopharmacol. 2000;10(2):129–32.
Kitagami T, et al. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. 2003;978(1–2):104–14.
Raison CL, et al. Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry. 2009;65(4):296–303.
Lichtblau N, et al. Cytokines as biomarkers in depressive disorder: current standing and prospects. Int Rev Psychiatry. 2013;25(5):592–603.
Schlaak JF, et al. Selective hyper-responsiveness of the interferon system in major depressive disorders and depression induced by interferon therapy. PLoS ONE. 2012;7(6):e38668.
Juengling FD, et al. Prefrontal cortical hypometabolism during low-dose interferon alpha treatment. Psychopharmacology. 2000;152(4):383–9.
Capuron L, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology. 2007;32(11):2384–92.
Bachen EA, Chesney MA, Criswell LA. Prevalence of mood and anxiety disorders in women with systemic lupus erythematosus. Arthritis Rheum. 2009;61(6):822–9.
Nery FG, et al. Prevalence of depressive and anxiety disorders in systemic lupus erythematosus and their association with anti-ribosomal P antibodies. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(3):695–700.
Utset TO, et al. Depressive symptoms in patients with systemic lupus erythematosus: association with central nervous system lupus and Sjogren’s syndrome. J Rheumatol. 1994;21(11):2039–45.
Santer DM, et al. Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J Immunol. 2009;182(2):1192–201.
Winfield JB, et al. Intrathecal IgG synthesis and blood-brain barrier impairment in patients with systemic lupus erythematosus and central nervous system dysfunction. Am J Med. 1983;74(5):837–44.
Shiozawa S, et al. Interferon-alpha in lupus psychosis. Arthritis Rheum. 1992;35(4):417–22.
Lee SW, et al. The efficacy of brain (18)F-fluorodeoxyglucose positron emission tomography in neuropsychiatric lupus patients with normal brain magnetic resonance imaging findings. Lupus. 2012;21(14):1531–7.
Morris G, et al. Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med. 2015;13:28.
Morris G, Maes M. Oxidative and nitrosative stress and immune-inflammatory pathways in patients with myalgic encephalomyelitis (ME)/chronic fatigue syndrome (CFS). Curr Neuropharmacol. 2014;12(2):168–85.
Lucas K, Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol. 2013;48(1):190–204.
Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. 2012;18(6):871–82.
Horton CG, Pan ZJ, Farris AD. Targeting Toll-like receptors for treatment of SLE. Mediators Inflamm. 2010. doi:10.1155/2010/498980.
Morris G, Maes M. Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med. 2013;11:205.
Wu T, et al. Metabolic disturbances associated with systemic lupus erythematosus. PLoS One. 2012;7(6):e37210.
Arriens C, et al. Placebo-controlled randomized clinical trial of fish oil’s impact on fatigue, quality of life, and disease activity in systemic lupus erythematosus. Nutr J. 2015;14(1):82.
Ben Menachem-Zidon O, et al. Astrocytes support hippocampal-dependent memory and long-term potentiation via interleukin-1 signaling. Brain Behav Immun. 2011;25(5):1008–16.
Wolf G, et al. Interleukin-1 signaling is required for induction and maintenance of postoperative incisional pain: genetic and pharmacological studies in mice. Brain Behav Immun. 2008;22(7):1072–7.
Yang YM, et al. Microglial TNF-alpha-dependent elevation of MHC class I expression on brain endothelium induced by amyloid-beta promotes T cell transendothelial migration. Neurochem Res. 2013;38(11):2295–304.
Schneider H, et al. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci USA. 1998;95(13):7778–83.
Goshen I, et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. 2007;32(8–10):1106–15.
Labrousse VF, et al. Impaired interleukin-1beta and c-Fos expression in the hippocampus is associated with a spatial memory deficit in P2X(7) receptor-deficient mice. PLoS ONE. 2009;4(6):e6006.
Yirmiya R, Winocur G, Goshen I. Brain interleukin-1 is involved in spatial memory and passive avoidance conditioning. Neurobiol Learn Mem. 2002;78(2):379–89.
Gibertini M. Cytokines and cognitive behavior. NeuroImmunoModulation. 1998;5(3–4):160–5.
Avital A, et al. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. 2003;13(7):826–34.
Goshen I, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry. 2008;13(7):717–28.
Beattie EC, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295(5563):2282–5.
Steinmetz CC, Turrigiano GG. Tumor necrosis factor-alpha signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. 2010;30(44):14685–90.
Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440(7087):1054–9.
Balschun D, et al. Interleukin-6: a cytokine to forget. Faseb J. 2004;18(14):1788–90.
