
Abstract
Polymorphonuclear leukocytes (PMNs) are the most abundant white cell in humans and an essential component of the innate immune system. PMNs are typically the first type of leukocyte recruited to sites of infection or areas of inflammation. Ingestion of microorganisms triggers production of reactive oxygen species and fusion of cytoplasmic granules with forming phagosomes, leading to effective killing of ingested microbes. Phagocytosis of bacteria typically accelerates neutrophil apoptosis, which ultimately promotes the resolution of infection. However, some bacterial pathogens alter PMN apoptosis to survive and thereby cause disease. Herein, we review PMN apoptosis and the ability of microorganisms to alter this important process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polymorphonuclear leukocytes (PMNs or neutrophils) are the most abundant leukocyte in humans and typically the first type of white cell recruited to sites of infection. Neutrophils are the primary cellular defense against bacterial and fungal infections. PMNs ingest microbes by a process known as phagocytosis, and the ingested microorganisms are destroyed by the combination of reactive oxygen species (ROS) and cytotoxic components of granules [1–4] (Fig. 1). This task is facilitated by the numerous receptors enriched on the cell surface and proinflammatory stimuli.

Neutrophil phagocytosis and microbicidal activity. Following phagocytosis, microbes are destroyed by ROS and antimicrobial proteins released from granules. See text for details. FcR, Fc receptor; CR, complement receptor; MPO, myeloperoxidase
On the other hand, PMNs contain or produce many cytotoxic molecules that can cause significant damage to host tissues/organs if the inflammatory response is not tightly regulated. Resolution of the inflammatory response is a complex process and includes production of host-derived anti-inflammatory mediators and apoptosis of PMNs. Specifically, PMN apoptosis and removal of apoptotic PMNs by macrophages is a mechanism to clear effete neutrophils and ultimately facilitate the resolution of inflammation.
This review highlights two areas of neutrophil biology related to immune system homeostasis and the innate host defense, namely, PMN apoptosis and the ability of microbes to manipulate this process, and the resolution of the acute inflammatory response, including clearance of apoptotic cells by macrophages.
Granulocytes, granulopoiesis, and the development of myeloid cells
Granulocytes (basophils, eosinophils, and neutrophils) can be differentiated physically from lymphocytes and other circulating leukocytes based upon multiple phenotypes, such as the presence of characteristic granules in the cytoplasm, a multilobed nucleus and specific cell surface markers (Fig. 2, top panels). Development of granulocytes involves transcription factors distinct from those that facilitate lymphocyte maturation [5–9]. Many of these transcription factors are required for granule protein maturation and ligands essential for PMN activation, as different granules appear during separate stages of PMN development ([9], reviewed in [1, 10]). Secondary or specific granules predominate in mature neutrophils, whereas the relative number of azurophilic or primary granules decreases during granulocyte maturation [11].

Neutrophil apoptosis. Human neutrophils isolated from peripheral blood have multilobed nuclei when fresh (0 h), whereas those cultured for 24 h in vitro have condensed nuclei of apoptotic cells (24 h). Left panels, transmission electron micrographs. Right panels, brightfield micrographs of PMNs strained with a modified Wright-Giemsa. Nucleus, n. Transmission electron microscopy was performed by David W. Dorward at Rocky Mountain Laboratories, NIAID
Circulating leukocytes and other cells of the immune system develop from pluripotent stem cells located in bone marrow [12]. Granulocytes differentiation proceeds through a concerted series of steps that include granule formation and ultimately exit of mature PMNs from bone marrow to peripheral blood [1, 9–11, 13–18]. This maturation and differentiation process takes ~6.5 days and is called granulopoiesis, i.e., the formation of granulocytes [11]. Although PMNs have a relatively short life span, the greatest percentage of hematopoiesis is committed to the production of neutrophils––nearly 60% of all leukocytes in bone marrow are granulocyte precursors. Mature PMNs are terminally differentiated cells and circulate in the bloodstream for ~10–24 h before migrating into tissues [15–18]. Neutrophils may function for an additional 1–2 days in tissues before undergoing apoptosis [11, 17] and are then cleared by macrophages [19, 20]. Remarkably, PMN turnover is on the order of 1011 cells per day in the average adult human [15].
Recruitment of neutrophils from the bloodstream
Neutrophil recruitment, priming, and extravasation
Neutrophils survey venules and lymphatic organs for signs of invading microorganisms and/or host tissue injury and inflammation. These “signs” are often chemoattractants, host- or microorganism-derived molecules recognized by receptors on the cell surface. Chemoattractants recruit PMNs to sites of infection or areas of tissue damage. Most chemoattractants, such as IL-8, lipopolysaccharide (LPS), and N-formylated peptides (e.g., fMLP), also prime neutrophils for enhanced function (see Table 1 in [21]).
Neutrophil priming was originally defined as the ability of a primary agonist such as LPS to enhance superoxide production by a secondary stimulus [22]. Secretory vesicles, gelatinase-containing granules, and a limited population of specific granules fuse with the plasma membrane during priming [23], thereby enriching the cell surface with receptors important for host defense and the inflammatory response. Primed cells can be differentiated from unprimed cells in part by distinct cell morphology, e.g., cell size and/or granularity by flow cytometry, or increased expression of surface receptors such as CD11b [24]. Some priming agents are agonists or ligands of TLRs, and in vitro and in vivo experiments have shown that TLRs are critical for PMN priming as well as pathogen recognition [25].
Neutrophils enter sites of infection by exiting peripheral circulation through the endothelial wall in a process called extravasation. This process is initiated by selectins, a C-type lectin family of glycoproteins that are expressed on activated endothelial cells (E- and P-selectin), platelets (P-selectin), and primed/activated PMNs (L-selectin, CD62). As part of the inflammatory response, E- and P-selectin become up-regulated on endothelial cells and interact with L-selectin expressed on the surface of neutrophils. This initial interaction, called “tethering,” facilitates subsequent rolling of PMNs along the surface of the endothelium. Rolling is followed by firm adhesion promoted by CD11b/CD18 [26] and CD54 [27], and transmigration is mediated in part by CD31 [28], CD54 [27], CD44 [29], and CD47 [30]. Following extravasation, there are several possible fates for PMNs related to apoptosis and turnover, and these are discussed in detail below.
PMN microbicidal activity
PMNs destroy pathogens through a series of well-coordinated steps that result in the production of ROS and enrichment of phagosomes with cytotoxic molecules from cytoplasmic granules (called degranulation) (Fig. 1). ROS are derived from superoxide produced by a multisubunit enzyme complex called NADPH oxidase (reviewed in [3]). The phagocyte NADPH oxidase is composed of at least 7 proteins that reside in cytosolic and membrane compartments in resting PMNs. During cell activation, cytosolic oxidase components translocate to the phagosome membrane and associate with the membrane component, flavocytochrome b 558, to form the assembled NADPH oxidase. NADPH oxidase assembly and activation is regulated by multiple signaling events, including phosphorylation of one of the cytosolic proteins, p47phox, and several SH3-domain interactions [3, 31]. NADPH oxidase catalyzes the formation of superoxide (O2 −), which dismutates rapidly to H2O2, although O2 − and H2O2 are weakly microbicidal [2].
As the phagosome forms/matures, granule components such as myeloperoxidase (MPO) accumulate in the phagosome [11]. MPO catalyzes a reaction with H2O2 and chloride to form hypochlorous acid (HOCl) and other secondarily derived ROS such as OH·and singlet oxygen [32]. Neutrophil cytoplasts––neutrophils lacking nuclei and depleted of cytoplasmic granules––retain a respiratory burst, but lack MPO. Odell and Segal reported that cytoplasts phagocytose Staphylococcus aureus, but bacteria are not killed unless they are coated with MPO [33]. MPO activity is inhibited by azide, and to a lesser extent cyanide, and this inhibition decreases killing of bacteria by neutrophils [34].
Landmark work by Rosen and Klebanoff [35] demonstrated that the key antimicrobial components of human PMNs, NADPH oxidase and the MPO-halide system, serve as redundant mechanisms to kill ingested microbes. Consistent with this notion, MPO deficiency in humans does not necessarily correlate with morbidity from infections (reviewed in [4]). PMNs from MPO-deficient individuals retain microbicidal activity against several bacterial pathogens, although at a lower rate than PMNs from normal donors [4]. Collectively, these findings suggest that a combination of mechanisms is necessary for optimal killing of microbes.
In addition to the intracellular proteins that degrade the ingested microbes, the surface of PMNs is decorated with multiple receptors that bind to foreign particles, including those that bind pathogen associated molecular patterns (PAMPs). Many pattern recognition receptors (PRRs) do not promote phagocytosis directly, although they do prime phagocytes for enhanced uptake/activation or function as co-receptors. For example, TLRs recognize numerous PAMPs [36], but there is little data available to show that TLRs promote phagocytosis directly (reviewed in [37]). By comparison, neutrophil dectin-1 [38], a C-type lectin PRR, binds β-glucan residues on fungi and this interaction triggers phagocytosis [39]. Phagocytosis of unopsonized β-glucan particles is morphologically different than phagocytosis of complement-opsonized zymosan, suggesting that signal transduction following these receptor-ligand interactions is distinct [39]. Importantly, dectin-1 mediated phagocytosis elicits ROS production and degranulation, and ultimately killing of fungi [39].
PMN apoptosis and the resolution of the acute inflammatory response
Normal turnover of cells, regardless of type or tissue location, is governed by apoptosis, and defects in apoptosis lead to profound and devastating conditions ranging from cancer to autoimmune diseases. Neutrophils undergo constitutive or spontaneous apoptosis as a mechanism to maintain immune system homeostasis [19]. Importantly, PMN apoptosis is accompanied by a general decrease in cell function and proinflammatory capacity, which is key to non-inflammatory removal of effete cells [40, 41]. Neutrophil apoptosis can be initiated by extrinsic pathway stimuli (extracellular, e.g., tumor necrosis factor-alpha (TNFα) or FAS ligand) or intrinsic pathway stimuli (intracellular, e.g., ROS and/or that mediated by mitochondria). Neutrophil apoptosis is a non-inflammatory process characterized by loss of cytoplasmic granules, rounding of the nucleus, and condensation of nuclear heterochromatin [19, 20] (Fig. 2, lower panels). PMNs also undergo apoptosis after phagocytosis, a process also known as phagocytosis-induced cell death (PICD, see below). Apoptotic neutrophils are subsequently ingested by macrophages, providing the means to resolve the inflammatory response without releasing cytotoxic molecules that would otherwise damage host tissues. Phagocytosis of apoptotic PMNs was first observed by Elie Metchnikoff in the 19th century [42].
Spontaneous (constitutive) PMN apoptosis
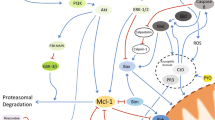
PMN apoptosis can be induced through multiple mechanisms (Fig. 3a, b). Spontaneous neutrophil apoptosis is an intrinsic process, meaning that the signal to initiate apoptosis originates from within the cell (Fig. 3a). Many proteins and/or molecules are known to regulate constitutive PMN apoptosis and only a few examples are provided here. For example, MCL1 is a key BCL2-family protein located in the nucleus and cytoplasm of PMNs [43–45]. As senescent neutrophils undergo apoptosis, MCL1 levels decline rapidly, presumably degraded by the proteasome (and there is a concurrent decrease in MCL1 mRNA), suggesting neutrophil survival is regulated by MCL1 expression [46].

Mechanisms of PMN apoptosis. (a) Intrinsic pathways of apoptosis result from intracellular signals, while extrinsic pathways (b) originate from extracellular signals
Src homology 2 domain-containing inositol 5′-phosphatase (SHIP-1) is an intracellular protein critical for signal transduction during PMN apoptosis [47]. The molecule is a negative regulator of immune receptor and cytokine and growth factor receptor signaling [48]. SHIP-1 is recruited to the plasma membrane of myeloid cells after ligation of CD18 [49], and this initiates an anti-apoptotic signaling cascade that requires the Src kinase Lyn and the anti-apoptotic protein, Akt [48]. Ship-1 −/− mice have increased numbers of myeloid cells and form lung granulomas after infection [50]. It was proposed that the granulomas fail to resolve because Ship-1 −/− cells display enhanced survival and proliferation [50].
BCL2A1 is another protein important for PMN survival. Like MCL1, BCL2A1 is a member of the BCL2 family of proteins, and BCL2A1 mRNA is constitutively expressed in human neutrophils [51]. PMN BCL2A1 is up-regulated by G-CSF or LPS, agonists known to promote neutrophil survival [51]. Promyelocytic HL-60 cells have increased BCL2A1 mRNA after neutrophilic differentiation with all-trans retinoic acid [51], suggesting that the protein has an important role in mature neutrophils.
BCL2-associated X protein (BAX) interacts with the mitochondria, an intracellular organelle known to regulate PMN survival [52, 53]. In contrast to BCL2A1, BAX is a BCL2-family member that promotes neutrophil apoptosis. Upon translocation to the mitochondria, BAX is cleaved by calpain-1 to an 18-kDa fragment unable to interact with BCL2 family members, thereby promoting apoptosis [54, 55]. Inhibition of BAX cleavage in vivo leads to increased and prolonged inflammation, indicating that the protein is required for the resolution of acute inflammation [52]. Using BAX antisense gene silencing, Dibbert et al. showed that PMN apoptosis is delayed in several human inflammatory diseases and there is an associated decrease in BAX protein expression [56].
Soluble extracellular factors also influence spontaneous PMN apoptosis [57]. For example, macrophage migration inhibitory factor is a proinflammatory cytokine that delays cleavage of BAX and ultimately neutrophil apoptosis [58]. PMNs stimulated with G-CSF or GM-CSF have decreased expression of BAX, suggesting the anti-apoptotic effect of these cytokines is in part related to inhibition of BAX. GM-CSF and G-CSF also preserve the chemotaxis and phagocytosis capacity of PMNs aged in vitro [40, 59], indicating inhibition of apoptosis is tied to the preservation of PMN function.
In addition to BAX activity, mitochondria regulate their transmembrane potential, which is critical for cell survival. As depicted in Fig. 3a, ROS and oxidative stress can lead to PMN apoptosis through disruption of mitochondria transmembrane potential [60–65]. Neutrophil mitochondria contain calpain-1 and its release can activate pro-apoptotic factors or degrade anti-apoptotic proteins [66]. Although human neutrophils express low levels of cytochrome c, it plays an important role in apoptosis [65]. As cytochrome c is released from the mitochondria, it associates with apoptotic protease-activating factor 1 (APAF-1) and initiates apoptosis [53].
