
Abstract
Genetic risk factors play a role in sudden unexpected infant death; either as a cause of death, such as in cases with medium-chain acyl-coenzyme A dehydrogenase deficiency and cardiac arrest due to long QT syndrome, or as predisposing factors for sudden infant death syndrome (SIDS). Most likely genetic predisposition to SIDS represent a polygenic inheritance pattern leading to sudden death when combined with other risk factors, such as a vulnerable developmental stage of the central nervous system and/or the immune system, in addition to environmental risk factors, such as a common cold or prone sleeping position. Genes involved in the regulation of the immune system, cardiac function, the serotonergic network and brain function and development have so far emerged as the most important with respect to SIDS. The purpose of the present paper is to survey current knowledge on SIDS and possible genetic contributions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
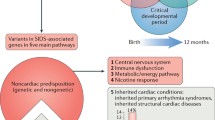
Sudden infant death may be divided into two main categories; explained and unexplained. Besides accidents, homicide, and infections, genetic disorders such as medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency and long QT-time syndrome (LQTS) represent explainable causes of death. In unexplained deaths, diagnosed as sudden infant syndrome (SIDS) an extensive autopsy, history of disease and investigation of the circumstances do not disclose a cause of death. Current theories imply that infants may die from SIDS when three conditions occur at the same time [1, 2]: a genetic predisposition, a vulnerable developmental stage of the CNS and/or the immune system, and a triggering event. Certain gene mutations may represent obvious causes of death, for example some LQTS mutations. However, there are also mutations and polymorphisms that may represent risk factors that may induce fatal situations when combined with environmental factors, e.g. some polymorphisms in LQTS genes that contribute to cardiac arrest when combined with acidosis (Fig. 1) [3].

Possible causes of sudden unexpected infant death
Over the last two decades there has been a focus on environmental risk factors for SIDS, and the hazards of prone sleeping, parental smoking and overheating are now well documented [4–7]. However, reports dealing with genetic risk factors for SIDS are growing in number, and the purpose of the present paper is to examine the relationship between SIDS and genetic factors.
Immune system
Many SIDS victims have an activated immune system, which may indicate that they are vulnerable to simple infections. Approximately half of the SIDS victims are reported to have symptoms of a slight upper airway infection prior to death [6], and the mucosal immune system is stimulated compared to victims of sudden violent death [8–10]. The observation by Vege et al. [11] that half of the SIDS victims had IL-6 levels in the cerebrospinal fluid (CSF) in the same range as infants that died from infections such as meningitis and septicemia supported the view that the brain is the target organ for a lethal mechanism initiated by an immune reaction [10, 12]. A link between the mucosal immune system and the central nervous system (CNS) was furthermore demonstrated by Vege et al. [13] showing that both increased expression of HLA-DR antigens in glandular epithelium and increased number of IgA immunocytes in the laryngeal mucosa were significantly related to high IL-6 levels in the CSF.
Recently Rognum et al. [14] demonstrated IL-6 receptors on serotonergic neurons in the arcuate nucleus in the brainstem of SIDS cases, and together with the studies by Vege et al. [11, 13] this indicates a connection between the mucosal immune system and the serotonergic network in the brainstem. The medullary serotonergic (5-HT) system in the brainstem is involved in the modulation of breathing, blood pressure and heart rate according to the level of arousal. It is proposed that SIDS results from an abnormality of the medullary 5-HT system that causes an inability to restore homeostasis following life-threatening challenges during sleep, and thus leads to sudden death in the critical first year of life, when homeostatic systems are still maturing [15]. A differential modulation of the serotonin transporter by pro and anti-inflammatory cytokines may represent a fine-tuned mechanism to communicate an immune response to the central nervous system [16], and one hypothesis may be that a simple upper airway infection in vulnerable infants may initiate an immune reaction in the laryngeal and tracheal mucosa, resulting in a fatal IL-6 liberation within the CNS. One reason for such vulnerability may be certain patterns of polymorphisms in genes encoding components in the immune system.
The first gene to be investigated in this regard was the gene encoding complement component C4, where an association between slight infection prior to death and deletions of either the C4A or the C4B gene was found in cases of SIDS [17, 18]. Individuals with complete deficiency of C4 demonstrate delayed complement activation and suboptimal immune response, but even a partial deficiency in one of the isotypes may lead to a situation in which neutralisation of the microorganism is impaired [19, 20].
A reason for increased vulnerability has also been looked for in innate immunity. Surfactant protein A (SP-A) and surfactant protein D (SP-D) are humoral molecules involved in the innate host defence against various bacterial and viral pathogens. Ten SNPs that might influence expression of the genes encoding these two surfactants have been investigated in 42 SIDS cases and 46 explained infant deaths [21]. No differences in genotype distributions between SIDS cases and controls were found, even though there was a tendency for the most common SP-A haplotype, 6A2/1A0, to be overrepresented in cases with low immunohistochemical SP-A expression.
The most likely candidates for explaining vulnerability to infections are the cytokine genes. The cytokines regulate the intensity and duration of the immune response by stimulating the different cells involved, and thus activates a network of interacting cells. The interleukin genes investigated with regard to SIDS so far are those encoding for IL-1α, IL-1β, IL-4, IL-6, IL-10, IL-12, IL-13, IL-16, IL-18, transforming growth factor (TGF), tumor necrosis factor α (TNFα), and interferon gamma (IFNγ) [22–34].
In the IL-10 gene, the ATA haplotype and the ATA/ATA genotype of the IL-10 gene is proposed to be associated with both infectious death and SIDS. Even though it has proven difficult to determine the exact relationship between IL-10 genotype and IL-10 production, several studies report that the ATA haplotype is associated with low IL-10 levels [35]. The association with SIDS was found in two smaller studies [22, 25], and in a larger study including 214 SIDS cases, 29 cases of infectious death and 163 controls (69 living infants and 67 diseased adults) the ATA haplotype and the ATA/ATA genotype were found to be associated with infectious death [23].
Two common polymorphisms relevant to IL-1β levels are the -511C/T polymorphism in the IL-1β gene and the 2018T/C polymorphism in the IL-1Ra gene. Both these polymorphisms have been investigated in cases of SIDS, however no differences in genotype distribution between SIDS cases and controls have been found [24]. Another study investigated the 89 bp VNTR in the IL-1Ra gene in 49 Australian SIDS cases and 103 living infant controls, and found that carriage of the 2/2 genotype was 4.85 times more likely to increase the risk for SIDS compared with the predominant 1/1 genotype [32]. Homozygous carriers of allele 2 have a more severe and prolonged proinflammatory immune response than do those with other IL-1Ra genotypes [36], which may contribute to the vulnerability to infection seen in SIDS.