Jankowsky JL, Derrick BE, Patterson PH. Cytokine responses to LTP induction in the rat hippocampus: a comparison of in vitro and in vivo techniques. Learn Mem. 2000;7(6):400–12.
Olmos G, Llado J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014;2014:861231.
Santello M, Volterra A. TNFalpha in synaptic function: switching gears. Trends Neurosci. 2012;35(10):638–47.
Vallieres L, et al. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci. 2002;22(2):486–92.
Nelson TE, et al. Altered synaptic transmission in the hippocampus of transgenic mice with enhanced central nervous systems expression of interleukin-6. Brain Behav Immun. 2012;26(6):959–71.
Paolicelli RC, Bisht K, Tremblay ME. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front Cell Neurosci. 2014;8:129.
Schwabe L, et al. Stress impairs spatial but not early stimulus-response learning. Behav Brain Res. 2010;213(1):50–5.
Schwabe L, Wolf OT, Oitzl MS. Memory formation under stress: quantity and quality. Neurosci Biobehav Rev. 2010;34(4):584–91.
Kempermann G, Song H, Gage FH. Neurogenesis in the adult hippocampus. Cold Spring Harb Perspect Biol. 2015;7:a018812.
Koehl M. Gene-environment interaction in programming hippocampal plasticity: focus on adult neurogenesis. Front Mol Neurosci. 2015;8:41.
Koehl M, Abrous DN. A new chapter in the field of memory: adult hippocampal neurogenesis. Eur J Neurosci. 2011;33(6):1101–14.
Amrein I. Adult hippocampal neurogenesis in natural populations of mammals. Cold Spring Harb Perspect Biol. 2015;5:a021295.
Aimone JB, Deng W, Gage FH. Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron. 2011;70(4):589–96.
Sahay A, Wilson DA, Hen R. Pattern separation: a common function for new neurons in hippocampus and olfactory bulb. Neuron. 2011;70(4):582–8.
Leuner B, et al. Learning enhances the survival of new neurons beyond the time when the hippocampus is required for memory. J Neurosci. 2004;24(34):7477–81.
Cameron HA, McKay RD. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol. 2001;435(4):406–17.
Ziv Y, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9(2):268–75.
Opendak M, Gould E. Adult neurogenesis: a substrate for experience-dependent change. Trends Cogn Sci. 2015;19(3):151–61.
Schoenfeld TJ, Gould E. Stress, stress hormones, and adult neurogenesis. Exp Neurol. 2012;233(1):12–21.
Conrad CD. A critical review of chronic stress effects on spatial learning and memory. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(5):742–55.
Kreisel T, et al. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry. 2014;19(6):699–709.
Chetty S, et al. Stress and glucocorticoids promote oligodendrogenesis in the adult hippocampus. Mol Psychiatry. 2014;19(12):1275–83.
Whitney NP, et al. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108(6):1343–59.
Inoue K, et al. Long-term mild, rather than intense, exercise enhances adult hippocampal neurogenesis and greatly changes the transcriptomic profile of the hippocampus. PLoS One. 2015;10(6):e0128720.
Biedermann SV, et al. The hippocampus and exercise: histological correlates of MR-detected volume changes. Brain Struct Funct. 2014. doi:10.1007/s00429-014-0976-5.
Suh H, et al. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell. 2007;1(5):515–28.
Ahlskog JE, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876–84.
Smith PJ, et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239–52.
Mouret A, et al. Learning and survival of newly generated neurons: when time matters. J Neurosci. 2008;28(45):11511–6.
Drapeau E, et al. Learning-induced survival of new neurons depends on the cognitive status of aged rats. J Neurosci. 2007;27(22):6037–44.
Winocur G, et al. Inhibition of neurogenesis interferes with hippocampus-dependent memory function. Hippocampus. 2006;16(3):296–304.
Jessberger S, et al. Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn Mem. 2009;16(2):147–54.
Stanojcic M, et al. Disturbed distribution of proliferative brain cells during lupus-like disease. Brain Behav Immun. 2009;23(7):1003–13.
Zimmermann N, et al. Global cognitive impairment in systemic lupus erythematosus patients: a structural MRI study. Clin Neuroradiol. 2015. doi:10.1007/s00062-015-0397-8.
Lauvsnes MB, et al. Association of hippocampal atrophy with cerebrospinal fluid antibodies against the NR2 subtype of the N-methyl-d-aspartate receptor in patients with systemic lupus erythematosus and patients with primary Sjogren’s syndrome. Arthritis Rheumatol. 2014;66(12):3387–94.
Hanly JG, et al. A prospective analysis of cognitive function and anticardiolipin antibodies in systemic lupus erythematosus. Arthritis Rheum. 1999;42(4):728–34.