Several distal effector proteins participate in apoptosis. Caspase 3, caspase 8, and caspase 9 trigger proteolytic cascades that result in the apoptotic death of human neutrophils [67] (Fig. 3b). The importance of neutrophil caspases in inflammation is illustrated in a recent study by Tanejo et al. [68], in which it was reported that individuals with bacterial sepsis have significant reduction of caspase-3 and caspase-9 activities and decreased neutrophil apoptosis.
These are only a few specific examples of the molecules known to regulate spontaneous neutrophil apoptosis and overlap exists with extrinsic pathway stimuli as described below.
FAS/FAS ligand-mediated apoptosis
In contrast to spontaneous PMN apoptosis, FAS-mediated, TNFα-mediated and TRAIL-mediated apoptosis proceed by an extrinsic pathway, as an extracellular signal initiates apoptosis (Fig. 3b). Extrinsic pathway signals are usually soluble protein factors that bind to membrane-bound receptors on the cell surface and trigger apoptosis. FAS (CD95) is the nidus of the death-inducing signaling complex (DISC) and is expressed on the surface of neutrophils as well as on virtually all other cells in humans [69]. Neutrophils also express FAS ligand (FASL, CD95L) [70], and FASL–FAS interaction initiates PMN apoptosis in a caspase-dependent manner. Because PMNs express FAS and soluble FASL, PMN apoptosis is triggered through autocrine and paracrine pathways. FAS-mediated PMN apoptosis can be suppressed by GM-CSF, G-CSF, interferon-gamma (IFNγ), TNFα, and the tyrosine kinase inhibitors herbimycin A and genistein [70], thus highlighting the complexity of signaling pathways involved in regulating neutrophil turnover.
FAS is a transmembrane protein containing a FASL-binding extracellular domain and FAS-associated death domain (FADD) on its intracellular region. Interaction of FASL with FAS leads to clustering of FADDs on the cytoplasmic side of the plasma membrane, which, in turn, leads to caspase activation and apoptosis. As with many other signaling receptor complexes, e.g., T- and B-cell receptors, current evidence indicates that FAS accumulates in cholesterol-rich membrane domains called lipid rafts and that clustering of these receptors leads to caspase-8 activation and, ultimately, cell death. Macrophages also secrete FASL in response to phagocytosis [71]. Secretion of FASL by macrophages induces leukocyte apoptosis in a paracrine manner and assists further with the resolution of inflammation [71]. For example, Jimenez et al. studied patients with systemic inflammatory response syndrome, and PMNs from these patients have decreased FAS-mediated apoptosis and accompanying tissue damage following bacterial infections, presumably from the accumulation of activated neutrophils that release tissue degrading enzymes [72].
TNFα receptor (TNFR)-mediated apoptosis
The TNFR is a transmembrane protein containing an extracellular TNFα binding domain and a TNFR-associated death domain (TRADD) in the cytoplasmic region of the protein [73] (Fig. 3b). Neutrophils express two TNFRs, TNFRSF1A (55-R, CD120a, or TNFRI) and TNFSFR1B (75-R, CD120b, or TNFRII), and each has a slightly different role in PMN apoptosis. Gon et al. showed that TNFRI is required for TNFα-mediated PMN apoptosis and its ability to promote apoptosis is enhanced by TNFRII [74]. Blocking TNFRI but not TNFRII with specific antibodies inhibits neutrophil apoptosis [74]. Additional work has shown that TNFRI is dominant using TNFR-selective mutants [75]. As with FADDs, TRADDs cluster in lipid rafts during TNFR–TNFα binding. This clustering of signaling molecules leads to the recruitment of adaptor molecules and activation of transcription factors. Although similar to FAS/FASL-mediated apoptosis in many ways, one major difference is that TNFα receptor-mediated apoptosis does not depend solely on caspase activation [76].
TNFα can elicit inflammatory or anti-inflammatory effects depending on the type of cell with which it interacts. Notably, TNFα has been shown to have both pro-apoptotic and anti-apoptotic effects toward neutrophils. Van den Berg et al. showed that this bi-polar effect is concentration dependent [77]. At low concentrations (≤0.1 ng/ml), TNFα delays PMN apoptosis and elicits production of proinflammatory cytokines, whereas at higher concentrations TNFα initiates apoptosis [77]. Consistent with these observations, high concentrations of TNFα (10–100 ng/ml) override the ability of IFNγ and GM-CSF to delay apoptosis [77]. Moreover, the ability of TNFα to induce neutrophil apoptosis was linked to ROS production, since PMNs from patients with chronic granulomatous disease (CGD, see below) and PMN cytoplasts fail to undergo apoptosis in the presence of high concentrations of TNFα [76, 77]. Individuals with TNFR-associated periodic syndrome (TRAPS) have a defect in the TNFR and, therefore, diminished TNFα-induced PMN apoptosis [78]. Patients with TRAPS experience recurrent fever attacks lasting >1 week that is associated with abdominal pain, severe arthromyalgias, rash, and periorbital edema. However, TRAPS has not been associated with increased infection [78].
TNF-related apoptosis-inducing ligand (TRAIL)
Our understanding of TRAIL-induced PMN apoptosis is relatively recent compared to that of FAS- or TNF-receptor-mediated apoptosis. Five TRAIL receptors (TRAIL-R1, TRAIL-R2, TRAIL-R3, TRAIL-R4, and TRAIL-R5) have been identified and PMNs express TRAIL-R2 and TRAIL-R3 constitutively [79, 80]. Of the five TRAIL receptors, only TRAIL-R1 and TRAIL-R2 have the ability to induce pro-apoptotic, caspase-dependent signaling (Fig. 3b), whereas TRAIL-R3 and TRAIL-R4 are decoy receptors that lack the ability to induce apoptosis [81]. TRAIL-R3 and TRAIL-R4 do not have intact death domains and these receptors have been proposed to serve as “decoys” that inhibit apoptosis either by sequestering TRAIL or by complexing with TRAIL-R1 or TRAIL-R2 upon binding to the ligand. Either mechanism would inhibit apoptosis, but there is paucity of data to provide conclusive support to this notion. TRAIL-induced apoptosis appears important for senescent PMNs that preferentially home to bone marrow after circulating in peripheral blood [82]. Trail −/− mice have an enhanced susceptibility to autoimmune diseases [83], and Trail blocked experimentally in animal models exacerbates inflammation caused by type 1 diabetes [84] and autoimmune encephalomyelitis [85]. PMNs also express and secrete TRAIL, which has been implicated in host defense against virus-infected cells and tumor cells [86]. IFNγ up-regulates TRAIL expression and IFNγ-activated PMNs store TRAIL in an intracellular pool that is mobilized following exposure to proinflammatory mediators [79, 87, 88]. TRAIL expressed and secreted by PMNs acts in an autocrine manner to induce apoptosis [87].
Down-regulation of proinflammatory capacity
PMNs produce numerous cytokines and chemokines following phagocytosis, including TNFα and IL-8, which contribute to the inflammatory response [89–91]. Production of these molecules is regulated in part at the level of gene expression [92, 93]. Microarray-based studies have shown that phagocytosis initially up-regulates neutrophil transcripts encoding proteins that play an important role in the acute inflammatory response, such as IL-1α, IL-1β, IL-1ε, IL-1RN, IL-8, IL-10, IL-12β, IL-15, TNFα, vascular endothelial growth factor (VEGF), oncostatin M (OSM), IL-6, GROβ, and GROγ [21, 94–97]. Taken together, these processes are critical for defense against invading microorganisms. However, once phagocytosis is complete and microbes are destroyed, these PMN functions are down-regulated coincident with the induction of apoptosis [95, 98–102]. For example, key proinflammatory or signal transduction molecules, including CXCR1 and CXCR2, are down-regulated during the initial stages of apoptosis [94]. Walcheck et al. have shown recently that phagocytosis-induced neutrophil apoptosis is accompanied by shedding of L-selectin and concomitant increased surface expression of ADAM17, a metalloprotease that likely plays an important role in the down-regulation of neutrophil function [103, 104]. In general, most primary PMN functions, including chemotaxis, phagocytosis, superoxide production, and degranulation, decrease significantly with apoptosis [40, 41].
IL-6 receptor (IL-6R) transsignaling
IL-6 signals through a receptor system composed of the IL-6R, an 80 kDa transmembrane protein that binds the signaling molecule gp130. Although the expression of IL-6R is restricted to a small subset of cells, gp130 is expressed ubiquitously. IL-6 can also elicit responses through a pathway involving soluble IL-6R (sIL-6R) and this phenomenon is known as IL-6R transsignaling. sIL-6R is generated by either shedding from the cell surface by proteolytic cleavage or possibly alternative splicing of IL-6R mRNA [105, 106]. Chalaris et al. demonstrated that apoptosis is accompanied by the release of IL-6R from cells, a phenomenon mediated by ADAM17 [103, 104]. sIL-6R recruits mononuclear phagocytes to the site of infection, which are then either directly involved in the removal of apoptotic PMNs or differentiate into macrophages that carry out this task [107]. This process links the acute inflammatory stage of disease and resolution of the inflammatory response.
Resolvins and lipoxins
Endogenous mediators termed resolvins and protectins actively participate in the dampening of host responses to orchestrate resolution of inflammation. These endogenous mediators lend more evidence that the resolution of inflammation is an actively regulated program rather than a passive termination of the immune response. After the initial inflammatory response, these molecules promote recruitment of macrophages to sites of inflammation and initiate the resolution of the acute inflammatory response [108]. One of these mediators is known as lipoxin A4 (LXA4), which is generated from arachidonic acid via the lipoxygenase pathways. Together with the aspirin-triggered 15-epi-LXA4 (ATL), these molecules are among the first to down-regulate PMN infiltration and begin the “slow down” during the course of inflammation [109–111]. For example, lipoxins drive the resolution of inflammation by stimulating nonphlogistic uptake of apoptotic PMNs by fixed-tissue macrophages [112].
Removal of apoptotic PMNs by macrophages
As with removal of undesirable, autoreactive T-cells in the thymus, apoptosis is a mechanism to clear effete PMNs. Apoptotic cells maintain membrane integrity for a fixed period of time and need to be cleared quickly to prevent secondary necrosis and the release of cytotoxic molecules that would otherwise cause inflammation and/or damage to host tissues. Clearance of apoptotic neutrophils has been studied mainly with human monocyte-derived macrophages [113] and mouse alveolar macrophages [114]. Although clearance of apoptotic cells is not completely understood, several macrophage receptors have been implicated in this process (Fig. 4). These molecules include the phosphatidylserine (PS) receptor [114, 115], complement receptors [116], scavenger receptors and lectins [117], the αvβ3/CD36/thrombospondin recognition system [118, 119], CD14 [119], and CD44 [121–123]. The redundancy of removal mechanisms suggests that clearance of apoptotic neutrophils is a critical process to preserve immune system homeostasis.

Receptors on macrophages that bind antigens on apoptotic cells. Apoptotic neutrophils are recognized by specific receptors present on mononuclear phagocytes. See text for details
Phosphatidylserine receptor (PSR)
Transposition of PS from the inner leaflet of the plasma membrane to the outer leaflet is a traditional hallmark of cells undergoing apoptosis. Recently, Park et al. have shown that stabilin-2 serves as a novel PSR and that blocking stabilin-2 on macrophages inhibits phagocytosis of apoptotic cells and causes release of proinflammatory cytokines [124]. Miyanishi et al. identified Tim4 as a PSR through the use of an antibody library and showed that blocking Tim4 in vivo resulted in an accumulation of apoptotic cells in the thymus and the development of autoantibodies [125]. The transcript encoding the Psr is expressed in several tissues in mice [126]. Human and mouse macrophages express the Psr and it is required for clearance of apoptotic cells in vitro and in vivo [126–128]. Consequently, mice genetically deficient in the gene encoding Psr have accumulation of apoptotic cells in the lung and brain during embryogenesis and develop autoimmune diseases [126, 129]. It is possible that the generation of autoimmune diseases in these models results from the pool of apoptotic cells that releases autoantigens over time.
Complement receptors
The human complement cascade can be activated by antibodies (classical pathway) or by oligosaccharide antigens on microbes or apoptotic cells (lectin and alternative pathways) (reviewed in [130]). Each of the complement cascades is a sequential, proteolytic cascade that results in opsonization of targets with C3b(i) and/or lysis of opsonized cell by the membrane attack complex (MAC). Complement proteins, specifically C3b and its breakdown products, bind to apoptotic cells, possibly through the exposure of PS [116], and altered lipid and carbohydrate moieties. This process facilitates removal of apoptotic cells from tissues (reviewed in [131]). The majority of complement receptor phagocytosis is mediated through one of the four receptors that bind to complement component C3; CR1 (CD35), CR2 (CD21), CR3 (CD11b/CD18), and CR4 (CD11c/CD18). Macrophages express CR3 and CR4 as well as the C1q receptor, which binds to the first component of the classical complement pathway. The importance of C3 and C1q in apoptotic cell clearance has been shown in mice with targeted deletion of the genes encoding these receptors [129, 132]. Grevnik et al. reported that although systemic lupus erythematosus patients have reduced total complement hemolytic activity in their sera, complement-mediated phagocytosis of apoptotic cells remained unaffected [133]. This observation can be explained by differential depletion of specific complement components in the patients, i.e., reduced levels of C9 and normal levels C3.
Vitronectin (αvβ3)/CD36/thrombospondin recognition system
Vitronectin (αvβ3 integrin), CD36, and thrombospondin are believed to function in a complex on macrophages and promote binding and uptake of apoptotic cells. Cooperation among these three antigens occurs during recognition of apoptotic PMNs by human monocyte-derived macrophages [118] and murine bone marrow-derived macrophages [113]. Thrombospondin functions somewhat like C3b in that thrombospondin binds to an unknown ligand on apoptotic cells, which then is recognized by the CD36/vitronectin complex expressed on macrophages. CD36 is an 88 kDa multifunctional glycoprotein, a B-type scavenger receptor, expressed by several cell types including monocytes, endothelial cells, epithelial cells, and several tumor cell lines [134]. In addition to thrombospondin, oxidized PS has recently been shown to be a ligand for CD36 [135]. Macrophage CD36, along with αvβ3 integrin, is important for the removal of apoptotic cells during the resolution of viral infections [119]. The domain on CD36 that is critical for the removal of apoptotic cells has been localized to amino acids 155–183 [136].
CD44
CD44 is a ~90 kDa surface protein that binds to hyaluronic acid. Ligation of CD44 generates intracellular signals that specifically augment clearance of apoptotic PMNs [121, 122]. For example, blocking CD44 with anti-CD44 Fab’ antibodies decreases macrophage-mediated phagocytosis of apoptotic bodies [121]. Furthermore, CD44-deficient mice have reduced capacity to clear apoptotic PMNs post-inflammation [137].