A British study that included common polymorphisms in the genes encoding IL-4, IL-6, IFNγ, TGF, and VEGF, showed significant differences for IL-6 and VEGF: the genotypes -174GG, and -1154AA being more frequent in SIDS cases than in controls [27]. Only 25 SIDS cases and 133 living adult controls were investigated, but the authors concluded that the gene polymorphism association suggests that the causation of SIDS is related to both fetal lung development and an infant’s innate ability to mount an inflammatory response to infection [27]. The findings with IL-6 were confirmed in a study of 19 Australian SIDS cases. The SIDS cases also had the -174GG genotype more often, associated with high IL-6 production [37], compared to adult controls [28]. However, this could not be confirmed in a Norwegian study of 175 SIDS cases [29]. Since then, Ferrante et al. have investigated in total 10 different SNPs in the genes encoding IL-1, IL-6, IL-8, IL-12, IL-13 IL-16, and IL-18, as well as one VNTRs in each of the genes encoding IL-1α and IL-1Ra [33, 34] in 204 Norwegian SIDS cases and in 131 adult diseased controls. The only association that was found with SIDS was for the gene combination IL-1α VNTR A1A1/IL-1β +4845TT (P < 0.01), a gene combination one could expect to result in a decreased level of IL-1α [33]. Associations were also found between genotype and sleeping position, as the genotypes IL-18 -251AA/AT, IL-8 -781 CT/TT, and IL-1β -511CC/CT were more frequent in SIDS cases found prone. For these cases, the alleles IL-8 -251A, IL-8 -781T and IL-1β -511T all led to increased expression of the gene [34]. In addition, Ferrante et al. have also shown an association between TNFα promoter polymorphism and SIDS [30]. The genotype -238GG, leading to a low expression of TNFα, was observed more often in SIDS cases than in controls, while the genotype -308GA was more frequent in borderline SIDS cases than in controls. For the SNP in bp -308, the A allele leads to a high production of TNFα [38]. Furthermore, four SNP profiles in the TNFα promoter had significantly different frequencies in SIDS cases and controls [30]. Taken together, these findings may indicate that a genetic determined disturbed homeostasis of the interleukin network is involved in SIDS.
Another explanation for increased vulnerability may be found in different gene variants of G-proteins, which are relevant to the immune response as an association between the Gβ3 825T allele and increased immune cell function has been reported [39]. The T-allele results in increased G protein mediated signal transduction compared to the C-allele [40]. The best investigated polymorphism in the Gβ3 gene is C825T. A study of the C825T polymorphism in 250 SIDS cases, 38 cases of infectious death, and 99 living infant controls revealed no difference in genotype frequency between SIDS cases and controls, but found that the cases of infectious death had the CC genotype more often, compared to controls [41]. This observation may indicate that the presence of the 825T allele exerts a protective effect towards serious infection, perhaps through enhanced G protein signalling.
Cardiac arrhythmia
The long QT syndrome (LQTS) is a cardiac disorder causing syncope, seizures, and sudden death, usually in young, otherwise healthy, individuals. LQTS is a genetically heterogeneous condition, caused by mutations in one or several of nine genes, LQT1–LQT9, encoding for cardiac ion channels. Since LQTS is a potentially lethal disorder, the condition should be excluded before a diagnosis of SIDS is made.
Several papers have investigated LQT related genes in SIDS cases [42–55]. Many of these have included only small numbers of SIDS cases [42, 43, 45–47, 53], but from the investigation of SIDS cohorts [44, 49–52, 54, 55], it may be assumed that 5–10% of the cases originally diagnosed as SIDS are either due to LQTS, or have mutations or functional polymorphisms in LQT genes that may represent a predisposing factor that may be lethal in combination with other risk factors such as hypoxemia [3]. The largest study of LQTS and SIDS so far, including 201 Norwegian SIDS cases and 182 controls (137 adults and 45 infants, all diseased), found that 9.5% of the SIDS victims had functionally significant genetic variants in the LQTS genes [3, 51].
The SCN5A S1103Y mutation has been reported to be of special importance in African-American SIDS cases [49, 52]. Plant et al. found an overrepresentation of this mutation in a cohort of 133 SIDS cases, suggesting a 24-fold increased risk for SIDS in infants with the YY genotype [49], while another study found that 22.5% of the investigated SIDS cases had the heterozygous SY genotype [52]. The authors showed that Y1103 channels demonstrated abnormal function when subjected to low intracellular pH, in comparison to S1103 channels [49].
The ryanodine receptor RyR2 is a receptor in cardiac muscle where it is component of calcium channels. It has been shown that “leaky” RyR2 channels may trigger fatal arrhythmias. The gene encoding for RyR2 has been investigated in 134 SIDS cases of different ethnicity, however the study did not include any controls [56]. Two SIDS cases with novel mutations were found, and the authors conclude that it is most likely that mutations alter the response of the channels to sympathetic nervous system stimulation, such that during stress the channels become “leaky” and trigger fatal cardiac arrhythmias.
Another gene in which mutations in SIDS cases have been reported is the glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L), and in a study by van Norstrand et al. two out of 221 investigated SIDS cases had a mutation in this gene; the study did not however include any controls [57]. Mutations in GPD1-L are associated with Brugada syndrome, and may be a pathogenic cause for a subset of SIDS via a secondary loss of-function whereby perturbations in GPD1-l precipitate a marked decrease in the peak sodium current.
The CAV3-encoded caveolin-3 is established as a LQTS-associated gene, with mutations producing a gain-of-function, and a LQT3-like molecular/cellular phenotype. The CAV3 gene has been investigated in Norwegian, Caucasian-American and African-American SIDS cases [51, 58]. Mutations in the CAV3 gene were found in two of 201 SIDS cases in the Norwegian study, as well as in one of 182 controls (137 adults and 45 infants, all diseased) [51], while in the study by Cronk et al. mutations were detected only in the African-American cases; in this group three out of 50 SIDS cases had a mutation but this study did not include controls [58].
A novel candidate for a genetic basis for LQT interval is the nitric oxide synthase 1 adaptor protein gene (NOS1AP) as a common SNP in this gene (rs10494366) is associated both with long QT interval and sudden cardiac death [59]. This SNP has been genotyped in 42 Japanese SIDS cases and in 210 adult living controls, and it was found that the TT genotype was associated with SIDS [60].