Menon S, et al. A longitudinal study of anticardiolipin antibody levels and cognitive functioning in systemic lupus erythematosus. Arthritis Rheum. 1999;42(4):735–41.
Waterloo K, et al. Neuropsychological function in systemic lupus erythematosus: a five-year longitudinal study. Rheumatology (Oxford). 2002;41(4):411–5.
Waterloo K, et al. Neuropsychological dysfunction in systemic lupus erythematosus is not associated with changes in cerebral blood flow. J Neurol. 2001;248(7):595–602.
Devinsky O, Petito CK, Alonso DR. Clinical and neuropathological findings in systemic lupus erythematosus: the role of vasculitis, heart emboli, and thrombotic thrombocytopenic purpura. Ann Neurol. 1988;23(4):380–4.
Ellis SG, Verity MA. Central nervous system involvement in systemic lupus erythematosus: a review of neuropathologic findings in 57 cases, 1955–1977. Semin Arthritis Rheum. 1979;8(3):212–21.
Hanly JG, Walsh NM, Sangalang V. Brain pathology in systemic lupus erythematosus. J Rheumatol. 1992;19(5):732–41.
Scolding NJ, Joseph FG. The neuropathology and pathogenesis of systemic lupus erythematosus. Neuropathol Appl Neurobiol. 2002;28(3):173–89.
Yajima N, et al. Elevated levels of soluble fractalkine in active systemic lupus erythematosus: potential involvement in neuropsychiatric manifestations. Arthritis Rheum. 2005;52(6):1670–5.
Hirohata S, Miyamoto T. Elevated levels of interleukin-6 in cerebrospinal fluid from patients with systemic lupus erythematosus and central nervous system involvement. Arthritis Rheum. 1990;33(5):644–9.
Trysberg E, Carlsten H, Tarkowski A. Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus. 2000;9(7):498–503.
Alcocer-Varela J, Aleman-Hoey D, Alarcon-Segovia D. Interleukin-1 and interleukin-6 activities are increased in the cerebrospinal fluid of patients with CNS lupus erythematosus and correlate with local late T-cell activation markers. Lupus. 1992;1(2):111–7.
Katsumata Y, et al. Diagnostic reliability of cerebral spinal fluid tests for acute confusional state (delirium) in patients with systemic lupus erythematosus: interleukin 6 (IL-6), IL-8, interferon-alpha, IgG index, and Q-albumin. J Rheumatol. 2007;34(10):2010–7.
George-Chandy A, Trysberg E, Eriksson K. Raised intrathecal levels of APRIL and BAFF in patients with systemic lupus erythematosus: relationship to neuropsychiatric symptoms. Arthritis Res Ther. 2008;10(4):R97.
Fragoso-Loyo H, et al. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. 2007;56(4):1242–50.
Trysberg E, et al. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheum. 2003;48(10):2881–7.
Trysberg E, et al. Intrathecal levels of matrix metalloproteinases in systemic lupus erythematosus with central nervous system engagement. Arthritis Res Ther. 2004;6(6):R551–6.
Hsu TC, et al. Beneficial effects of treatment with cystamine on brain in NZB/W F1 mice. Eur J Pharmacol. 2008;591(1–3):307–14.
Tomita M, Holman BJ, Santoro TJ. Aberrant cytokine gene expression in the hippocampus in murine systemic lupus erythematosus. Neurosci Lett. 2001;302(2–3):129–32.
McLean BN, Miller D, Thompson EJ. Oligoclonal banding of IgG in CSF, blood-brain barrier function, and MRI findings in patients with sarcoidosis, systemic lupus erythematosus, and Behcet’s disease involving the nervous system. J Neurol Neurosurg Psychiatry. 1995;58(5):548–54.
Nishimura K, et al. Blood-brain barrier damage as a risk factor for corticosteroid-induced psychiatric disorders in systemic lupus erythematosus. Psychoneuroendocrinology. 2008;33(3):395–403.
Bertsias GK, Boumpas DT. Pathogenesis, diagnosis and management of neuropsychiatric SLE manifestations. Nat Rev Rheumatol. 2010;6(6):358–67.
Sidor MM, et al. Elevated immunoglobulin levels in the cerebrospinal fluid from lupus-prone mice. J Neuroimmunol. 2005;165(1–2):104–13.
Schenatto CB, et al. Raised serum S100B protein levels in neuropsychiatric lupus. Ann Rheum Dis. 2006;65(6):829–31.
Hirohata S, et al. Blood-brain barrier damages and intrathecal synthesis of anti-N-methyl-d-aspartate receptor NR2 antibodies in diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Arthritis Res Ther. 2014;16(2):R77.
Sled JG, et al. Time course and nature of brain atrophy in the MRL mouse model of central nervous system lupus. Arthritis Rheum. 2009;60(6):1764–74.