CD14
CD14 is a 53–55 kDa protein expressed on monocytes, on macrophages, and at relatively low levels on granulocytes. Although CD14 is a co-receptor for LPS/LPS-binding protein [138], recent evidence indicates CD14 also binds intercellular adhesion molecule-3 (ICAM-3) on leukocytes [139]. CD14 can be expressed in soluble form (sCD14) and may bind apoptotic cells and subsequently react with a sCD14 receptor on phagocytes [140]. Little is known about the role of CD14 in the removal of apoptotic PMNs; however, there is good evidence that CD14 is crucial for the removal of apoptotic lymphocytes [141, 142].
Scavenger receptors and lectins
In addition to the flipping of PS from the inner leaflet to the outer leaflet of the plasma membrane, sugars and oxidized lipids that are exposed on the surface of apoptotic cells are modified and recognized as “non-self” by macrophages. These modifications are recognized by scavenger receptors and lectins expressed on the surface of macrophages and fibroblasts [143].
Non-inflammatory removal of apoptotic PMNs
Neutrophil apoptosis and the removal of apoptotic neutrophils is a nonphlogistic process. Phagocytosis of apoptotic cells by macrophages, monocytes, or dendritic cells causes these cell types to produce anti-inflammatory cytokines [144, 145]. For example, macrophages exposed to apoptotic cells secrete TGF-β, which suppresses LPS-mediated release of inflammatory cytokines by infected cells [146]. Macrophages also secrete factors that contribute to PMN apoptosis (e.g., soluble FASL [71]) and increase expression of anti-inflammatory factors [147]. For example, in the airway (bronchi and lungs), apoptotic PMNs are ingested by alveolar macrophages without release of inflammatory mediators (reviewed in [148]). Moreover, engagement of apoptotic cells by CD36 on macrophages inhibits production of proinflammatory cytokines TNFα, IL-1β, and IL-12, but increases secretion of TGF-β and IL-10, both anti-inflammatory cytokines [149].
In addition to the autologous signals that contribute to the anti-inflammatory process, steroids––specifically glucocorticoids––promote a non-inflammatory environment during phagocytosis of apoptotic cells. Human and murine macrophages exposed to glucocorticoids for 24 h have enhanced ability to phagocytose apoptotic leukocytes [150]. Compared to macrophages, peripheral blood monocytes have a much lower phagocytic capacity toward apoptotic cells, but incubation of human monocytes with glucocorticoids induces the phenotype of monocyte-derived macrophages much more competent to ingest apoptotic cells [19, 151]. The ability of monocytes to phagocytose apoptotic leukocytes after incubation with glucocorticoids has been linked to 11β-hydroxysteroid dehydrogenase (11β-HSD), a molecule not present in monocytes [152, 153]. Cytokines also influence phagocytic capacity of macrophages toward apoptotic cells. For instance, IFNγ inhibits the glucocorticoid-induced ability of macrophages to clear apoptotic PMNs [154].
Factors that prolong neutrophil survival
Although neutrophil apoptosis is critical for granulocyte homeostasis and the resolution of inflammation, many proinflammatory molecules extend PMN survival during the initial stages of the inflammatory response. For example, neutrophils that migrate to sites of tissue inflammation have a prolonged lifespan and become resistant to both FAS- and TNF-induced apoptosis [155]. This delay in apoptosis likely promotes robust early response to infection or inflammatory insult and is important because it would increase the window of time during which neutrophils can be recruited to such sites and remain fully functional.
Host-derived factors as well as bacterial and fungal products serve to delay neutrophil apoptosis. Cytokines, such as IL-1β, TNFα, GM-CSF, G-CSF, and IFNγ, recruit PMNs to sites of infection and each of these molecules can delay PMN apoptosis [57, 156, 157]. Notably, most of the host-derived cytokines that delay apoptosis also prime neutrophils for enhanced function to a second agonist. Complement component C5 is cleaved to form C5b, a member of the membrane attack complex, and C5a, a powerful chemotactic agent for neutrophils [158]. C5a protects PMNs from apoptosis and this process involves BCL2-antagonist of cell death (BAD) and phosphoinositol-3 kinase [159]. Bacterial products, especially LPS and lipoteichoic acid (LTA), are potent inhibitors of PMN apoptosis [57].
In addition to soluble factors, the extracellular environment can signal PMN apoptosis. For instance, acute hypoxia enhances the inflammatory response and causes a decrease in PMN apoptosis that has an additive effect with GM-CSF [160]. This effect is also BCL2-independent, suggesting that another intracellular, pro-survival signaling molecule is involved in this process [161]. Hypoxia also increases TLR4 expression and enhances the pro-survival effects of LPS toward human PMNs [162].
Role of toll-like receptors (TLRs) in PMN apoptosis
TLRs in combination with other receptors, e.g., complement receptors, promote rapid and efficient recognition of pathogens by neutrophils [163, 164]. To date, ten TLRs are expressed in human tissues and all but TLR3 are expressed in neutrophils [165]. Ligands for TLRs include lipoproteins (TLR1/TLR2) LTA (TLR6), LPS (TLR4), flagellin (TLR5), double-stranded DNA (TLR3), single-stranded RNA (TLR7), and unmethylated CpG DNA of bacteria and viruses (TLR9) [166]. TLR1, TLR2, TLR4, TLR5, and TLR6 are expressed on the surface of PMNs, whereas TLR79 is localized to intracellular compartments and recruited to the plasma membrane during PMN priming/activation [166–169].
Although all TLRs play an important role in the innate immune response, only TLR2, TLR4, and TLR6 seem to modulate PMN apoptosis directly. Of these TLRs, TLR4 seems to be more tightly coupled to PMN survival than either TLR2 or TLR6. TLR4 binds to LPS and this interaction up-regulates IL-8 mRNA and leads to prolonged neutrophil survival that involves monocytes [170]. This observation can be explained in part by the ability of monocyte CD14 to bind LPS, thereby signaling release of a factor that acts in a synergistic manner with TLR4 to prolong PMN survival [171]. The monocyte-dependent delay in PMN apoptosis can be reconstituted by adding peripheral blood mononuclear cells (PBMCs) to highly purified PMNs [172]. Sta. aureus peptidoglycan and LTA, agonists for TLR2, also delay PMN apoptosis [173, 174]. In PMN preparations that have been depleted of monocytes by magnetic bead isolation, TLR4 ligation to LPS is the principal TLR survival signal. However, the LPS-mediated delay in apoptosis in the absence of monocytes is relatively short (~4 h), as opposed to the 22-h delay observed in the presence of monocytes [43, 175].
Phagocytosis-induced cell death (PICD)
Neutrophil apoptosis differentiation program
Although a widely accepted view was that mature PMNs have little biosynthetic capacity, recent evidence clearly indicates that this previous view is incorrect. Human PMNs express ~12,000–14,000 transcripts and have significant biosynthetic potential. Changes in PMN transcript levels (and associated proteins) during phagocytosis are an important component to the resolution of the inflammatory response [89, 92, 93, 95, 97, 176–180]. Furthermore, phagocytosis of bacterial pathogens, including Burkholderia cepacia, Borrelia hermsii, Sta. aureus, Str. pyogenes, and Listeria monocytogenes, is followed by global changes in neutrophil gene expression concurrent with induction of apoptosis [94]. These findings provide an explanation for the increased RNA synthesis in neutrophils following phagocytosis that was originally reported by Martin Cline in the 1960s [181]. Bacteria-induced apoptosis or PICD correlates well with alterations in transcripts encoding factors that promote or repress cell death, influencing the fate of PMNs [94]. Furthermore, PICD is accompanied by down-regulation of genes encoding proinflammatory molecules [94, 177]. Based on these findings, it was proposed that bacteria induce an apoptosis differentiation program in human PMNs that facilitates timely neutrophil turnover during infection. The neutrophil apoptosis differentiation program represents the final stage of neutrophil maturation and regulates in part multiple processes in human neutrophils, including apoptosis and associated decreased function.
Bacteria-induced cell death
Phagocytosis-induced cell death (PICD) is a mechanism to clear tissues of spent neutrophils containing dead or partially digested microbes, thereby facilitating the resolution of infection [94, 182–185]. As with spontaneous neutrophil apoptosis, timely removal of neutrophils––although in this case after phagocytosis––would prevent release of cytotoxic molecules into surrounding host tissues, a phenomenon mediated by necrotic lysis [186]. This process is important because neutrophils and neutrophil products are known to contribute to inflammatory diseases [187–191]. The mechanism of neutrophil PICD remains incompletely defined, but progress has been made.
Watson et al. were the first to demonstrate that phagocytosis accelerates PMN apoptosis [192]. Since that time, many bacteria have been shown to trigger neutrophil apoptosis or PICD (Table 1). Importantly, the work by Watson et al. linked PMN ROS with apoptosis [192] and ROS play a crucial role in neutrophil apoptosis [3, 47, 60–62, 95, 161, 182, 188, 193–199]. The bacteria: PMN ratio dictates in part the level of ROS produced in vitro, which in turn influences cell fate. For example, interaction of E. coli with human PMNs at a 1:1 ratio (1 bacteria per PMN) delays PMN apoptosis, whereas increasing this ratio to 10:1 triggers apoptosis/PICD (see Fig. 2a of ref. [192]). Simons et al. found similar results with Neisseria gonorrhoeae using low (1:1) bacteria: PMN ratios, although the readout was caspase activity rather than ROS production [200]. These results underscore the delicate balance between factors that delay apoptosis (e.g., LPS) and those that trigger PICD (ROS). Inasmuch as pathogen-induced apoptosis is complex process involving a multitude of signaling molecules and bacterial components, the process is likely dictated by the specific microbe and/or number of microbes ingested.
Coxon et al. showed that neutrophils from Cd11b/Cd18 −/− mice have decreased ROS production and delayed apoptosis [195]. Work by Kobayashi et al. used PMNs from 6 X-linked CGD patients to demonstrate that ROS are essential for PICD and a microarray-based approach was used to investigate the role of ROS in PICD [188]. Zhang et al. reported that ROS production triggered by ligation of CD11b/CD18 ultimately activates initiator caspases 3 and 8 in PMNs and that PICD occurs independent of typical death receptors (e.g., FAS/FASL) [60]. Additional links between ROS production and PMN apoptosis were identified in myeloperoxidase-deficient mice and may involve generation of hypochlorous acid [64, 201]. The importance of ROS in the resolution of infection is exemplified by studies in CGD mice, which revealed that neutrophils fail to undergo apoptosis after interaction with pathogens and are thus not removed from the site of infection [202]. Together, these studies revealed that NADPH oxidase-derived ROS are an important intracellular link between the destruction of bacteria and the resolution of infection.
Although most proinflammatory cytokines and many bacteria-derived factors delay apoptosis [57, 174, 203–206], a complex signaling system exists such that bacteria-induced apoptosis or PICD overrides any delay in apoptosis due to these factors [207, 208].
Modulation of neutrophil cell death by bacterial pathogens
While phagocytosis-induced neutrophil apoptosis or PICD is desirable for the resolution of infection and prevention of excessive or chronic inflammation, many pathogens have devised means to alter apoptosis and promote pathogenesis [99]. Here, we provide a few examples of this phenomenon and refer the reader to recent reviews on the topic for a more comprehensive overview [99, 209].
Community-associated methicillin-resistant Sta. aureus (CA-MRSA) and Streptococcus pyogenes cause direct PMN lysis and/or accelerate bacteria-induced apoptosis to the point of secondary necrosis [94, 99, 204, 210–213]. The most prominent CA-MRSA strains produce leukotoxins known to cause lysis of human neutrophils, but at sublytic concentrations these toxins induce apoptosis [214]. It has been suggested that lysis of neutrophils by certain Sta. aureus strains promotes severe pathologic states such as necrotizing pneumonia or fatal sepsis, although this hypothesis remains to be tested. Inasmuch as PMNs are the primary cellular defense against staphylococcal infections, depletion of neutrophils seems a likely component of CA-MRSA pathogenesis [212, 215, 216]. However, Sta. aureus leukotoxins such as Panton-Valentine leukocidin (PVL) play little or no unique role in causing cell lysis after phagocytosis [213], a phenomenon that occurs rapidly after uptake of the most prominent CA-MRSA strains [212].
Str. pyogenes is associated with human infections ranging from mild pharyngitis to life-threatening necrotizing fasciitis [217]. Notably, Str. pyogenes accelerates neutrophil PICD, thereby causing necrosis [211, 217–220]. As with CA-MRSA, Str. pyogenes kills neutrophils to survive within the human host.
Pathogens that delay neutrophil apoptosis
In contrast to macrophages, few bacteria are known to survive and replicate within neutrophils. A. phagocytophilum is an obligate intracellular bacterium that delays neutrophil apoptosis to survive within PMNs [176, 221–225]. Although progress has been made, the mechanism(s) by which A. phagocytophilum delays apoptosis are not well understood. The pathogen is ingested by caveolae-mediated endocytosis and survives within inclusions/vacuoles that do not contain critical components of the NADPH oxidase [226, 227]. Moreover, A. phagocytophilum actively alters normal neutrophil granule trafficking and inhibits NADPH oxidase activation [176, 228, 229]. Notably, the pathogen fails to induce neutrophil apoptosis and dysregulates neutrophil gene expression to maximize survival and dissemination within the mammalian host [221–223, 230]. By prolonging the lifespan of neutrophils after uptake, A. phagocytophilum survives and disseminates in the host [223].
PMNs readily phagocytose C. pneumoniae and a small fraction of internalized bacteria remain viable for up to 90 h after uptake [224]. C. pneumoniae multiplies within neutrophils and delays constitutive apoptosis such that both phases of the Chlamydia biphasic life cycle occur within PMN inclusions [224, 231]. Delay of neutrophil apoptosis by C. pneumoniae can be as long as 3 days and is associated with decreased caspase-3 activity [224]. Prolonged PMN survival may be mediated by LPS and autocrine production of IL-8 [224]. Intracellular growth of Chlamydia is essential for pathogenesis, because the host cell supplies nutrients and shields the microbe from antimicrobial serum factors.
Mechanisms of cell death
Overview
The resolution of infection is complete when bacteria and spent host cells are cleared from tissues. Although phagocyte apoptosis or PICD has been reported after phagocytosis of many types of bacteria, the mechanism of cell death remains incompletely defined. For example, it is not always apparent if bacteria cause apoptosis after phagocytosis or if the observed cell death is a more cursory form of host cell lysis. Delineating the mechanism of host cell death is an important first step toward the development of better therapeutics for inflammatory disorders [232].