Mutations in the gene encoding α1-syntrophin are another novel cause of LQTS. The α1-syntrophin gene has been investigated in 292 SIDS cases, and it was found in a total of six different mutations in eight of the cases [61]. The study did not include any controls. After functionally testing only three of the mutations were found to be functionally relevant, and the authors concluded that mutations in the α1-syntrophin gene may be a pathogenic mechanism for a subset of channelopathic SIDS.
Serotonergic network
Serotonin influences a broad range of physiological systems, including the regulation of the respiratory and cardiovascular systems, temperature, and the sleep-wake cycle, and it has been speculated that a dysregulation of the serotonergic network may play a role in SIDS [15, 62, 63]. The complex serotonergic neuronal system is under a ‘bottleneck’ control by a single protein, the serotonin transporter (5-HTT). A variable number of tandem repeat regions (VNTR) in the promoter of this gene have been investigated in several SIDS populations, and an association between the long alleles (L allele and LL genotype) and SIDS has been found in African-American [64], Japanese [65], Norwegian [66] and Italian [67] SIDS populations, but not in a Californian mixed SIDS population [68] or in Swiss SIDS cases [69]. In the VNTR intron 2, the 12-allele has been reported to be associated with SIDS only in an African-American SIDS population, and not in Caucasian SIDS cases [66, 70]. For both polymorphisms, the longest allele is the more effective promoter, associated with increased midbrain serotonin transporters; synaptic serotonin levels would be expected to be lower in those with long alleles. These discrepant results in different geographical populations may indicate that ethnicity is important in determining the significance of the LL-genotype in SIDS, or alternatively it may merely be a reflection of different diagnostic criteria for the diagnosis of SIDS.
A study by Lavezzi et al. correlated genotypes of the 5-HTT promoter VNTR with brainstem pathology [71]. They included 28 SIDS cases and 17 controls (11 infants and 6 fetal deaths, all with known causes of death). The study revealed a correlation between the L-allele, hypoplasia of the raphe nuclei, and maternal smoking during pregnancy in both SIDS and sudden fetal death, and the authors suggest that this combination may result in failure in mediating vital functions, triggering a lethal mechanism in both intrauterine and postnatal life.
An additional polymorphism in the 5-HTT gene is in a putative polyadenylation site in the untranslated region in the 3′ end of the gene. This polymorphism is expected to be a useful tool for assessing the contribution of genetic variation of the serotonin transporter gene to disease, and has been investigated in the same Caucasian and American-African SIDS cases as the other polymorphisms in the 5-HTT gene [72]. However, the genotype distribution for this polymorphism did not differ between SIDS cases and controls.
Several studies have investigated genetic variation in serotonergic receptors in SIDS cases. Rand et al. investigated the gene encoding HTR2A in 96 SIDS cases and 96 living infant controls, and found a total of 21 HTR2A gene variations, however none of them showed a significant association with SIDS [73]. Another study analyzing the coding region of the gene encoding the receptor HTR1A also did not reveal any association between genotype and SIDS risk [74].
The human fifth Ewing variant (FEV) gene is specifically expressed in central 5-HT neurons in the brain, with a predicted role in specification and maintenance of serotonergic neuronal phenotype. One may hypothesize that variations of the FEV gene may underlie abnormalities in the 5-HT system in SIDS cases. This gene has been investigated in 49 African-American and 47 Caucasian SIDS cases, and infant control subjects that were matched for ethnicity and gender to SIDS cases, with a 1:1 match ratio. An association with SIDS was found only in the African-American cases, which had a higher frequency of both the insertion mutation IVS2-191_insA and a higher total variation [75]. A study by Broabeldt et al. however found this mutation in 14% of a cohort of 78 African-American SIDS cases, as well as in 32% of African-American controls (59 diseased infants and 296 living adults from the Human Variation Panel), and concluded that IVS2-191_insA seems to be a common, likely non-pathogenic, variant in the African-American population [76].
Brain development
The serotonergic network imbalance hypothesis clearly indicates a relationship between autonomic nervous system dysregulation and SIDS. In accordance with this, Weese-Mayer et al. investigated genes with relevance to early embryologic development of the autonomic nervous system [77]. Their findings included 11 rare mutations and 4 common polymorphisms in the genes investigated. When investigating the genes separately the allele frequencies did not differ between the 92 SIDS cases and 92 living infant controls that were included in the study, even though in total 15% of the SIDS cases had a rare mutation, compared to only 2% of the controls. This may suggest that specific polymorphisms in these genes may confer a risk of SIDS, perhaps due to a disturbed development of the central nervous system.
Another neuropeptide that plays an important role in the developing nervous system is pituitary adenylate cyclase-activation polypeptide (PACAP). The PACAP gene has been investigated in 46 Caucasian and 46 African-American SIDS, as well as in 92 living infant controls matched for ethnicity [78]. No associations between PACAP and SIDS were found in Caucasians, whereas in the African-American SIDS cases the G allele of rs2856966, leading to the substitution of aspartic acid with glycine, was significantly associated with SIDS. The functional effect of this SNP is unknown.
The PHOX2B gene is also of interest, as it is a key gene in the development of central serotonergic neurons. The entire PHOX2B gene has been investigated in two SIDS populations [79, 80]. Kijima et al. investigated 23 Japanese SIDS cases and did not find any mutations, concluding that PHOX2B gene variants is not likely to be associated with SIDS [79]. Rand et al. investigated 91 American SIDS cases and 91 living infant controls, and found an association to SIDS both for a specific genotype in intron 2, and for the total number of mutations in exon 3 [80]. These authors concluded that the findings suggests that PHOX2b polymorphisms may confer some increased risk for SIDS, most likely in combination with other specific polymorphisms in the cascade of genes mediating autonomic nervous system development.
Another gene of interest with regard to SIDS the TPSYL gene, which is a testis specific Y-like gene on chromosome 6, and is assumed to play a role in brain development. A deleterious mutation in this gene was discovered in a large Amish family that had lost 21 infants in a syndrome named sudden infant death with dysgenesis of the testes syndrome, SIDDT [81]. The TPSYL gene has been investigated in a population of 126 German SIDS cases and 261 adult living controls [82]. Five sequence variations were found, but there were no significant differences between SIDS cases and controls. Thus it seems that genetic analysis of the TPSYL gene is of limited significance in SIDS cases.