Abbott NJ, Mendonca LL, Dolman DE. The blood-brain barrier in systemic lupus erythematosus. Lupus. 2003;12(12):908–15.
O’Carroll SJ, et al. Pro-inflammatory TNFalpha and IL-1beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J Neuroinflammation. 2015;12:131.
Stock AD, Wen J, Putterman C. Neuropsychiatric lupus, the blood brain barrier, and the TWEAK/Fn14 pathway. Front Immunol. 2013;4:484.
Rekvig OP, et al. Autoantibodies in lupus: culprits or passive bystanders? Autoimmun Rev. 2012;11(8):596–603.
Jeltsch-David H, Muller S. Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat Rev Neurol. 2014;10(10):579–96.
Hu C, et al. Autoantibody profiling on human proteome microarray for biomarker discovery in cerebrospinal fluid and sera of neuropsychiatric lupus. PLoS One. 2015;10(5):e0126643.
Gaynor B, et al. Peptide inhibition of glomerular deposition of an anti-DNA antibody. Proc Natl Acad Sci USA. 1997;94(5):1955–60.
Faust TW, et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc Natl Acad Sci USA. 2010;107(43):18569–74.
Huerta PT, et al. Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci USA. 2006;103(3):678–83.
Kowal C, et al. Cognition and immunity; antibody impairs memory. Immunity. 2004;21(2):179–88.
Chang EH, et al. Selective impairment of spatial cognition caused by autoantibodies to the N-Methyl-d-Aspartate receptor. EBioMedicine. 2015;2(7):755–64.
Omdal R, et al. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol. 2005;12(5):392–8.
Arinuma Y, Yanagida T, Hirohata S. Association of cerebrospinal fluid anti-NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2008;58(4):1130–5.
Fragoso-Loyo H, et al. Serum and cerebrospinal fluid autoantibodies in patients with neuropsychiatric lupus erythematosus. Implications for diagnosis and pathogenesis. PLoS One. 2008;3(10):e3347.
Yoshio T, et al. Association of IgG anti-NR2 glutamate receptor antibodies in cerebrospinal fluid with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2006;54(2):675–8.
Massardo L, et al. Anti-N-methyl-d-aspartate receptor and anti-ribosomal-P autoantibodies contribute to cognitive dysfunction in systemic lupus erythematosus. Lupus. 2015;24(6):558–68.
Mackay M, et al. Brain metabolism and autoantibody titres predict functional impairment in systemic lupus erythematosus. Lupus Sci Med. 2015;2(1):e000074.
Elkon KB, Parnassa AP, Foster CL. Lupus autoantibodies target ribosomal P proteins. J Exp Med. 1985;162(2):459–71.
Briani C, et al. Neurolupus is associated with anti-ribosomal P protein antibodies: an inception cohort study. J Autoimmun. 2009;32(2):79–84.
Hirohata S, et al. Association of cerebrospinal fluid anti-ribosomal p protein antibodies with diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Arthritis Res Ther. 2007;9(3):R44.
Karassa FB, et al. Accuracy of anti-ribosomal P protein antibody testing for the diagnosis of neuropsychiatric systemic lupus erythematosus: an international meta-analysis. Arthritis Rheum. 2006;54(1):312–24.
Hanly JG, et al. Autoantibodies as biomarkers for the prediction of neuropsychiatric events in systemic lupus erythematosus. Ann Rheum Dis. 2011;70(10):1726–32.
Reichlin M. Autoantibodies to the ribosomal P proteins in systemic lupus erythematosus. Clin Exp Med. 2006;6(2):49–52.
Matus S, et al. Antiribosomal-P autoantibodies from psychiatric lupus target a novel neuronal surface protein causing calcium influx and apoptosis. J Exp Med. 2007;204(13):3221–34.
Segovia-Miranda F, et al. Pathogenicity of lupus anti-ribosomal P antibodies: role of cross-reacting neuronal surface P antigen in glutamatergic transmission and plasticity in a mouse model. Arthritis Rheumatol. 2015;67(6):1598–610.
Bravo-Zehnder M, et al. Anti-ribosomal P protein autoantibodies from patients with neuropsychiatric lupus impair memory in mice. Arthritis Rheumatol. 2015;67(1):204–14.
Katzav A, et al. Anti-P ribosomal antibodies induce defect in smell capability in a model of CNS -SLE (depression). J Autoimmun. 2008;31(4):393–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mackay, M. Lupus brain fog: a biologic perspective on cognitive impairment, depression, and fatigue in systemic lupus erythematosus. Immunol Res 63, 26–37 (2015). https://doi.org/10.1007/s12026-015-8716-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-015-8716-3