Initially, cell death was thought to proceed through either apoptosis or necrosis. However, necrosis is really the end result/equilibrium of cell death and thus not a cell death mechanism [233]. Several unique types of cell death are now known to occur depending on specific conditions, including the environment and state of the cell (Table 2). These conditions include extracellular factors, e.g., the presence of inflammatory markers or pH of the extracellular fluid, and intracellular factors such as the abundance of ATP [234]). For example, under conditions where ATP is depleted or limiting, cell death proceeds by oncosis, which entails cell swelling and lysis. However, when ATP is abundant, cell death will proceed via the more organized and non-inflammatory forms of cell death, such as apoptosis or autophagy. Importantly, a cohort of cell death mechanisms has been identified in cell populations in organs under stress [235]. Therefore, the resolution of bacterial infections through host cell death/turnover may encompass multiple mechanisms and not occur solely via apoptosis. As the different mechanisms of cell death receive more attention, the subcellular markers that distinguish these mechanisms have become more defined. Each of the mechanisms can be distinguished from the others based on physical characteristics in combination with biochemical assays (Table 2).
Apoptosis
Apoptosis, also referred to as type I cell death, is defined as a non-inflammatory form of cell death characterized by membrane blebbing, nuclear condensation, and cytoplasmic condensation. As stated above, apoptosis can be initiated by extracellular signals or by intracellular signals. The end result is an orderly destruction of the infected/damaged cell that does not cause inflammation in the surrounding tissues.
Typically, apoptosis is detected by DNA degradation, caspase activation, and/or early exposure of PS on the cell surface. Nuclear fragmentation or DNA “laddering” can be visualized by transmission electron microscopy (condensation and fragmentation of the nucleus) or agarose gel electrophoresis. Caspases are expressed initially as pro-proteins that undergo activation by proteolytic cleavage. As such, activated caspases can be detected by Western blot or flow cytometry analysis (using a fluorophore labeled reagent that binds only to the activated caspase). The traditional marker for neutrophil apoptosis is nuclear condensation [19], as membrane blebbing does not appear to occur in vitro (Fig. 2).
Although other assays have been utilized to monitor apoptosis, several reports have shown that these methods may not be specific for apoptotic cells. For example, apoptotic DNA damage has also been monitored by terminal deoxyribonucleotidyl transferase-mediated dUTP nick end labeling (TUNEL). However, Grasl-Kraupp et al. reported that TUNEL staining does not discriminate between apoptotic and late oncotic cells [236]. The plasma membrane of oncotic cells is extremely porous and TUNEL probably stains DNA via large holes in the cell membrane. During apoptosis the inner leaflet of the plasma membrane containing PS becomes exposed to the extracellular space and the exposed PS can be detected using labeled annexin V. As with TUNEL, recent evidence has shown that late oncotic cells stain positive for annexin V, thereby bringing into question the specificity of annexin V staining apoptotic cells [237, 238].
Autophagy-induced cell death
Autophagy, also known as Type II cell death, is a process whereby cells catabolize damaged components so that the essential building blocks, amino acids, lipids, carbohydrates, and etc., can be utilized for cellular repair, especially under times of cellular stress. Cells have the capacity to catabolize damaged proteins as well as entire organelles, including mitochondria. Autophagy was first observed in yeast and several genetic markers correlate with the formation of autophagic vacuoles in yeast and mammalian cells.
Autophagy-induced cell death is a programmed cell death that is distinct from apoptosis. Specifically, autophagy-induced cell death is induced under conditions of cellular starvation or endoplasmic reticulum (ER) stress [239]. Although the mechanism of autophagic cell death is not understood, two genetic markers, beclin 1 and ATG7, are critical for this non-apoptotic death pathway in mammalian cells ([240, 241] and reviewed in [242]). During autophagy-induced cell death, caspases are not activated, although there is cytoplasmic condensation and a reduction in cell size. The most defining feature of autophagy-induced cell death is the formation of multilamellar autophagosomes that engulf intracellular components. These large vacuoles are easily distinguished from other subcellular compartments using transmission electron microscopy. The formation of these large multilamellar vacuoles may be cell type dependent and more studies need to be done with phagocytes to determine if these vacuoles form in vitro or in vivo. After autophagosome formation, intracellular components are degraded during autophagosome––lysosome fusion [243].
Pyroptosis
Pyroptosis is a programmed cell death mechanism that results in cell lysis, tissue inflammation, and recruitment of host professional phagocytes. As with apoptosis, nuclear DNA is cleaved during pyroptosis, although this event is dependent on caspase-1-stimulated nuclease activity and not poly(ADP-ribose) polymerase [244]. Caspase-1 activation is a key event of pyroptosis and the enzyme activates IL-1β and IL-18. These cytokines in turn recruit macrophages and PMNs to the site of infection. Caspase-1 activity also leads to the formation of pores in the eukaryotic cell membrane, allowing an influx of extracellular ions that causes cell swelling and lysis [244]. Shao et al. found that caspase-1 has enzymatic activity toward several proteins in the glycolytic pathway, suggesting a link between metabolism/ATP production and pyroptosis [245].
Analogous to the apoptosome complex that mediates apoptosis, pyroptosis is controlled by the formation of a large (~700 kDa) multiprotein complex known as the inflammasome. The inflammasome is composed of a sensor (NOD-like receptor) [246], an adaptor molecule (apoptosis-associated speck-like protein containing a CARD, ASC, or PYCARD), caspase-1, and caspase-5 [247]. Engagement of sensor molecules with ligands such as PAMPs triggers formation of the inflammasome complex [248]. It is interesting to note that activation of the inflammasome occurs in the host cell cytosol after bacteria escape from the phagosome. Bacteria that activate the inflammasome complex include Shigella [249], Francisella tularensis (reviewed in [250]), L. monocytogenes [251], Yersinia [252], and Salmonella enterica serovar Typhimurium [244, 253, 254].
Oncosis
Like pyroptosis, oncosis is a form of cell death characterized by cell swelling and lysis. Earlier research had defined cell swelling and lysis as necrosis, but necrosis is now widely accepted as the end result of cell death. Necrosis not only includes the lysis of infected cells but also the equilibrium or damage that is caused to surrounding tissues after a cell has lysed [233]. Oncosis, also known as Type III cell death, occurs through multiple mechanisms, but two of the most prominent involve pore forming toxins of pathogenic bacteria and complement-mediated lysis. Pore forming toxins such as alpha-hemolysin of Sta. aureus have the ability to lyse a variety of host cell types, and complement-mediated cell lysis occurs after the formation of the complement membrane attack complex on C3b-targeted cells. As a result, intracellular contents and inflammatory cytokines are released into the extracellular milieu, causing tissue damage and inflammation. Oncosis occurs when membrane integrity is damaged and an efflux of inorganic ions (e.g., Ca2+, Na+, Cl−) increases the intracellular and intraorganelle osmotic pressure [255, 256]. Depletion of ATP and loss of mitochondrial transmembrane potential accompanies this efflux of ions into the cell.
Bacteria such as Brucella abortus, Campylobacter jejuni, Pseudomonas aeruginosa, and Salmonella enterica serovar Typhimurium can cause host cell death after phagocytosis [257–260]. These bacteria escape from phagosomes and then alter cell metabolism through the expression of toxins that disrupt membrane integrity, thus causing oncosis. The invasive ability of Cam. jejuni has been associated with the expression of its cytolethal distending toxin and subsequent ability to cause oncosis of epithelial cells [258]. Dacheux et al. showed that P. aeruginosa cystic fibrosis isolates induce rapid (within 60 minutes) oncosis of macrophages and PMNs that is type III secretion-dependent [259]. Expression of flagella proteins is required for Sal. enterica-mediated oncosis of and escape from human macrophages [260].
NETosis
A novel form of PMN death named NETosis has been described recently [261, 262] (Table 2). Neutrophil extracellular traps (NETs) are extrusions of plasma membrane and nuclear material composed of granule components and histones. NETs were initially reported to occur with viable PMNs [261, 262] and this finding was confirmed by others [263]. However, subsequent studies by Fuchs et al. suggest that the process of NET formation involves a type of cell death the authors called NETosis [261, 262]. NETosis occurs in an NADPH oxidase dependent manner [259] and bacteria deficient in DNAses are susceptible to killing by NETs [264]. Although the physiological relevance in vivo has been debated, as has the issue of whether NETosis is distinct from other types of cell lysis, it is clear that in vitro NETs bind and kill extracellular microorganisms. Further research is needed to better understand these interesting structures.
Summary of neutrophil death mechanisms
Although there are many fates for phagocytes after interactions with microorganisms, the manner of cell death has serious repercussions that can impact severity and duration of disease. Pyroptosis and oncosis cause inflammation that recruits leukocytes to the site of infection, but if unchecked, these processes can cause necrosis that leads to the damage of surrounding cell and tissues. In contrast, apoptosis and autophagy are non-inflammatory forms of cell death that recruit phagocytes to the site of infection. A comprehensive understanding neutrophil apoptosis, death, and turnover is critical for a full understanding of bacterial pathogenesis mechanisms.
A paradigm for the resolution of infection
Neutrophil apoptosis is essential for maintaining normal immune system homeostasis and this process facilitates the resolution of inflammation. PMN apoptosis is mediated by both intrinsic and extrinsic mechanisms, and apoptotic cells are subsequently cleared by macrophages through multiple receptor-ligand interactions.
Neutrophil fate is altered by interaction with microorganisms. In most cases, the outcome of this interaction dictates whether the ingested organism will ultimately cause disease. Based upon our current knowledge, there are two possible outcomes for neutrophil-bacteria interactions (Fig. 5) [99]. On one hand, phagocytosis and killing of bacteria culminate with induction of apoptosis/PICD and the subsequent removal by macrophages, ultimately resulting in the resolution of infection (Fig. 5, top). Alternatively, pathogens such as A. phagocytophilum, C. pneumoniae, Str. pyogenes, and Sta. aureus alter neutrophil apoptosis to survive, and thereby disseminate and cause disease [176, 217, 219, 221, 222, 224] (Fig. 5, bottom). This relatively simple model will need refining and revision as we continue to accumulate new information.

A model schematic illustrating two possible outcomes of bacteria-neutrophil interaction. See text for details
Concluding comment
The use of genomics-based technologies has dramatically enhanced our discovery of mechanisms of bacterial virulence and fundamental immunology. A better understanding of innate host defense, host cell death, and bacterial pathogenesis will likely provide information important for development of vaccines, treatments, and prophylactic agents designed to prevent and/or control infections.
References
Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5:1317–27.
Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625.
Quinn MT, Ammons MC, DeLeo FR. The expanding role of NADPH oxidases in health and disease: no longer just agents of death and destruction. Clin Sci (Lond). 2006;111:1–20.
Nauseef WM. How human neutrophils kill and degrade microbes: an integrated review. Immunol Rev. 2007;219:88–102.
Cowland JB, Borregaard N. Isolation of neutrophil precursors from bone marrow for biochemical and transcriptional analysts. J Immunol Methods. 1999;232:191–200.
Ward AC, Loeb DM, Soede-Bobok AA, Touw IP, Friedman AD. Regulation of granulopoiesis by transcription factors and cytokine signals. Leukemia. 2000;14:973–90.
Bjerregaard MD, Jurlander J, Klausen P, Borregaard N, Cowland JB. The in vivo profile of transcription factors during neutrophil differentiation in human bone marrow. Blood. 2003;101:4322–32.
Friedman AD. Transcriptional regulation of granulocyte and monocyte development. Oncogene. 2002;21:3377–90.
Theilgaard-Monch K, Jacobsen LC, Borup R, Rasmussen T, Bjerregaard MD, Nielsen FC, et al. The transcriptional program of terminal granulocytic differentiation. Blood. 2005;105:1785–96.
Borregaard N, Theilgaard-Monch K, Sorensen OE, Cowland JB. Regulation of human neutrophil granule protein expression. Curr Opin Hematol. 2001;8:23–7.
Bainton DF, Ullyot JL, Farquhar MG. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med. 1971;134:907–34.
Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci USA. 1992;89:2804–8.
Killmann SA, Cronkite EP, Fliedner TM, Bond VP. Mitotic indices of human bone marrow cells. I. Number and cytologic distribution of mitoses. Blood. 1962;19:743–50.
Borregaard N, Sehested M, Nielsen BS, Sengelov H, Kjeldsen L. Biosynthesis of granule proteins in normal human bone marrow cells. Gelatinase is a marker of terminal neutrophil differentiation. Blood. 1995;85:812–7.
Athens JW, Haab OP, Raab SO, Mauer AM, Ashenbrucker H, Cartwright GE, et al. Leukokinetic studies. IV. The total blood, circulating and marginal granulocytes pools and the granulocytes turnover rate in normal subjects. J Clin Invest. 1961;40:989–95.
Cline MJ. Production, destruction, and distribution of neutrophilic granulocytes. The white cell. Cambridge: Harvard University Press; 1975. p. 24–38.
Cronkite EP, Fliedner TM. Granulocytopoiesis. N Engl J Med. 1964;270:1347–52.
Fliedner TM, Cronkite EP, Robertson JS. Granulocytopoiesis. I. Senescence and random loss of neutrophilic granulocytes in human beings. Blood. 1964;24:402–14.
Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation: programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–75.
Savill JS, Henson PM, Haslett C. Phagocytosis of aged human neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J Clin Invest. 1989;84:1518–27.
Kobayashi SD, Voyich JM, Burlak C, DeLeo FD. Neutrophils in the innate immune response. Arch Immunol Ther Exp. 2005;53:505–17.
McPhail LC, Clayton CC, Snyderman R. The NADPH oxidase of human polymorphonuclear leukocytes. Evidence for regulation by multiple signals. J Biol Chem. 1984;259:5768–75.
DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, et al. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest. 1998;101:455–63.
Sengelov H, Kjeldsen L, Diamond MS, Springer TA, Borregaard N. Subcellular localization and dynamics of Mac-1 (αmβ2) in human neutrophils. J Clin Invest. 1993;92:1467–76.
Kurt-Jones EA, Mandell L, Whitney C, Padgett A, Gosselin K, Newburger PE, et al. Role of toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood. 2002;100:1860–8.
Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–71.
Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, et al. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18). J Cell Biol. 1990;111:3129–39.
Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–60.
Khan AI, Kerfoot SM, Heit B, Liu L, Andonegui G, Rufell B, et al. Role of CD44 and hyaluronan in neutrophil recruitment. J Immunol. 2004;173:7594–601.
Cooper D, Lindberg FP, Gamble JR, Brown EG, Vadas MA. Transendothelial migration of neutrophils involves integrin-associated protein (CD47). Proc Natl Acad Sci USA. 1995;92:3978–82.
de Mendez I, Adams AG, Sokolic RA, Malech HL, Leto TL. Multiple SH3 domain interactions regulate NADPH oxidase assembly in whole cells. EMBO J. 1996;15:1211–20.
Clark RA, Klebanoff SJ. Myeloperoxidase––H2O2––halide system: cytotoxic effect on human blood leukocytes. Blood. 1977;50:65–70.