Aquaporin-4
Aquaporins provide a way of rapid transporting water across plasma membranes. Aquaporin-4 (AQP4) is the major water channel in the brain and spinal cord, and it has been shown that AQP4 plays a significant role in the development of cererbal edema. Several studies report findings of increased brain weight in SIDS [83, 84], which might be due to a disturbed development of water homeostasis in SIDS. APQ4 gene variation has been investigated in 141 Norwegian SIDS cases and 179 controls (36 diseased infants and 143 diseased adults) [85]. An association between the genotypes CT/TT in rs2075575 and SIDS was found: 84.4% of the SIDS cases had these genotypes, compared to 74.3% of the controls (P < 0.01). An association between brain/body weight ratio and rs2075575 genotype in SIDS cases aged 0.3–12 weeks was also found (median ratio CC 10.6, median ratio CT/TT 12.1, P = 0.014). These findings indicate that rs2075575 may be of significance as a predisposing factor for SIDS, and that the CT/TT genotypes are associated with an increased brain/body weight ratio in infants dying in SIDS during the vulnerable period from birth to 3 months [85].
Tyrosine hydroxylase
Catecholamines, most notably noradrenalin, act as neurotransmitters in the brain stem and basal ganglia, and are, together with serotonin, of importance with regard to respiratory regulation and arousal. Both mechanisms may be impaired in SIDS. Genetic variants of the tyrosine hydroxylase gene influence the production of noradrenalin, and of particular importance is a short tandem repeat marker in intron 1 (THO1). In a study of 172 German Caucasian SIDS cases, the THO1 9.3 allele was shown to be more frequent in SIDS cases than in 390 living infant controls, which may indicate that noradrenalinergic neuronal activity may contribute to the mechanism of death in a subset of SIDS victims [86]. The 9.3 allele results in increased activity of tyrosine hydroxylase [87], and may contribute to an impairment of breathing control or arousal.
Sodium/proton exchanger NHE3
The sodium/proton exchanger NHE3 has an important role in neural control of breathing and it has been shown that over-expression may lead to apnea, which may be a risk factor for SIDS. Preliminary studies demonstrated a significantly higher NHE3 expression in the medulla oblongata in SIDS cases compared to controls [88]. The gene encoding NHE3 has been investigated in 251 German SIDS cases and 220 controls (50 diseased infants and 170 living adults), and three polymorphisms were found (C2403T, G1131A and C1197T). In total, 49% of the SIDS cases had a mutation, compared to 30% of controls [89]. The authors conclude that disturbed breathing control due to a genetic determined over-expression of NHE3 in the brainstem may be involved in SIDS.
Fatty acid metabolism
The most investigated gene with regard to sudden infant death is the MCAD gene, which catalyses the first step in the β-oxidation of fatty acids. The most prevalent mutation causing MCAD deficiency is A985G, and this mutation has been investigated in at least nine studies including 2587 SIDS cases and 4636 controls [90–98]. In this series 16 SIDS cases had the A985G mutation (0.6%), and only two of the cases were homozygous for the mutation, and thus had MCAD deficiency as the cause of death. The G583A mutation has also been investigated, however the mutation was not identified in any cases [99]. Other rare mutations are the splice mutation IVS3-1G in the MCAD gene, detected in one sudden death in an Arabic infant [100], and a deletion mutation in the carnitine transporter OCTN2 gene, detected in two SIDS cases belonging to Hungarian Roma families [101]. Altogether the findings indicate that MCAD deficiency is not a frequent cause of sudden unexpected infant death.
Apolipoprotein E
Apolipoprotein E (ApoE) is a protein primarily involved in lipoprotein and cholesterol metabolism. ApoE is expressed in large amounts in brain tissue, and there is growing evidence that there is an interaction between ApoE, lipid metabolism and immune responses in the central nervous system. The ApoE genotype is controlled by three alleles at a single locus, designated e2, e3 and e4. Becher et al. investigated the ApoE gene in 167 SIDS cases and 371 living healthy newborns, but found that children dying from SIDS had the same allele distribution as controls [102].
Glucose metabolism
Glucokinase and glucose-6-phosphatase are two key enzymes in blood glucose homeostasis, and it has been hypothesised that mutations in their genes may be involved in SIDS. Forsyth et al. have investigated the genes encoding these two enzymes in SIDS cases and controls [103] and found four polymorphisms in the glucokinase gene, and one polymorphism in the glucose-6-phosphatase gene, none of which were associated with SIDS. Another study investigated the promoter region of the gene encoding glucose-6-phosphate transporter (G6PT1) in 170 SIDS cases and 64 diseased infant controls [104]. A novel promoter polymorphism, -259C/T, was found, and the -259T allele frequency was greater in term SIDS infants compared to both term control infants and preterm SIDS infants. The T allele results in a decreased expression of the gene, which the authors speculated makes the infants vulnerable to repeated hypoglycemic episodes [104].
mtDNA
Apathy, increased sleepiness, and a lower activity score have been reported in infants that later died from SIDS [105–107]. A mitochondriopathy resulting in decreased ATP production has therefore been hypothesized in SIDS. A higher substitution frequency observed in the D-loop in SIDS cases compared to controls indicates that mtDNA instability may play a role [108, 109], and at least 60 different mutations have been observed in coding regions of mtDNA in victims of sudden death [110–117]. However, so far no predominant mtDNA mutation has been found to be associated with SIDS, which makes it difficult to conclude that mtDNA is of great importance with regard to sudden unexpected death.
Overheating
Overheating is a well known risk factor for SIDS [118, 119], and it may be speculated that this is due to a genetic predisposition to inadequate thermal regulation. Two proteins important for thermal regulation have been investigated in cases of SIDS: uncoupling protein 1 (UCP1) and heat shock proteins (hsp) [120, 121]. Only one polymorphism in the UCP1 gene has been investigated so far, which was not found to be associated with SIDS [121]. For the heat shock proteins an association between loss of a MspI restriction site in the hsp60 gene and SIDS has been observed, however, only 12 SIDS cases were included in the study [120]. A study sequencing the genes encoding the mitochondrial hsp60/hsp10 chaperone complex in 61 SIDS cases and 50 living adult controls did not disclose any evidence that these genes play a major role in SIDS, even though some variation was identified that may represent genetic modifiers with subtle effects [122].
Detoxification processes
Maternal smoking is a key risk factor for SIDS. Consequently, genes involved in nicotine metabolism have been identified as possible candidate genes for further study of the genetic basis for SIDS. Glutathione S-transferase theta 1 (GSTT1), and cytochrome p-450 1A1 (CYP1A1) are two enzymes involved in metabolic detoxification processes. Polymorphisms in both genes have been reported to impact upon the detoxification process for cigarette smoke, and have also been associated with low birth weight [123–125]. However, when investigating known polymorphisms in these genes in 106 SIDS cases and 106 infant controls matched for gender and ethnicity, no associations were found between any of the polymorphisms and SIDS [126].