Odell EW, Segal AW. The bactericidal effects of the respiratory burst and the myeloperoxidase system isolated in neutrophil cytoplasts. Biochim Biophys Acta. 1988;971:266–74.
Klebanoff SJ. Myeloperoxidase: contribution to the microbicidal activity of intact leukocytes. Science. 1970;169:1095–7.
Rosen H, Klebanof SJ. Bactericidal activity of a superoxide anion-generating system. J Exp Med. 1979;149:27–39.
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511.
Underhill DM, Gantner B. Integration of toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. 2004;6:1368–73.
Brown GD, Gordon S. Immune recognition: a new receptor for β-glucans. Nature. 2001;413:36–7.
Kennedy AD, Willment JA, Dorward DW, Williams DL, Brown GD, DeLeo FR. Dectin-1 promotes fungicidal activity on human neutrophils. Eur J Immunol. 2007;37:467–78.
Kobayashi SD, Voyich JM, Whitney AR, DeLeo FR. Spontaneous neutrophil apoptosis and modulation of cell survival by granulocyte macrophage-colony stimulating factor. J Leukoc Biol. 2005;78:1408–18.
Whyte MK, Meagher LC, MacDermot J, Haslett C. Impairment of function in aging neutrophils is associated with apoptosis. J Immunol. 1993;150:5124–34.
Metchnikoff E. Lecture VII. Lectures on the comparative pathology of inflammation. In: Starling FA, Starling EH, editors. London: Kegan, Paul, Trench, Trubner; 1893. p. 107–31.
Leuenroth SJ, Grutkoski PS, Ayala A, Simms HH. The loss of Mcl-1 expression in human polymorphonuclear leukocytes promotes apoptosis. J Leukoc Biol. 2000;68:158–66.
Moulding DA, Quayle JA, Hart CA, Edwards SW. Mcl-1 expression in human neutrophils: regulation by cytokines and correlation with cell survival. Blood. 1998;92:2495–502.
Kato T, Kutsuna H, Oshitani N, Kitagawa S. Cyclic AMP delays neutrophil apoptosis via stabilization of Mcl-1. FEBS Lett. 2006;580:4582–6.
Moulding DA, Akgul C, Derouet M, White MRH, Edwards SW. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J Leukoc Biol. 2001;70:783–92.
Gardai S, Whitlock BB, Helgason C, Ambruso D, Fadok V, Bratton D, et al. Activation of SHIP by NADPH oxidase-stimulated lyn leads to enhanced apoptosis in neutrophils. J Biol Chem. 2002;277:5236–46.
Baran CP, Tridanapani S, Helgason CD, Humphries K, Krystal G, Marsh CB. The inositol 5′-phosphate SHIP-1 and the src kinase lyn negatively regulate macrophage colony-stimulating factor-induced akt activity. J Biol Chem. 2003;278:38628–36.
Cox D, Dale BM, Kashiwada M, Helgason CD, Greenberg S. A regulatory role for src homology 2 domain-containing inositol 5′-phosphatase (SHIP) in phagocytosis mediated by Fcγ receptors and complement receptor 3 (αMβ2; CD11b/CD18). J Exp Med. 2001;193:61–71.
Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–20.
Chuang PI, Yee E, Karsan A, Winn RK, Harlan JM. A1 is a constitutive and inducible Bcl-2 homologue in mature human neutrophils. Biochem Biophys Res Commun. 1998;249:361–5.
Sawatzky DA, Willoughby DA, Colville-Nash PR, Rossi AG. The involvement of the apoptosis-modulating proteins ERK 1/2, Bcl, xL and Bax in the resolution of acute inflammation in vivo. Am J Pathol. 2006;168:33–41.
Murphy BM, O’Neill AJ, Adrain C, Watson RWG, Martin SJ. The apoptosome pathway to caspase activation in primary human neutrophils exhibits dramatically reduced requirements for cytochrome c. J Exp Med. 2003;197:625–32.
Maianski NA, Mul FP, van Buul JD, Roos D, Kuijpers TW. Granulocyte colony-stimulating factor inhibits the mitochondria-dependent activation of caspase-3 in neutrophils. Blood. 2002;99:672–9.
Altznauer F, Conus S, Cavalli A, Folkers G, Simon H-U. Calpain regulates bax and subsequent smac-dependent caspase-3 activation in neutrophil apoptosis. J Biol Chem. 2004;279:5947–57.
Dibbert B, Weber M, Nikolaizik WH, Vogt P, Schoni MH, Blaser K, et al. Cytokine-mediated Bax deficiency and consequent delayed neutrophil apoptosis: a general mechanism to accumulate effector cells in inflammation. Proc Natl Acad Sci USA. 1999;96:13330–5.
Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:2012–20.
Baumann R, Casaulta C, Simon D, Conus S, Yousefi S, Simon H-U. Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB J. 2003;17:2221–30.
Wolach B, van der Laan LJ, Maianski NA, Tool AT, van Bruggen R, Roos D, et al. Growth factors G-CSF and GM-CSF differentially preserve chemotaxis of neutrophils aging in vitro. Exp Hematol. 2007;35:541–50.
Zhang B, Hiranhashi J, Cullere X, Mayadas TN. Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis: cross-talk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J Biol Chem. 2003;278:28443–54.
Yamamoto A, Taniuchi S, Tsuji S, Hasui M, Kobayashi Y. Role of reactive oxygen species in neutrophil apoptosis following ingestion of heat-killed Staphylococcus aureus. Clin Exp Immunol. 2002;129:479–84.
Kasahara Y, Iwai K, Yachie A, Ohta K, Konno A, Seki H, et al. Involvement of reactive oxygen intermediates in spontaneous and CD95 (APO-1)-mediated apoptosis of neutrophils. Blood. 1997;89:1748–53.
Fadeel B, Ahlin A, Henter J-I, Orrenius S, Hampton MB. Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood. 1998;92:4808–18.
Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92:3007–17.
Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004;11:143–53.
Knepper-Nicolai B, Savill J, Brown SB. Constitutive apoptosis in human neutrophils requires synergy between calpains and the proteasome downstream of caspases. J Biol Chem. 1998;273:30530–6.
Scheel-Toellner D, Wang K, Assi LK, Webb PR, Craddock RM, Salmon M, et al. Clustering of death receptors in lipid rafts initiates neutrophil spontaneous apoptosis. Biochem Soc Trans. 2004;32:679–81.
Taneja R, Parodo J, Jia SH, Kapus A, Rotstein OD, Marshall JC. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med. 2004;32:1460–9.
Scheel-Toellner D, Wang K, Craddock R, Webb PR, McGettrick HM, Assi LK, et al. Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood. 2004;104:2557–64.
Liles WC, Kiener PA, Ledbetter JA, Aruffo A, Klebanoff SJ. Differential expression of Fas (CD95) and Fas Ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med. 1996;184:429–40.
Brown SB, Savill J. Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J Immunol. 1999;162:480–5.
Jimenez MF, Watson RW, Parodo J, Evans D, Foster D, Steinberg M, et al. Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch Surg. 1997;132:480–5.
Harper N, Hughes M, MacFarlane M, Cohen GM. Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem. 2003;278:25534–41.
Gon S, Gatanaga T, Sendo F. Involvement of two types of TNF receptor in TNF-α induced neutrophil apoptosis. Microbiol Immunol. 1996;40:463–5.
Murray J, Barbar JAJ, Dunkley SA, Lopez AF, Van Ostade X, Condliffe AM, et al. Regulation of neutrophil apoptosis by tumor necrosis factor-α: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood. 1997;90:2772–83.
Maianski NA, Roos D, Kuijpers TW. Tumor necrosis factor α induces a caspase-independent death pathway in human neutrophils. Blood. 2003;101:1987–95.
van den Berg JM, Weyer S, Weening JJ, Roos D, Kuijpers TW. Divergent effects of tumor necrosis factor α on apoptosis of human neutrophils. J Leukoc Biol. 2001;69:467–73.
D’Osualdo A, Ferlito F, Prigione I, Obici L, Meini A, Zulian F, et al. Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis. Arthritis Rheum. 2006;54:998–1008.
Kamohara H, Matsuyama W, Shimozato O, Abe K, Galligan C, Hashimoto S-I, et al. Regulation of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptor expression in human neutrophils. Immunology. 2004;111:186–94.
Renshaw SA, Parmar JS, Singleton V, Rowe SJ, Dockrell DH, Dower SK, et al. Acceleration of human neutrophil apoptosis by TRAIL. J Immunol. 2003;170:1027–33.
Walczak H, Krammer PH. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res. 2000;256:58–66.
Lum JJ, Bren G, McClure R, Badley AD. Elimination of senescent neutrophils by TNF-relating apoptosis-inducing ligand. J Immunol. 2005;175:1232–8.
Lamhamedi-Cherradi SE, Zheng SJ, Maguschak KA, Peschon J, Chen YH. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL−/− mice. Nat Immunol. 2003;4:255–60.
Lamhamedi-Cherradi SE, Zheng S, Tisch RM, Chen YH. Critical roles of tumor necrosis factor-related apoptosis-inducing ligand in type 1 diabetes. Diabetes. 2003;52:2274–8.
Hilliard B, Wilmen A, Seidel C, Liu TS, Goke R, Chen Y. Roles of TNF-related apoptosis-inducing ligand in experimental autoimmune encephalomyelitis. J Immunol. 2001;166:1314–9.
Koga Y, Matsuzaki A, Suminoe A, Hattori H, Hara T. Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): a novel mechanism of antitumor effect by neutrophils. Cancer Res. 2004;64:1037–43.
Cassatella MA, Huber V, Calzetti F, Margotto D, Tamassia N, Peri G, et al. Interferon-activated neutrophils store a TNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) intracellular pool that is readily mobilizable following exposure to proinflammatory mediators. J Leukoc Biol. 2006;79:123–32.
Simons MP, Leidal KG, Nauseef WM, Griffith TS. TNF-related apoptosis-inducing ligand (TRAIL) is expressed throughout myeloid development, resulting in a broad distribution among neutrophil granules. J Leukoc Biol. 2008;83:621–9.
Bovolenta C, Gasperini S, Cassatella MA. Granulocyte colony-stimulating factor induces the binding of STAT1 and STAT3 to the IFNγ response region within the promoter of the FcγRI/CD64 gene in human neutrophils. FEBS Lett. 1996;386:239–42.
Scapini P, Laudanna C, Pinardi C, Allavena P, Mantovani A, Sozzani S, et al. Neutrophils produce biologically active macrophage inflammatory protein-3α (MIP-3α)/CCL20 and MIP-3β/CCL19. Eur J Immunol. 2001;31:1981–8.
Bazzoni F, Cassatella MA, Rossi F, Ceska M, Dewald B, Baggiolini M. Phagocytosing neutrophils produce and release high amounts of the neutrophil-activating peptide 1/interleukin 8. J Exp Med. 1991;173:771–4.
Gasperini S, Marchi M, Calzetti F, Laudanna C, Vincentini L, Olsen H, et al. Gene expression and production of the monokine induced by IFN-γ (MIG), IFN-inducible T cell α chemoattractant (I-TAC), and IFN-γ-inducible protein-10 (IP-10) chemokines by human neutrophils. J Immunol. 1999;162:4928–37.
Lapinet JA, Scapini P, Calzetti F, Perez O, Cassatella MA. Gene expression and production of tumor necrosis factor alpha, interleukin 1-β, (IL-1β), IL-8, macrophage inflammatory protein 1α (MIP-1α), MIP-1β, and gamma interferon-inducible protein 10 by human neutrophils stimulated with group b meningococcal outer membrane vesicles. Infect Immun. 2000;68:6917–23.
Kobayashi SD, Braughton KR, Whitney AR, Voyich JM, Schwan TG, Musser JM. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci USA. 2003;100:10948–53.
Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci USA. 2002;99:6901–6.
Quinn MT, Gauss KA. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with non-phagocyte oxidases. J Leukoc Biol. 2004;76:760–81.
Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The neutrophil as a cellular source of chemokines. Immunol Rev. 2000;177:195–203.
Cohen JJ, Duke RC, Fadok VA, Sellins KS. Apoptosis and programmed cell death in immunity. Ann Rev Immunol. 1992;10:267–93.
DeLeo FR. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis. 2004;9:399–413.
Kobayashi SD, Voyich JM, Somerville GA, Braughton KB, Malech HL, Musser JM, et al. An apoptosis-differentiation programme in human polymorphonuclear leukocytes facilitates resolution of inflammation. J Leukoc Biol. 2003;73:315–22.
Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death Differ. 1998;5:563–8.
Savill J. Apoptosis in resolution of inflammation. J Leukoc Biol. 1997;61:375–80.
Walcheck B, Herrera AH, St.Hill C, Mattila PE, Whiteney AR, DeLeo FR. ADAM17 activity during human neutrophil activation and apoptosis. Eur J Immunol. 2006;36:968–76.
Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, et al. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748–55.
Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine. 1992;4:96–100.
Matthews V, Schuster B, Schutze S, Bubmeyer I, Ludwig A, Hunghausen C, et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J Biol Chem. 2003;278:38829–39.
Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, et al. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14:705–14.
Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–55.
Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA. 1995;92:9475–9.
Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–61.
el Kebir D, Jozsef L, Khreiss T, Pan W, Petasis NA, Serhan CN, et al. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid a in human neutrophils: a novel mechanism for resolution of inflammation. J Immunol. 2007;179:616–22.
Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. 2000;164:1663–7.
Newman SL, Henson JE, Henson PM. Phagocytosis of senescent neutrophils by human monocyte-derived macrophages and rabbit inflammatory macrophages. J Exp Med. 1982;156:430–42.
Yamaryo T, Oishi K, Yoshimine H, Tsuchihashi Y, Matsushima K, Nagatake T. Fourteen-member macrolides promote the phosphatidylserine receptor-dependent phagocytosis of apoptotic neutrophils by alveolar macrophages. Antimicrob Agents Chemother. 2003;47:48–53.
Fadok VA, Haslett JSC, Bratton DL, Doherty DE, Cambell PA, Henson PM. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J Immunol. 1992;149:4029–35.
Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med. 1998;188:2313–20.
Platt N, Suzuki H, Kurihara Y, Kodama T, Gordon S. Role of the class A macrophage scavenger receptor in the phagocytosis of apoptotic thymocytes in vitro. Proc Natl Acad Sci USA. 1996;93:12456–60.
Savill J, Dransfield I, Hogg N, Haslett C. Vitronectin receptor-mediated phagocytosis of cells undergoing apoptosis. Nature. 1990;343:170–3.
Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest. 1992;90:1513–22.
Devitt A, Moffatt OD, Raykundalia C, Capra JD, Simmonda DL, Gregory CD. Human CD14 mediates recognition and phagocytosis of apoptotic cells. Nature. 1998;392:505–9.