Another enzyme metabolising nicotine is flavin-monooxygenase 3 (FMO3). This gene has been investigated in 159 German SIDS cases and 170 living adult controls [127]. It was found that the 472AA genotype occurred more frequently in SIDS cases than in controls (P = 0.005) and was more frequent in those SIDS cases whose mothers reported heavy smoking (P = 0.008). The 472A allele results in reduced enzyme activity [128] and the authors concluded that the polymorphism G472A could act as an additional genetic risk factor for SIDS in children whose mothers smoke.
Copy number variation
Copy number variation (CNV) is duplication or deletion of large segments of DNA, and approximately 2000 CNVs have been described in the human genome. CNVs that effect critical developmental genes may cause disease. Toruner et al. investigated 27 SIDS cases, and found CNV variation in three of them, however the study did not include any controls [129]. One case had a three megabase (Mb) duplication on chromosome 8, as well as a 4.4 Mb deletion on chromosome 22, and based on this, as well as a detailed family history, it is clear in retrospect that this infant had a syndromic genetic disease. Cases 2 and 3 had a 240 kb duplication and a 1.9 Mb deletion, both on chromosome 6. These areas on chromosome 6 include the major cluster of histone genes, and it is plausible that dosage of histone-encoding genes has an impact on their expression, and thus in turn on the global regulation of gene expression. The observed CNVs should however, according to the authors, not be regarded as a causative factor for SIDS, but rather as a predisposing genetic factor [129].
Conclusion
Studies of gene variants in SIDS support the view that there are genetic risk factors predisposing to sudden unexpected death in infants. However, only when a gene has been investigated in large cohorts of SIDS cases from different countries can conclusions with regard to casual effects be considered valid. Furthermore, it is important to distinguish between lethal mutations leading to disease and possibly death, and polymorphisms that are normal gene variants that might be suboptimal in critical situations and thus predispose to sudden infant death (Fig. 1) [130]. Most likely the genetic predisposition to SIDS operates as a polygenic inheritance pattern that predisposes to sudden infant death in combination with environmental risk factors. It appears most likely that the genes involved in the immune system, in the regulation of cardiac function, in the serotonergic network, and in brain function and development are the most important with regard to SIDS [130–132].
Key points
-
1.
In sudden unexpected infant death, it is important to search for potentially lethal mutations. Cases with such mutations should be excluded from the SIDS group.
-
2.
Infants may die from SIDS when three conditions occur at the same time: a genetic predisposition, a vulnerable developmental stage of the CNS and/or the immune system, and a trigger event.
-
3.
The genetic predisposition to SIDS most likely represents a polygenic inheritance.
-
4.
Genes involved in the regulation of the immune system, cardiac function, the serotonergic network and brain function and development have so far emerged as the most important with regard to SIDS.
References
Rognum TO, Saugstad OD. Biochemical and immunological studies in SIDS victims. Clues to understanding the death mechanism. Acta Paediatr Suppl. 1993;82(Suppl 389):82–5.
Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biol Neonate. 1994;65:194–7.
Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115:368–76.
Schoendorf KC, Kiely JL. Relationship of sudden infant death syndrome to maternal smoking during and after pregnancy. Pediatrics. 1992;90:905–8.
Markestad T, Skadberg B, Hordvik E, Morild I, Irgens LM. Sleeping position and sudden infant death syndrome (SIDS): effect of an intervention programme to avoid prone sleeping. Acta Paediatr. 1995;84:375–8.
Arnestad M, Andersen M, Vege A, Rognum TO. Changes in the epidemiological pattern of sudden infant death syndrome in southeast Norway, 1984–1998: implications for future prevention and research. Arch Dis Child. 2001;85:108–15.
Blair PS, Sidebotham P, Berry PJ, Evans M, Fleming PJ. Major epidemiological changes in sudden infant death syndrome: a 20-year population-based study in the UK. Lancet. 2006;367:314–9.
Stoltenberg L, Saugstad OD, Rognum TO. Sudden infant death syndrome victims show local immunoglobulin M response in tracheal wall and immunoglobulin A response in duodenal mucosa. Pediatr Res. 1992;31:372–5.
Thrane PS, Rognum TO, Brandtzaeg P. Up-regulated epithelial expression of HLA-DR and secretory component in salivary glands: reflection of mucosal immunostimulation in sudden infant death syndrome. Pediatr Res. 1994;35:625–8.
Vege A, Rognum TO. Sudden infant death syndrome, infection and inflammatory responses. FEMS Immunol Med Microbiol. 2004;42:3–10.
Vege A, Rognum TO, Scott H, Aasen AO, Saugstad OD. SIDS cases have increased levels of interleukin-6 in cerebrospinal fluid. Acta Paediatr. 1995;84:193–6.
Guntheroth WG. Interleukin-1 as intermediary causing prolonged sleep apnea and SIDS during respiratory infections. Med Hypotheses. 1989;28:121–3.
Vege A, Rognum TO, Anestad G. IL-6 cerebrospinal fluid levels are related to laryngeal IgA and epithelial HLA-DR response in sudden infant death syndrome. Pediatr Res. 1999;45:803–9.
Rognum IJ, Haynes RL, Vege A, Yang M, Rognum TO, Kinney HC. Interleukin-6 and the serotonergic system of the medulla oblongata in the sudden infant death syndrome. Acta Neuropathol. 2009;118:519–30.
Duncan JR, Paterson DS, Hoffman JM, Mokler DJ, Borenstein NS, Belliveau RA, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. 2010;303:430–7.
Mossner R, Daniel S, Schmitt A, Albert D, Lesch KP. Modulation of serotonin transporter function by interleukin-4. Life Sci. 2001;68:873–80.
Schneider PM, Wendler C, Riepert T, Braun L, Schacker U, Horn M, et al. Possible association of sudden infant death with partial complement C4 deficiency revealed by post-mortem DNA typing of HLA class II and III genes. Eur J Pediatr. 1989;149:170–4.
Opdal SH, Vege A, Stave AK, Rognum TO. The complement component C4 in sudden infant death. Eur J Pediatr. 1999;158:210–2.
Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. 1991;4:359–95.
Isenman DE, Young JR. Covalent binding properties of the C4A and C4B isotypes of the fourth component of human complement on several C1-bearing cell surfaces. J Immunol. 1986;136:2542–50.