Hart SP, Dougherty GJ, Haslett C, Dransfield I. CD44 regulates phagocytosis of apoptotic neutrophil granulocytes, but not apoptotic lymphocytes, by human macrophages. J Immunol. 1997;159:919–25.
Vivers S, Heasman SJ, Hart SP, Dransfield I. Divalent cation-dependent and -independent augmentation of macrophage phagocytosis of apoptotic neutrophils by CD44 antibody. Clin Exp Immunol. 2004;138:447–52.
Vivers S, Dransfield I, Hart SP. Role of macrophage CD44 in the disposal of inflammatory cell corpses. Clin Sci. 2002;103:441–9.
Park S-Y, Jung M-Y, Kim H-J, Lee S-J, Kim S-Y, Lee B-H, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Diff. 2008;15:192–201.
Miyanishi M, Taka K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–9.
Li MO, Sarkisian MR, Mehal WZ, Rakic P, Flavell RA. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science. 2003;302:1560–3.
Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RAB, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90.
Arur S, Uche UE, Rezaqul K, Fong M, Scranton V, Cowan AE, et al. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. 2003;4:587–98.
Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–9.
Thielens NM, Tacnet-Delorme P, Arlaud GJ. Interaction of C1q and mannan-binding lectin with viruses. Immunobiology. 2002;205:563–74.
Ogden CA, Elkon KB. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr Dir Autoimmun. 2006;9:120–42.
Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–66.
Grevink ME, Horst G, Limburg PC, Kallenberg CGM, Bijl M. Levels of complement in sera from inactive SLE patients, although decreased, do not influence in vitro uptake of apoptotic cells. J Autoimmm. 2005;24:329–36.
Greenwalt DE, Lipsky RH, Ockenhouse CF, Ikeda H, Tandon NN, Jamieson GA. Membrane glycoprotein CD36: a review of its roles in adherence, signal transduction, and transfusion medicine. Blood. 1992;80:1105–15.
Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserin-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203:2613–25.
Navazo MDP, Daviet L, Savill J, Ren Y, Leung LLK, McGregor JL. Identification of a domain (155–183) on CD36 implicated in the phagocytosis of apoptotic neutrophils. J Biol Chem. 1996;271:15381–5.
Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, et al. Resolution of lung inflammation by CD44. Science. 2002;296:155–8.
Wright SD, Ramos RA, Tobias PS, Ulevitch RT, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–3.
Moffat OD, Devitt A, Bell ED, Simmons DL, Gregory CD. Macrophage recognition of ICAM-3 on apoptotic leukocytes. J Immunol. 1999;162:6800–10.
Gregory CD. CD14-dependent clearance of apoptotic cells: relevance to the immune system. Curr Opin Immunol. 2000;12:27–34.
Schlegel RA, Krahling S, Callahan MK, Williamson P. CD14 is a component of multiple recognition systems used by macrophages to phagocytose apoptotic lymphocytes. Cell Death Diff. 1999;6:583–92.
Bottcher A, Gaipl US, Furnrohr BG, Herrmann M, Girkontaite I, Kalden JR, et al. Involvement of phosphatidylserine, αvβ3, CD14, CD36, and complement C1q in the phagocytosis of primary necrotic lymphocytes by macrophages. Arth Rheum. 2006;54:927–38.
Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–8.
Huynh M-L, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50.
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine-paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest. 1998;101:890–8.
Lucas M, Stuart LM, Zhang A, Hodivala-Dilke K, Febbraio M, Silverstein R, et al. Requirements for apoptotic cell contact in regulation of macrophage responses. J Immunol. 2006;177:4047–54.
Xing L, Remick DG. Neutrophils as firemen, production of anti-inflammatory mediators by neutrophils in a mixed cell environment. Cell Immunol. 2004;231:126–32.
Bianchi SM, Dockrell DH, Renshaw SA, Sabroe I, Whyte MKB. Granulocyte apoptosis in the pathogenesis and resolution of lung disease. Clin Sci. 2006;110:293–304.
Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1.
Liu Y, Cousin JM, Hughes J, Damme JV, Seckle JR, Haslette C, et al. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol. 1999;162:3639–46.
Giles KM, Ross K, Rossi AG, Hotchin NA, Haslett C, Dransfield I. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol. 2001;167:976–86.
Thieringer R, Le Grand CB, Carbin L, Cai TQ, Wong B, Wright SD, et al. 11β-hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J Immunol. 2001;167:30–5.
Gilmour JS, Coutinho AE, Cailhier J-F, Man TY, Clay M, Thomas G, et al. Local amplification of glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol. 2006;176:7605–11.
Heasman SJ, Giles KM, Rossi AG, Allen JE, Haslett C, Dransfield I. Interferon γ suppresses glucocorticoid augmentation of macrophage clearance of apoptotic cells. Eur J Immunol. 2004;34:1752–61.
Hotta K, Niwa M, Hara A, Ohno T, Wang X, Matsuno H, et al. The loss of susceptibility to apoptosis in exudated tissue neutrophils is associated with their nuclear factor-κB activation. Eur J Pharmacol. 2001;433:17–27.
Daffern PJ, Jagels MA, Hugli TE. Multiple epithelial cell-derived factors enhance neutrophil survival; regulation by glucocorticoids and tumor necrosis factor-α. Am J Respir Cell Mol Biol. 1999;21:259–67.
Kotone-Miyahara Y, Yamashita K, Lee K-K, Yonehara S, Uchiyama T, Sasada M, et al. Short-term delay of Fas-stimulated apoptosis by GM-CSF as a result of temporary suppression of FADD recruitment in neutrophils: evidence implicating phosphatidylinositol 3-kinase and MEK1-ERK1/2 pathways downstream of classical protein kinase C. J Leukoc Biol. 2004;76:1047–56.
Larsen GL, Mitchell BC, Harper TB, Henson PM. The pulmonary response of C5a deficient mice to Pseudomonas aeruginosa. Am Rev Respir Dis. 1982;126:306–11.
Perianayagam MC, Balakrishnan VS, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils via an extracellular signal-regulated kinase and Bad-mediated signalling pathway. Eur J Clin Invest. 2004;34:50–6.
Tamura DY, Moore EE, Partrick DA, Johnson JL, Offner PJ, Silliman CC. Acute hypoxemia in humans enhances the neutrophil inflammatory response. Shock. 2002;17:269–73.
Hannah S, Mecklenburgh K, Rahman I, Bellingan GJ, Greening A, Haslett C, et al. Hypoxia prolongs neutrophil survival in vitro. FEBS Lett. 1995;372:233–7.
Molloy EJ, O’Neill AJ, Doyle BT, Grantham JJ, Taylor CT, Sheridan-Pereira M, et al. Effects of heat shock and hypoxia on neonatal neutrophil lipopolysaccharide responses: altered apoptosis, toll-like receptor-4 and CD11b expression compared with adults. Biol Neonate. 2006;90:34–9.
Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, et al. The contribution of the toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol. 2004;172:3059–69.
Ehlenberger AG, Nussenzweig V. The role of membrane receptors for C3b and C3d in phagocytosis. J Exp Med. 1997;145:357–71.
Hayashi F, Means TK, Luster AD. Toll-like receptors stimulate human neutrophil function. Blood. 2003;102:2660–9.
Takeda K, Akira S. Microbial recognition by toll-like receptors. J Dermatol Sci. 2004;34:73–82.
Ahmad-Nejad P, Hacker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Bacterial CpG-DNA and lipopolysaccharides activate toll-like receptors at distinct cellular compartments. Eur J Immunol. 2002;32:1958–68.
Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, et al. The toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8, and 9 subfamily. Eur J Immunol. 2003;33:2987–97.
Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, et al. Subcellular localization of toll-like receptor 3 in human dendritic cells. J Immunol. 2003;171:3154–62.
Sabroe I, Jones EC, Usher LR, Whyte MKB, Dower SK. Toll-like receptor (TLR)2 and TLR4 in human peripheral blood granulocytes: a critical role for monocytes in leukocyte lipopolysaccharide responses. J Immunol. 2002;168:4701–10.
Sabroe I, Prince LR, Dower SK, Chilvers ER, Whyte MKB. What can we learn from highly purified neutrophils? Biochem Soc Trans. 2004;32:468–9.
Watson RW, Rotstein OD, Parodo J, Bitar R, Marshall JC. The IL-1 beta-converting enzyme (caspase-1) inhibits apoptosis of inflammatory neutrophils through activation of IL-1 beta. J Immunol. 1998;161:957–62.
Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, et al. Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J Immunol. 2003;170:5628–75.
Lotz S, Aga E, Wilde I, van Zandbergen G, Hartung T, Solbach W, et al. Highly purified lipoteichoic acid activates neutrophil granulocytes and delays their spontaneous apoptosis via CD14 and TLR2. J Leukoc Biol. 2004;75:467–77.
Feterowski C, Wieighardt H, Emmanuilidis K, Hartung T, Holzmann B. Immune protection against septic peritonitis in endotoxin-primed mice is related to reduced neutrophil apoptosis. Eur J Immunol. 2001;31:1268–77.
Borjesson DL, Kobayashi SD, Whitney AR, Voyich JM, Argue CM, DeLeo FR. Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J Immunol. 2005;174:6364–72.
Kobayashi SD, Voyich JM, Braughton KR, DeLeo FR. Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J Immunol. 2003;170:3357–68.
McDonald P, Russo MP, Ferrini S, Cassatella MA. Interleukin-15 (IL-15) induces NF-κB activation and IL-8 production in human neutrophils. Blood. 1998;92:4828–35.
McDonald PP, Gasperini S, Calzetti F, Cassatella MA. Modulation of interferon-γ of the production and gene expression of IL-1 receptor antagonist in human neutrophils. Cell Immunol. 1998;184:45–50.
McDonald PP, Bald A, Cassatella MA. Activation of the NF-κB pathway by inflammatory stimuli in human neutrophils. Blood. 1997;9:3421–33.
Cline MJ. Phagocytosis and synthesis of ribonucleic acid in human granulocytes. Nature. 1966;212:1431–3.
Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol. 1996;156:3986–92.
Matsuda T, Saito H, Inoue T. Ratio of bacteria to polymorphonuclear neutrophils (PMNs) determines PMN fate. Shock. 1999;12:365–72.
Perskvist N, Long M, Stendahl O, Zheng L. Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase-3 and altering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J Immunol. 2002;168:6358–65.
Lundqvist-Gustafsson H, Norman S, Nilsson J, Wilsson A. Involvement of p38-mitogen-activated protein kinase in Staphylococcus aureus-induced neutrophil apoptosis. J Leukoc Biol. 2001;70:642–8.
Weiss JP. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–76.
Firestein GS. Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J Clin Rheumatol. 2005;11:39–44.
Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WN, Malech HL, et al. Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol. 2004;172:636–43.
Guide SV. Reinfection, rather than persistent infection, in patients with chronic granulomatous disease. J Infect Dis. 2003;187:845–53.
Firestein GS. Inhibiting inflammation in rheumatoid arthritis. N Engl J Med. 2006;354:80–2.
Rosenzweig SD, Holland SM. Phagocyte immunodeficiencies and their infections. J Allergy Clin Immunol. 2004;113:620–6.
William R, Watson G, Redmond PH, Wang JH, Condron C, Bouchier-Hayes D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol. 1996;1996:3986–92.
Oishi K, Machida K. Inhibition of neutrophil apoptosis by antioxidants in culture medium. Scand J Immunol. 1997;45:21–7.
Rollet-Labelle E, Grange MJ, Elbim C, Marquetty C, Gougerot-Pociadalo MA, Pasquier C. Hydroxyl radical as a potential intracellular mediator of polymorphonuclear neutrophil apoptosis. Free Rad Biol Med. 1998;24:563–72.
Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, et al. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–66.
Gamberale R, Giordano M, Trevani AS, Andonegui G, Geffner JR. Modulation of human neutrophil apoptosis by immune complexes. J Immunol. 1998;161:3666–74.
Ottonello L, Frumento G, Arduino N, Dapino P, Tortolina G, Dallegri F. Immune complex stimulation of neutrophil apoptosis: investigating the involvement of oxidative and nonoxidative pathways. Free Rad Biol Med. 2001;30:161–9.
Schettini J, Salamone G, Trevani A, Raiden S, Gamberale R, Vermeulen M, et al. Stimulation of neutrophil apoptosis by immobilized IgA. J Leukoc Biol. 2002;72:685–91.
Nauseef WM, Clark RA. Basic principles in the diagnosis and management of infectious diseases. In: Mandell GL, Bennett JE, Dolin R, editors. Granulocytes. New York: Churchill Livingstone; 2000. p. 89–112.
Simons MP, Nauseef WM, Griffith TS, Apicella MA. Neisseria gonorrhoeae delays the onset of apoptosis in polymorphonuclear leukocytes. Cell Microbiol. 2006;8:1780–90.
Tsurubuchi T, Aratani Y, Maeda N, Koyama H. Retardation of early-onset PMA-induced apoptosis in mouse neutrophils deficient in myeloperoxidase. J Leukoc Biol. 2001;70:52–8.
Hampton MB, Vissers MCM, Keenan JI, Winterbourn CC. Oxidant-mediated phosphatidylserine exposure and macrophage uptake of activated neutrophils: possible impairment in chronic granulomatous disease. J Leukoc Biol. 2002;71:775–81.
Liles WC, Thomsen AR, O’Mahoney DS, Klebanoff SJ. Stimulation of human neutrophils and monocytes by staphylococcal phenol-soluble modulin. J Leukoc Biol. 2001;70:96–102.
Moulding DA, Walter C, Hart CA, Edwards SW. Effects of staphylococcal enterotoxins on human neutrophil functions and apoptosis. Infect Immun. 1999;67:2312–8.
Hofman V, Ricci V, Mograbi B, Brest P, Luciano F, Boquet P, et al. Helicobacter pylori lipopolysaccharide hinders polymorphonuclear leucocyte apoptosis. Lab Invest. 2001;81:375–84.
Liu J, Akahoshi t, Sasahana T, Kitasato H, Namai R, Sasaki T, et al. Inhibition of neutrophil apoptosis by verotoxin 2 derived from Escherichia coli O157:H7. Infect Immun. 1999;67:6203–5.
Engelich G, White M, Hartshorn KL. Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J Leukoc Biol. 2001;69:50–6.
Watson RW, Redmond HP, Wang JH, Bouchier-Hayes D. Bacterial ingestion, tumor necrosis factor-alpha, and heat induce programmed cell death in activated neutrophils. Shock. 1996;5:47–51.
Mayer-Scholl A, Averhoff P, Zychlinsky A. How do neutrophils and pathogens interact? Curr Opin Cell Microbiol. 2004;7:62–6.