Stray-Pedersen A, Vege A, Opdal SH, Moberg S, Rognum TO. Surfactant protein A and D gene polymorphisms and protein expression in victims of sudden infant death. Acta Paediatr. 2009;98:62–8.
Summers AM, Summers CW, Drucker DB, Hajeer AH, Barson A, Hutchinson IV. Association of IL-10 genotype with sudden infant death syndrome. Hum Immunol. 2000;61:1270–3.
Opdal SH, Opstad A, Vege A, Rognum TO. IL-10 gene polymorphisms are associated with infectious cause of sudden infant death. Hum Immunol. 2003;64:1183–9.
Moscovis SM, Gordon AE, Hall ST, Gleeson M, Scott RJ, Roberts-Thomsom J, et al. Interleukin 1-beta responses to bacterial toxins and sudden infant death syndrome. FEMS Immunol Med Microbiol. 2004;42:139–45.
Korachi M, Pravica V, Barson AJ, Hutchinson IV, Drucker DB. Interleukin 10 genotype as a risk factor for sudden infant death syndrome: determination of IL-10 genotype from wax-embedded postmortem samples. FEMS Immunol Med Microbiol. 2004;42:125–9.
Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, et al. Interleukin-10 and sudden infant death syndrome. FEMS Immunol Med Microbiol. 2004;42:130–8.
Dashash M, Pravica V, Hutchinson IV, Barson AJ, Drucker DB. Association of sudden infant death syndrome with VEGF and IL-6 gene polymorphisms. Hum Immunol. 2006;67:627–33.
Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, et al. IL6 G-174C associated with sudden infant death syndrome in a Caucasian Australian cohort. Hum Immunol. 2006;67:819–25.
Opdal SH, Rognum TO. The IL6–174G/C polymorphism and sudden infant death syndrome. Hum Immunol. 2007;68:541–3.
Ferrante L, Opdal SH, Vege A, Rognum TO. TNF-alpha promoter polymorphisms in sudden infant death. Hum Immunol. 2008;69:368–73.
Perskvist N, Skoglund K, Edston E, Backstrom G, Lodestad I, Palm U. TNF-alpha and IL-10 gene polymorphisms versus cardioimmunological responses in sudden infant death. Fetal Pediatr Pathol. 2008;27:149–65.
Highet AR, Berry AM, Goldwater PN. Distribution of interleukin-1 receptor antagonist genotypes in sudden unexpected death in infancy (SUDI); unexplained SUDI have a higher frequency of allele 2. Ann Med. 2010;42:64–9.
Ferrante L, Opdal SH, Vege A, Rognum TO. IL-1 gene cluster polymorphisms and sudden infant death syndrome. Hum Immunol. 2010;71:402–6.
Ferrante L, Opdal SH, Vege A, Rognum TO. Cytokine gene polymorphisms and sudden infant death syndrome. Acta Paediatr 2010; (in press).
Reuss E, Fimmers R, Kruger A, Becker C, Rittner C, Hohler T. Differential regulation of interleukin-10 production by genetic and environmental factors–a twin study. Genes Immun. 2002;3:407–13.
Witkin SS, Gerber S, Ledger WJ. Influence of interleukin-1 receptor antagonist gene polymorphism on disease. Clin Infect Dis. 2002;34:204–9.
Rivera-Chavez FA, Peters-Hybki DL, Barber RC, O’Keefe GE. Interleukin-6 promoter haplotypes and interleukin-6 cytokine responses. Shock. 2003;20:218–23.
Hohjoh H, Tokunaga K. Allele-specific binding of the ubiquitous transcription factor OCT-1 to the functional single nucleotide polymorphism (SNP) sites in the tumor necrosis factor-alpha gene (TNFA) promoter. Genes Immun. 2001;2:105–9.
Virchow S, Ansorge N, Rosskopf D, Rubben H, Siffert W. The G protein beta3 subunit splice variant Gbeta3-s causes enhanced chemotaxis of human neutrophils in response to interleukin-8. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:27–32.
Rosskopf D, Koch K, Habich C, Geerdes J, Ludwig A, Wilhelms S, et al. Interaction of Gbeta3 s, a splice variant of the G-protein Gbeta3, with Ggamma- and Galpha-proteins. Cell Signal. 2003;15:479–88.
Opdal SH, Melien O, Rootwelt H, Vege A, Arnestad M, Rognum T. The G protein beta3 subunit 825C allele is associated with sudden infant death due to infection. Acta Paediatr. 2006;95:1129–32.
Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Molecular diagnosis in a child with sudden infant death syndrome. Lancet. 2001;358:1342–3.
Piippo K, Swan H, Pasternack M, Chapman H, Paavonen K, Viitasalo M, et al. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: implications for estimation of disease prevalence and molecular diagnostics. J Am Coll Cardiol. 2001;37:562–8.
Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001;286:2264–9.
Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation. 2001;104:1158–64.
Bajanowski T, Rossi L, Biondo B, Ortmann C, Haverkamp W, Wedekind H, et al. Prolonged QT interval and sudden infant death–report of two cases. Forensic Sci Int. 2001;115:147–53.
Christiansen M, Tonder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutations in the HERG K+-ion channel: a novel link between long QT syndrome and sudden infant death syndrome. Am J Cardiol. 2005;95:433–4.
Arnestad M, Vege A, Rognum TO, Isaksen CV. Sudden infant death syndrome not caused by Norwegian Jervell and Lange-Nielsen mutations. Am J Med Genet A. 2005;134:459–60.
Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest. 2006;116:430–5.
Wedekind H, Bajanowski T, Friederich P, Breithardt G, Wulfing T, Siebrands C, et al. Sudden infant death syndrome and long QT syndrome: an epidemiological and genetic study. Int J Legal Med. 2006;120:129–37.
Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–7.
Van Norstrand DW, Tester DJ, Ackerman MJ. Overrepresentation of the proarrhythmic, sudden death predisposing sodium channel polymorphism S1103Y in a population-based cohort of African-American sudden infant death syndrome. Heart Rhythm. 2008;5:712–5.
Turillazzi E, La Rocca G, Anzalone R, Corrao S, Neri M, Pomara C, et al. Heterozygous nonsense SCN5A mutation W822X explains a simultaneous sudden infant death syndrome. Virchows Arch. 2008;453:209–16.
Millat G, Kugener B, Chevalier P, Chahine M, Huang H, Malicier D, et al. Contribution of long-QT syndrome genetic variants in sudden infant death syndrome. Pediatr Cardiol. 2009;30:502–9.
Otagiri T, Kijima K, Osawa M, Ishii K, Makita N, Matoba R, et al. Cardiac ion channel gene mutations in sudden infant death syndrome. Pediatr Res. 2008;64:482–7.
Tester DJ, Dura M, Carturan E, Reiken S, Wronska A, Marks AR, et al. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart Rhythm. 2007;4:733–9.
Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116:2253–9.
Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4:161–6.
Aarnoudse AJ, Newton-Cheh C, de Bakker PI, Straus SM, Kors JA, Hofman A, et al. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation. 2007;116:10–6.
Osawa M, Kimura R, Hasegawa I, Mukasa N, Satoh F. SNP association and sequence analysis of the NOS1AP gene in SIDS. Leg Med (Tokyo). 2009;11(Suppl 1):S307–8.
Cheng J, Van Norstrand DW, Medeiros-Domingo A, Valdivia C, Tan BH, Ye B, et al. Alpha1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current. Circ Arrhythm Electrophysiol. 2009;2:667–76.
Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol. 2009;4:517–50.
Paterson DS, Hilaire G, Weese-Mayer DE. Medullary serotonin defects and respiratory dysfunction in sudden infant death syndrome. Respir Physiol Neurobiol. 2009;168:133–43.
Weese-Mayer DE, Berry-Kravis EM, Maher BS, Silvestri JM, Curran ME, Marazita ML. Sudden infant death syndrome: association with a promoter polymorphism of the serotonin transporter gene. Am J Med Genet. 2003;117A:268–74.
Narita N, Narita M, Takashima S, Nakayama M, Nagai T, Okado N. Serotonin transporter gene variation is a risk factor for sudden infant death syndrome in the Japanese population. Pediatrics. 2001;107:690–2.
Opdal SH, Vege A, Rognum TO. Serotonin transporter gene variation in sudden infant death syndrome. Acta Paediatr. 2008;97:861–5.
Nonnis Marzano F, Maldini M, Filonzi L, Lavezzi AM, Parmigiani S, Magnani C, et al. Genes regulating the serotonin metabolic pathway in the brain stem and their role in the etiopathogenesis of the sudden infant death syndrome. Genomics. 2008;91:485–91.
Paterson DS, Trachtenberg FL, Thompson EG, Belliveau RA, Beggs AH, Darnall R, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. Jama. 2006;296:2124–32.
Haas C, Braun J, Bar W, Bartsch C. No association of serotonin transporter gene variation with sudden infant death syndrome (SIDS) in Caucasians. Leg Med (Tokyo). 2009;11(Suppl 1):210–2.
Weese-Mayer DE, Zhou L, Berry-Kravis EM, Maher BS, Silvestri JM, Marazita ML. Association of the serotonin transporter gene with sudden infant death syndrome: a haplotype analysis. Am J Med Genet. 2003;122A:238–45.
Lavezzi AM, Casale V, Oneda R, Weese-Mayer DE, Matturri L. Sudden infant death syndrome and sudden intrauterine unexplained death: correlation between hypoplasia of raphe nuclei and serotonin transporter gene promoter polymorphism. Pediatr Res. 2009;66:22–7.
Maher BS, Marazita ML, Rand C, Zhou L, Berry-Kravis EM, Weese-Mayer DE. 3′ UTR polymorphism of the serotonin transporter gene and sudden infant death syndrome: haplotype analysis. Am J Med Genet A. 2006;140:1453–7.
Rand CM, Berry-Kravis EM, Fan W, Weese-Mayer DE. HTR2A variation and sudden infant death syndrome: a case-control analysis. Acta Paediatr. 2009;98:58–61.
Morley ME, Rand CM, Berry-Kravis EM, Zhou L, Fan W, Weese-Mayer DE. Genetic variation in the HTR1A gene and sudden infant death syndrome. Am J Med Genet A. 2008;146:930–3.
Rand CM, Berry-Kravis EM, Zhou L, Fan W, Weese-Mayer DE. Sudden infant death syndrome: rare mutation in the serotonin system FEV gene. Pediatr Res. 2007;62:180–2.
Broadbelt KG, Barger MA, Paterson DS, Holm IA, Haas EA, Krous HF, et al. Serotonin-related FEV gene variant in the sudden infant death syndrome is a common polymorphism in the African-American population. Pediatr Res. 2009;66:631–5.
Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Curran ME, Silvestri JM, et al. Sudden infant death syndrome: case-control frequency differences at genes pertinent to early autonomic nervous system embryologic development. Pediatr Res. 2004;56:391–5.
Cummings KJ, Klotz C, Liu WQ, Weese-Mayer DE, Marazita ML, Cooper ME, et al. Sudden infant death syndrome (SIDS) in African Americans: polymorphisms in the gene encoding the stress peptide pituitary adenylate cyclase-activating polypeptide (PACAP). Acta Paediatr. 2009;98:482–9.
Kijima K, Sasaki A, Niki T, Umetsu K, Osawa M, Matoba R, et al. Sudden infant death syndrome is not associated with the mutation of PHOX2B gene, a major causative gene of congenital central hypoventilation syndrome. Tohoku J Exp Med. 2004;203:65–8.
Rand CM, Weese-Mayer DE, Zhou L, Maher BS, Cooper ME, Marazita ML, et al. Sudden infant death syndrome: Case-control frequency differences in paired like homeobox (PHOX) 2B gene. Am J Med Genet A. 2006;140:1687–91.
Puffenberger EG, Hu-Lince D, Parod JM, Craig DW, Dobrin SE, Conway AR, et al. Mapping of sudden infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification of TSPYL loss of function. Proc Natl Acad Sci USA. 2004;101:11689–94.
Hering R, Frade-Martinez R, Bajanowski T, Poets CF, Tschentscher F, Riess O. Genetic investigation of the TSPYL1 gene in sudden infant death syndrome. Genet Med. 2006;8:55–8.
Aranda FJ, Teixeira F, Becker LE. Assessment of growth in sudden infant death syndrome. Neuroepidemiology. 1990;9:95–105.
Kadhim H, Sebire G, Khalifa M, Evrard P, Groswasser J, Franco P, et al. Incongruent cerebral growth in sudden infant death syndrome. J Child Neurol. 2005;20:244–6.
Opdal SH, Vege A, Stray-Pedersen A, Rognum TO. Aquaporin-4 gene variation and sudden infant death syndrome. Pediatr Res 2010;68:48–51.
Klintschar M, Reichenpfader B, Saternus KS. A functional polymorphism in the tyrosine hydroxylase gene indicates a role of noradrenalinergic signaling in sudden infant death syndrome. J Pediatr. 2008;153:190–3.