Aleman M, Garcia A, Saab MA, De La Barrera SS, Finiasz M, Abbate E, et al. Mycobacterium tuberculosis-induced activation accelerates apoptosis in peripheral blood neutrophils from patients with active tuberculosis. Am J Respir Cell Mol Biol. 2002;27:583–92.
Zysk G, Gejo L, Schneider-Wald BK, Nau R, Heinz H. Induction of necrosis and apoptosis of neutrophil granulocytes by Streptococcus pneumoniae. Clin Exp Immunol. 2000;122:61–6.
Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol. 2005;175:3907–19.
Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, et al. Is Panton-Valentine Leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis. 2006;194:1761–70.
Genestier A-L, Michallet M-C, Prevost G, Bellot G, Chalabreysse L, Peyrol S, et al. Staphylococcus aureus panton-valentine leukocidin directly targets mitochondria and induced Bax-independent apoptosis of human neutrophils. J Clin Invest. 2005;115:3117–27.
Fridkin SK, Hageman JC, Morrison M, Sanza LT, Como-Sabetti K, Jernigan JA, et al. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352:1436–44.
Miller LG, Perdreau-Remington F, Rieg G, Mehdi S, Perlroth J, Bayer AS, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352:1445–53.
Musser JM, DeLeo FR. Toward a genome-wide systems biology analysis of host-pathogen interactions in group A Streptococcus. Am J Path. 2005;167:1461–72.
Voyich JM, Musser JM, DeLeo FR. Streptococcus pyogenes and human neutrophils: a paradigm for evasion of innate host defense by bacterial pathogens. Microbes Infect. 2004;6:1117–23.
Voyich JM, Braughton KR, Sturdevant DE, Vuong C, Kobayashi SD, Porcella SF, et al. Engagement of the pathogen survival response used by group A streptococcus to avert destruction by innate host defense. J Immunol. 2004;173:1194–201.
Miyoshi-Akiyama T, Takamatsu D, Koyanagi M, Zhao J, Imanishi K, Uchiyama T. Cytocidal effect of Streptococcus pyogenes on mouse neutrophils in vivo and the critical role of streptolysin S. J Infect Dis. 2005;192:107–16.
Ge Y, Rikihisa Y. Anaplasma phagocytophilum delays spontaneous human neutrophil apoptosis by modulation of multiple apoptotic pathways. Cell Micro. 2006;8:1406–16.
Lee HC, Goodman JL. Anaplasma phagocytophilum causes global induction of antiapoptosis in human neutrophils. Genomics. 2006;88:496–503.
Scaife H, Woldehiwet Z, Hart CA, Edwards SW. Anaplasma phagocytophilum reduces neutrophil apoptosis in vivo. Infect Immun. 2003;71:1995–2001.
van Zandbergen G, Gieffers J, Kothe H, Rupp J, Bollinger A, Aga E, et al. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J Immunol. 2004;172:1768–76.
Yoshiie K, Kim HY, Mott J, Rikihisa Y. Intracellular infection by the human granulocyte Ehrlichiosis agent inhibits human neutrophil apoptosis. Infect Immun. 2000;68:1125–33.
Carlyon JA, Latif DA, Pypaert M, Lacy P, Fikrig E. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect Immun. 2004;72:4772–83.
Webster P, Ijdo JW, Chicoine LM, Fikrig E. The agent of human granulocytic ehrlichiosis resides in an endosomal compartment. J Clin Invest. 1998;101:1932–41.
Ijdo JW, Mueller AC. Neutrophil NADPH oxidase is reduced at the Anaplasma phagocyophilum phagosome. Infect Immun. 2004;72:5392–401.
Mott J, Rikihisa Y, Tsunawaki S. Effects of Anaplasma phagocytophila on NADPH oxidase components in human neutrophils and HL-60 cells. Infect Immun. 2002;70:1359–66.
Ge Y, Yoshiie K, Kuribayashi F, Lin M, Rikihisa Y. Anaplasma phagocytophilum inhibits human neutrophil apoptosis via upregulation of bfl-1, maintenance of mitochondrial membrane potential and prevention of caspase 3 activation. Cell Microbiol. 2005;7:29–38.
Airenne S, Surcel HM, Tuukkanen J, Leinonen M, Saikku P. Chlamydia pneumoniae inhibits apoptosis in human epithelial and monocyte cell lines. Scand J Immunol. 2002;55:390–8.
Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–64.
Levin S, Bucci TJ, Cohen SM, Fix AS, Hardisty JF, Legrand EK, et al. The nomenclature of cell death: recommendations of an ad hoc committee of the society of toxicologic pathologists. Toxicol Pathol. 1999;27:490.
Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–6.
Kostin S, Pool L, Elsasser A, Hein S, Drexler HCA, Arnon E, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–24.
Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–8.
Lecoeur H, Prevost M-C, Gougeon M-L. Oncosis is associated with exposure of phosphatidylserine residues on the outside layer of the plasma membrane: a reconsideration of the specificity of the annexin V/propidium iodide assay. Cytometry. 2001;44:65–72.
Krysko O, de Ridder L, Cornelissen M. Phosphatidylserine exposure during early primary necrosis (oncosis) in JB6 cells as evidenced by immunogold labeling technique. Apoptosis. 2004;9:495–500.
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Diff. 2007;14:230–9.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6.
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2.
Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75.
Saftig P, Beersten W, Eskelinen E-L. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy. 2008;4:510–2.
Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–25.
Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem. 2007;282:36321–9.
Kanneganti T-D, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–59.
Fernandes-Alnemri T, Wu J, Yu J-W, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Diff. 2007;14:1590–604.
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32.
Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111.
Henry T, Monack DM. Activation of the inflammasome upon Francisella tularensis infection: interplay of innate immune pathways and virulence factors. Cell Microbiol. 2007;9:2543–51.
Cervantes J, Nagata T, Uchijima M, Shibata K, Koide Y. Intracytosolic listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cell Microbiol. 2008;10:41–52.
Bergsbaken T, Cookson BT. Macrophage activation redirects Yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Path. 2007;3:1570–82.
Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA. 2008;105:4312–7.
Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007;9:2562–70.
Trump BF, Berezesky IK. The role of altered [Ca2+]i regulation in apoptosis, oncosis, and necrosis. Biochem Biophys Acta. 1996;1313:173–8.
Perez JF, Chemello ME, Liprandi F, Ruiz M-C, Michelangeli F. Oncosis in MA104 cells is induced by rotavirus infection through an increase in intracellular Ca2+ concentration. Virology. 1998;252:17–27.
Pei J, Turse JE, Wu O, Ficht TA. Brucella abortus rough mutants induce macrophage oncosis that requires bacterial protein synthesis and direct interaction with the macrophage. Infect Immun. 2006;74:2667–75.
Kalischuk LD, Inglis GD, Buret AG. Strain-dependent induction of epithelial cell oncosis by Campylobacter jejuni, is correlated with invasion ability and is independent of cytolethal distending toxin. Microbiology. 2007;153:2952–63.
Dacheux D, Toussaint B, Richard M, Brochier G, Croize J, Attree I. Pseudomonas aeruginosa cystic fibrosis isolates induced rapid, type III secretion-dependent, but ExoU-independent, oncosis on macrophages and polymorphonuclear neutrophils. Infect Immun. 2000;68:2916–24.
Sano G-I, Takada Y, Goto S, Maruyama K, Shindo Y, Oka K, et al. Flagella facilitate escape of Salmonella from oncotic macrophages. J Bacteriol. 2007;189:8224–32.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–9.
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16:396–400.
Choi K-S, Park JT, Dumler JS. Anaplasma phagocytophilum delay of neutrophil apoptosis through the p38 mitogen-activated protein kinase signal pathway. Infect Immun. 2005;73:8209–18.
Abramson T, Kedem H, Relman DA. Proinflammatory and proapoptotic activities associated with Bordetella pertussis filamentous hemagglutinin. Infect Immun. 2001;69:2650–8.
Basler M, Masin J, Osicka R, Sebo P. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect Immun. 2006;74:2207–14.
Gueirard P, Druilhe A, Pretolani M, Guiso N. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect Immun. 1998;66:1718–25.
Khelef N, Zychlinsky A, Guiso N. Bordetella pertussis induces apoptosis in macrophages: role of adenylate cyclase hemolysin. Infect Immun. 1993;61:4064–71.
Khelef N, Guiso N. Induction of macrophage apoptosis by Bordetella pertussis adenylate cyclase-hemolysin. FEMS Microbiol Lett. 1995;134:27–32.
Khelef N, DeShazer D, Friedman RL, Guiso N. In vivo and in vitro analysis of Bordetella pertussis catalase and Fe-superoxide dismutase mutants. FEMS Microbiol Lett. 1996;142:231–5.
Khelef N, Gounon P, Guiso N. Internalization of Bordetella pertussis adenylate cyclase-hemolysin into endocytic vesicles contributes to macrophage cytotoxicity. Cell Microbiol. 2001;3:721–30.
Harvill ET, Cotter PA, Miller JF. Pregenomic comparative analysis between Bordetella bronchiseptica RB50 and Bordetella pertussis tohama I in murine models of respiratory tract infection. Infect Immun. 1999;67:6109–18.
Boyd AP, Ross PJ, Conroy H, Mahon N, Lavelle EC, Mills KHG. Bordetella pertussis adenylate cyclase toxin modulates innate and adaptive immune responses: distinct roles for acylation and enzymatic activity in immunomodulation and cell death. J Immunol. 2005;175:730–8.
Bylund J, Campsall PA, Ma RC, Conway BA, Speert DP. Burkholderia cenocepacia induces neutrophil necrosis in chronic granulomatous disease. J Immunol. 2005;174:3562–9.
Hutchison ML, Poxton IR, Govan JR. Burkholderia cepacia produces a hemolysin that is capable of inducing apoptosis and degranulation of mammalian phagocytes. Infect Immun. 1998;66:2033–9.
Punj V, Sharma R, Zaborina O, Chakrabarty AM. Energy-generating enzymes of Burkholderia cepacia and their interactions with macrophages. J Bacteriol. 2003;185:3167–78.
Carratelli CR, Rizzo A, Catania MR, Galle F, Losi E, Hasty DL, et al. Chlamydia pneumoniae infections prevent the programmed cell death of THP-1 cell line. FEMS Microbiol Lett. 2002;215:69–74.
Miyairi I, Byrne GI. Chlamydia and programmed cell death. Curr Opin Microbiol. 2006;9:102–8.
Yaraei K, Campbell LA, Zhu X, Liles WC, Kuo C-C, Rosenfeld ME. Chlamydia pneumoniae augments the oxidized low-density lipoprotein-induced death of mouse macrophages by a caspase-independent pathway. Infect Immun. 2005;73:4315–22.
Goth SR, Stephens RS. Rapid, transient phosphatidylserine externalization induced in host cells by infection with Chlamydia spp. Infect Immun. 2001;69:1109–19.
Ying S, Seiffert BM, Hacker G, Fischer SF. Broad degradation of proapoptotic proteins with the conserved Bcl-2 homology domain 3 during infection with Chlamydia trachomatis. Infect Immun. 2005;73:1399–403.
Mahida YR, Galvin A, Makh S, Hyde S, Sanfilippo L, Borriello SP, et al. Effect of Clostridium difficile toxin A on human colonic lamina propria cells: early loss of macrophages followed by T-cell apoptosis. Infect Immun. 1998;66:5462–9.
Solomon K, Webb J, Ali N, Robins RA, Mahida YR. Highly sensitive to Clostridium difficile toxin A-induced apoptotic and nonapoptotic cell death. Infect Immun. 2005;73:1625–34.
Brest P, Betis F, Cuburu N, Selva E, Herrant M, Servin A, et al. Increased rate of apoptosis and diminished phagocytic ability of human neutrophils infected with Afa/Dr diffusely adhering Escherichia coli strains. Infect Immun. 2004;72:5741–9.
Fernandez-Prada C, Tall BD, Elliott SE, Hoover DL, Nataro JP, Venkatesan MM. Hemolysin-positive enteroaggregative and cell-detaching Escherichia coli strains cause oncosis of human monocyte-derived macrophages and poptosis of murine J774 cells. Infect Immun. 1998;66:3918–24.
Hacker H, Furmann C, Wagner H, Hacker G. Caspase-9/-3 activation and apoptosis are induced in mouse macrophages upon ingestion and digestion of Escherichia coli bacteria. J Immunol. 2002;169:3172–9.
Lai XH, Arencibia I, Johansson A, Wai SN, Oscarsson J, Kalfas S, et al. Cytocidal and apoptotic effects of the ClyA protein from Escherichia coli on primary and cultured monocytes and macrophages. Infect Immun. 2000;68:4363–7.
Lai XH, Xu JG, Melgar S, Uhlin BE. An apoptotic response by J774 macrophage cells is common upon infection with diarrheagenic Escherichia coli. FEMS Microbiol Lett. 2006;172:29–34.
Rodrigues VS, Vidotto MC, Felipe I, Santos DS, Gaziri LC. Apoptosis of murine peritoneal macrophages induced by an avian pathogenic strain of Escherichia coli. FEMS Microbiol Lett. 1999;179:73–8.
Stravodimos KG, Singhal PC, Sharma S, Reddy K, Smith AD. Escherichia coli promotes macrophage apoptosis. J Endourol. 1999;13:273–7.
Yagi Y, Shiono H, Shibahara T, Chikayama Y, Makamura I, Ohnuma A. Increase in apoptotic polymorphonuclear neutrophils in peripheral blood after intramammary infusion of Escherichia coli lipopolysaccharide. Vet Immunol Immunopathol. 2002;89:115–25.
Gille C, Leiber A, Spring B, Kempf VAJ, Loeffler J, Poets C, et al. Diminished phagocytosis-induced cell death (PICD) in neonatal monocytes upon infection with Escherichia coli. Pediatr Res. 2008;63:33–8.
Narayanan S, Stewart GC, Chengappa MM. Fusobacterium necrophorum leukotoxin induces activation and apoptosis of bovine leukocytes. Infect Immun. 2002;70:4609–20.
Jewett A, Hume WR, Le H, Huynh TN, Han YW, Cheng G, et al. Induction of apoptotic cell death in peripheral blood mononuclear and polymorphonuclear cells by an oral bacterium, Fusobacterium nucleatum. Infect Immun. 2000;68:1893–8.
Yang YF, Sylte MJ, Czuprynski CJ. Apoptosis: a possible tactic of Haemophilus somnus for evasion of killing by bovine neutrophils? Microb Pathog. 1998;24:351–9.
Betten A, Bylund J, Cristophe T, Boulay F, Romero A, Hellstrand K, et al. A proinflammatory peptide from Helicobacter pylori activates monocytes to induce lymphocyte dysfunction and apoptosis. J Clin Invest. 2001;108:1221–8.
Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on Gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori Infection. J Immunol. 2006;176:3000–9.
Gobert aP, Cheng Y, Wang JY, Boucher JL, Iyer RK, Cederbaum SD, et al. Helicobacter pylori induces macrophage apoptosis by activation of arginase II. J Immunol. 2002;168:4692–700.
Kim JS, Kim JM, Jung HC, Song IS, Kim CY. Inhibition of apoptosis in human neutrophils by Helicobacter pylori water-soluble surface proteins. Scand J Gastroenterol. 2001;36:589–600.
Barsig J, Kaufmann SH. The mechanism of cell death in Listeria monocytogenes-infected murine macrophages is distinct form apoptosis. Infect Immun. 1997;65:4075–81.
Guzman CA, Domann E, Rohde M, Bruder D, Darji A, Weiss S, et al. Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol Microbiol. 1996;20:119–26.
Vadyvaloo V, Arous S, Gravesen A, Hechard Y, Chauhan-Haubrock R, Hastings JW, et al. Cell-surface alteration in class IIa bacteriocin-resistant Listeria monocytogenes strains. Microbiology. 2004;150:3025–33.
Warren SE, Mao DP, Rodriguez AE, Miao EA, Aderem A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J Immunol. 2008;180:7558–64.
Zwaferink H, Stockinger S, Reipert S, Decker T. Stimulation of inducible nitric oxide synthase expression by beta interferon increases necrotic death of macrophages upon Listeria monocytogenes infection. Infect Immun. 2008;76:1649–56.
Birmingham CL, Higgins DE, Brumell JH. Avoiding death by autophagy: interactions of listeria monocytogenes with the macrophage autophagy system. Autophagy. 2008;4:368–71.
Cudd LA, Ownby CL, Clarke CR, Sun Y, Clinkenbeard KD. Effects of Mannheimia haemolytica leukotoxin on apoptosis and oncosis of bovine neutrophils. Am J Vet Res. 2001;62:136–41.
Stevens PK, Czuprynski CJ. Pasteurella haemolytica leukotoxin induces bovine leukocytes to undergo morphologic changes consistent with apoptosis in vitro. Infect Immun. 1996;64:2687–94.
Thumbikat P, Dileepan T, Kannan MS, Maheswaran SK. Mechanisms underlying Mannheimia haemolytica leukotoxin-induced oncosis and apoptosis of bovine alveolar macrophages. Microb Pathog. 2005;38:161–72.
Ahmad A, Khan M, Raykundalia C, Catty D. Study of the mechanisms of killing of Mycobacterium bovis BCG by apoptosis in J774 murine macrophages. J Pak Med Assoc. 1999;49:273–8.
Gutierrez-Pabello JA, McMurray DN, Adams LG. Upregulation of thymosin beta-10 by Mycobacterium bovis infection of bovine macrophages is associated with apoptosis. Infect Immun. 2002;70:2121–7.
Kremer L, Estaquier J, Brandt E, Ameisen JC, Locht C. Mycobacterium bovis Bacillus Calmette Guerin infection prevents apoptosis of resting human monocytes. Eur J Immunol. 1997;27:2450–6.
Suttmann H, Lehan N, Bohle A, Brandau S. Stimulation of neutrophil granulocytes with Mycobacterium bovis bacillus Calmette-Guerin induces changes in phenotype and gene expression and inhibits spontaneous apoptosis. Infect Immun. 2003;71:4647–56.
Krzyzowska M, Schollenberger A, Pawlowski A, Hamasur B, Winnicka A, Augustynowicz-Kopec E, et al. Lipoarabinomannan as a regulator of the monocyte apoptotic response to Mycobacterium bovis BCG Danish strain 1331 infection. Pol J Microbiol. 2007;56:89–96.
Vega-Manriquez X, Lopez-Vidal Y, Moran J, Adams LG, Gutierrez-Pabello JA. Apoptosis-inducing factor participation in bovine macrophage Mycobacterium bovis-induced caspase-independent cell death. Infect Immun. 2007;75:1223–8.
Aleman M, Schierloh P, De La Barrera SS, Musella RM, Saab MA, Baldini M, et al. Mycobacterium tuberculosis triggers apoptosis in peripheral neutrophils involving toll-like receptor 2 and p38 mitogen protein kinase in tuberculosis patients. Infect Immun. 2004;72:5150–8.
Ciaramella A, Martino A, Cicconi R, Colizzi V, Fraziano M. Mycobacterial 19-kDa lipoprotein mediates Mycobacterium tuberculosis-induced apoptosis in monocytes/macrophages at early stages of infection. Cell Death Diff. 2000;7:1270–2.
Ciaramella A, Cavone A, Santucci MB, Amicosante M, Martino A, Auricchio G, et al. Proinflammatory cytokines in the course of Mycobacterium tuberculosis-induced apoptosis in monocytes/macrophages. J Infect Dis. 2002;186:1277–82.
Danelishvili L, McGarvey J, Li YJ, Bermudez LE. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol. 2003;5:649–60.
Duan L, Gan H, Arm J, Remold HG. Cytosolic phospholipase A2 participates with TNF-alpha in the induction of apoptosis of human macrophages infected with Mycobacterium tuberculosis H37Ra. J Immunol. 2001;166:7469–76.
Duan L, Gan H, Golan DE, Remold HG. Critical role of mitochondrial damage in determining outcome of macrophage infection with Mycobacterium tuberculosis. J Immunol. 2002;169:5181–7.
Durrbaum-Landmann I, Gercken J, Flad HD, Ernst M. Effect of in vitro infection of human monocytes with low numbers of Mycobacterium tuberculosis bacteria on monocytes apoptosis. Infect Immun. 1996;64:5384–9.
Kasahara K, Sato I, Ogura K, Takeuchi H, Kobayashi K, Adachi M. Expression of chemokines and induction of rapid cell death in human blood neutrophils by Mycobacterium tuberculosis. J Infect Dis. 1998;178:127–37.
Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. 1997;65:298–304.
Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–20.
Keane J, Shurtleff B, Kornfeld H. TNF-dependent BALB/c murine macrophage apoptosis following Mycobacterium tuberculosis infection inhibits bacillary growth in an IFN-gamma independent manner. Tuberculosis. 2002;82:55–61.
Lopez M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol. 2003;170:2409–16.
Mustafa T, Phyu S, Nilsen R, Bjune G, Jonsson R. Increased expression of Fas ligand on Mycobacterium tuberculosis infected macrophages: a potential novel mechanism of immune evasion by Mycobacterium tuberculosis? Inflammation. 1999;23:507–21.
Oddo M, Renno T, Attinger A, Bakker T, MacDonald HR, Meylan PR. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J Immunol. 1998;160:5448–54.
Placido R, Mancino G, Amendola A, Mariani F, Vendetti S, Piacentini M, et al. Apoptosis of human monocytes/macrophages in Mycobacterium tuberculosis infection. J Pathol. 1997;181:31–8.
Rojas M, Barrera LF, Garcia LF. Induction of apoptosis in murine macrophages by Mycobacterium tuberculosis is reactive oxygen intermediates-independent. Biochem Biophys Res Commun. 1998;247:436–42.
Rojas M, Olivier M, Gros P, Barrera LF, Garcia LF. TNF-alpha and IL-10 modulate the induction of apoptosis by virulent Mycobacterium tuberculosis in murine macrophages. J Immunol. 1999;162:6122–31.
Rojas M, Garcia LF, Nigou J, Puzo G, Olivier M. Mannosylated lipoarabinomannan antagonizes Mycobacterium tuberculosis-induced macrophage apoptosis by altering Ca2+-dependent cell signalling. J Infect Dis. 2000;182:240–51.
Santucci MB, Amicosante M, Cicconi R, Montesano C, Casarini M, Giosue S, et al. Mycobacterium tuberculosis-induced apoptosis in monocytes/macrophages: early membrane modifications and intracellular mycobacterial viability. J Infect Dis. 2000;181:1506–9.
Sly LM, Hingley-Wilson SM, Reiner NE, McMaster WR. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J Immunol. 2003;170:430–7.
Spira A, Carroll JD, Liu G, Aziz Z, Shah V, Kornfeld H, et al. Apoptosis genes in human alveolar macrophages infected with virulent or attenuated Mycobacterium tuberculosis: a pivotal role for tumor necrosis factor. Am J Respir Cell Mol Biol. 2003;29:545–51.
Arcila ML, Sanchez MD, Ortiz B, Barrera LF, Garcia LF, Rojas M. Activation of apoptosis, but not necrosis, during Mycobacterium tuberculosis infection correlated with decreased bacterial growth: role of TNF-alpha, IL-10, caspases and phospholipase A2. Cell Immunol. 2007;249:80–93.
do Vale A, Marques F, Silva MT. Apoptosis of sea bass (Dicentrarchus labrax L.) neutrophils and macrophages induced by experimental infection with Photobacterium damselae subsp. piscicida. Fish Shellfish Immunol. 2003;15:129–44.
Ozaki K, Hanazawa S. Porphyromonas ginigivalis fimbraie inhibit caspase-3-mediated apoptosis of monocytic THP-1 cells under growth factor deprivation via extracellular signal-regulated kinase-dependent expression of p21 Cip/WAF1. Infect Immun. 2001;69:4944–50.
Preshaw PM, Schifferle RE, Walters JD. Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro. J Periodontal Res. 1999;34:197–202.
Dacheux D, Goure J, Chabert J, Usson Y, Attree I. Pore-forming activity of type III system-secreted proteins leads to oncosis of Pseudomonas aeruginosa-infected macrophages. Mol Microbiol. 2001;40:76–85.
Dacheux D, Attree I, Schneider C, Toussaint B. Cell death of human polymorphonuclear neutrophils induced by a Pseudomonas aeruginosa cystic fibrosis isolate requires a functional type III secretion system. Infect Immun. 1999;67:6164–7.
Hauser aR, Engel JN. Pseudomonas aeruginosa induces type-III-secretion-mediated apoptosis of macrophages and epithelial cells. Infect Immun. 1999;67:5530–7.
Yt Huang, Jeng CR, Cheng CH, Chueh LL, Liu JJ, Pang VF. Morphological and immunological evidence of a unique selective production and endoplasmic reticular accumulation of interleukin-1alpha in rat peritoneal macrophages induced by Pseudomonas aeruginosa exotoxin A. Cell Immunol. 2003;221:143–56.
Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, et al. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun. 2003;71:5785–93.
Usher LR, Lawson RA, Geary I, Taylor CJ, Bingle CD, Taylor GW, et al. Induction of neutrophil apoptosis by the Pseudomonas aeruginosa exotoxin pyocyanin: a potential mechanism of persistent infection. J Immunol. 2002;168:1861–8.
Worgall S, Martushova K, Busch A, Lande L, Crystal RG. Apoptosis induced by Pseudomonas aeruginosa in antigen presenting cells is diminished by genetic modification with CD40 ligand. Pediatr Res. 2002;52:636–44.
Zaborina O, Dhiman N, Ling Chen M, Kostal J, Holder IA, Chakrabarty AM. Secreted products of a nonmucoid Pseudomonas aeruginosa strain induce two modes of macrophage killing: external-ATP-dependent, P2Z-receptor-mediated necrosis and ATP-independent, caspase-mediated apoptosis. Microbiology. 2000;146:2521–30.
Zhang J, Takayama H, Matsuba T, Jiang R, Tanaka Y. Induction of apoptosis in macrophage cell line, J774, by the cell-free supernatant from Pseudomonas aeruginosa. Microbiol Immunol. 2003;47:199–206.
Raqib R, Ekberg C, Sharkar P, Bardhan PK, Zychlinsky A, Sansonetti PJ, et al. Apoptosis in acute shigellosis is associated with increased production of Fas/Fas ligand, perforin, caspase-1, and caspase-3 but reduced production of Bcl-2 and interleukin-2. Infect Immun. 2002;70:3199–207.
Lee S-Y, Lee M-S, Cherla RP, Tesh VL. Shiga toxin1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008;10:770–80.
Nilsdotter-Augustinsson ASA, Wilsson ASA, Larsson JENN, Stendahl OLLE, Ohman LENA, Lundqvist-Gustafsson HELE. Staphylococcus aureus, but not Staphylococcus epidermidis, modulates the oxidative response and induces apoptosis in human neutrophils. APMIS. 2004;112:109–18.
Schnaith A, Kashkar H, Leggio SA, Addicks K, Kronke M, Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem. 2007;282:2695–706.
Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, et al. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS ONE. 2008;3:e1409.
Dockrell DH, Whyte MKB. Regulation of phagocyte lifespan in the lung during bacterial infection. J Leukoc Biol. 2006;79:904–8.
Ali F, Lee ME, Iannelli F, Pozzi G, Mitchell TJ, Read RC, et al. Streptococcus pneumoniae-associated human macrophage apoptosis after bacterial internalization via complement and Fcgamma receptors correlates with intracellular bacterial load. J Infect Dis. 2003;188:1119–31.
Dockrell DH, Lee M, Lynch DH, Read RC. Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J Infect Dis. 2001;184:713–22.
Kirby AC, Raynes JG, Kaye PM. The role played by tumor necrosis factor during localized systemic infection with Streptococcus pneumoniae. J Infect Dis. 2005;191:1538–47.
Marriott HM, Dockrell DH. Streptococcus pneumoniae: the role of apoptosis in host defense and pathogenesis. Int J Biochem Cell Biol. 2006;38:1848–54.
Marriott HM, Hellewell PG, Cross SS, Ince PG, Whyte MKB, Dockrell DH. Decreased alveolar macrophage apoptosis is associated with increased pulmonary inflammation in a murine model of pneumococcal pneumonia. J Immunol. 2006;177:6480–8.
Farnworth SL, Henderson NC, MacKinnon AC, Atkinson KM, Wilkinson T, Dhaliwal K, et al. Galectin-3 reduces the severity of pneumococcal pneumonia by augmenting neutrophil function. Am J Pathol. 2008;172:395–405.
Srivastava A, Henneke P, Visintin A, Morse SC, Martin V, Watkins C, et al. The apoptotic response to pneumolysin is toll-like receptor 4 dependent and protects against pneumococcal disease. Infect Immun. 2005;73:6479–87.
Staali L, Bauer S, Morgelin M, Bjorck L, Tapper H. Streptococcus pyogenes bacteria modulate membrane traffic in human neutrophils and selectively inhibit azurophilic granule fusion with phagosomes. Microbiology. 2006;8:690–703.
Medina E, Goldmann O, Toppel AW, Chhatwal GS. Survival of Streptococcus pyogenes within host phagocytic cells: a pathogenic mechanism for persistence and systemic invasion. J Infect Dis. 2003;187:597–603.
Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kennedy, A.D., DeLeo, F.R. Neutrophil apoptosis and the resolution of infection. Immunol Res 43, 25–61 (2009). https://doi.org/10.1007/s12026-008-8049-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-008-8049-6