Zhang L, Rao F, Wessel J, Kennedy BP, Rana BK, Taupenot L, et al. Functional allelic heterogeneity and pleiotropy of a repeat polymorphism in tyrosine hydroxylase: prediction of catecholamines and response to stress in twins. Physiol Genomics. 2004;19:277–91.
Wiemann M, Frede S, Tschentscher F, Kiwull-Schone H, Kiwull P, Bingmann D, et al. NHE3 in the human brainstem: implication for the pathogenesis of the sudden infant death syndrome (SIDS)? Adv Exp Med Biol. 2008;605:508–13.
Poetsch M, Nottebaum BJ, Wingenfeld L, Frede S, Vennemann M, Bajanowski T. Impact of sodium/proton exchanger 3 gene variants on sudden infant death syndrome. J Pediatr 2010;156:44–8.
Miller M, Brooks J, Forbes N, Insel R. Frequency of G-985 mutation in medium chain acyl-coenzyme A dehydrogenase (MCAD) deficiency in sudden infant death syndrome (SIDS). Prog Clin Biol Res. 1992;375:495–8.
Arens R, Gozal D, Jain K, Muscati S, Heuser ET, Williams JC, et al. Prevalence of medium-chain acyl-coenzyme A dehydrogenase deficiency in the sudden infant death syndrome. J Pediatr. 1993;122(5 Pt 1):715–8.
Lundemose JB, Gregersen N, Kolvraa S, Norgaard Pedersen B, Gregersen M, Helweg-Larsen K, et al. The frequency of a disease-causing point mutation in the gene coding for medium-chain acyl-CoA dehydrogenase in sudden infant death syndrome. Acta Paediatr. 1993;82:544–6.
Dundar M, Lanyon WG, Connor JM. Scottish frequency of the common G985 mutation in the medium-chain acyl-CoA dehydrogenase (MCAD) gene and the role of MCAD deficiency in sudden infant death syndrome (SIDS). J Inherit Metab Dis. 1993;16:991–3.
Penzien JM, Molz G, Wiesmann UN, Colombo JP, Buhlmann R, Wermuth B. Medium-chain acyl-CoA dehydrogenase deficiency does not correlate with apparent life-threatening events and the sudden infant death syndrome: results from phenylpropionate loading tests and DNA analysis. Eur J Pediatr. 1994;153:352–7.
Opdal SH, Vege A, Saugstad OD, Rognum TO. Is the medium-chain acyl-CoA dehydrogenase G985 mutation involved in sudden infant death in Norway? Eur J Pediatr. 1994;154:166–7.
Santer R, Gregersen N, Tanaka K, Hinck-Kneip C, Krawinkel M, Schaub J. The prevalence of the G985 allele of medium-chain acyl-CoA dehydrogenase deficiency among sudden infant death victims and healthy newborns in northern Germany. Eur J Pediatr. 1995;154:497.
Lecoq I, Mallet E, Bonte JB, Travert G. The A985 to G mutation of the medium-chain acyl-CoA dehydrogenase gene and sudden infant death syndrome in Normandy. Acta Paediatr. 1996;85:145–7.
Boles RG, Buck EA, Blitzer MG, Platt MS, Cowan TM, Martin SK, et al. Retrospective biochemical screening of fatty acid oxidation disorders in postmortem livers of 418 cases of sudden death in the first year of life. J Pediatr. 1998;132:924–33.
Ryan A, McGill J, Mountain H. Rapid testing for the MCAD G583A mutation, by PCR-mediated site directed mutagenesis, in an Australian population of SIDS patients. Dis Markers. 1997;13:131–4.
Korman SH, Gutman A, Brooks R, Sinnathamby T, Gregersen N, Andresen BS. Homozygosity for a severe novel medium-chain acyl-CoA dehydrogenase (MCAD) mutation IVS3–1G > C that leads to introduction of a premature termination codon by complete missplicing of the MCAD mRNA and is associated with phenotypic diversity ranging from sudden neonatal death to asymptomatic status. Mol Genet Metab. 2004;82:121–9.
Melegh B, Bene J, Mogyorosy G, Havasi V, Komlosi K, Pajor L, et al. Phenotypic manifestations of the OCTN2 V295X mutation: sudden infant death and carnitine-responsive cardiomyopathy in Roma families. Am J Med Genet A. 2004;131:121–6.
Becher JC, Keeling JW, Bell J, Wyatt B, McIntosh N. Apolipoprotein E e4 and its prevalence in early childhood death due to sudden infant death syndrome or to recognised causes. Early Hum Dev. 2008;84:549–54.
Forsyth L, Hume R, Howatson A, Busuttil A, Burchell A. Identification of novel polymorphisms in the glucokinase and glucose-6-phosphatase genes in infants who died suddenly and unexpectedly. J Mol Med. 2005;83:610–8.
Forsyth L, Scott HM, Howatson A, Busuttil A, Hume R, Burchell A. Genetic variation in hepatic glucose-6-phosphatase system genes in cases of sudden infant death syndrome. J Pathol. 2007;212:112–20.
Kahn A, Groswasser J, Rebuffat E, Sottiaux M, Blum D, Foerster M, et al. Sleep and cardiorespiratory characteristics of infant victims of sudden death: a prospective case-control study. Sleep. 1992;15:287–92.
Andersen M, Arnestad M, Rognum TO, Vege A. Cot death in eastern part of Norway 1984–92. Tidsskr Nor Laegeforen. 1995;115:34–7.
Kelmanson IA. An assessment of behavioural characteristics in infants who died of sudden infant death syndrome using the Early Infancy Temperament Questionnaire. Acta Paediatr. 1996;85:977–80.
Hofmann S, Jaksch M, Bezold R, Mertens S, Aholt S, Paprotta A, et al. Population genetics and disease susceptibility: characterization of central European haplogroups by mtDNA gene mutations, correlation with D loop variants and association with disease. Hum Mol Genet. 1997;6:1835–46.
Opdal SH, Rognum TO, Vege A, Stave AK, Dupuy BM, Egeland T. Increased number of substitutions in the D-loop of mitochondrial DNA in the sudden infant death syndrome. Acta Paediatr. 1998;87:1039–44.
Santorelli FM, Schlessel JS, Slonim AE, DiMauro S. Novel mutation in the mitochondrial DNA tRNA glycine gene associated with sudden unexpected death. Pediatr Neurol. 1996;15:145–9.
Ogle RF, Christodoulou J, Fagan E, Blok RB, Kirby DM, Seller KL, et al. Mitochondrial myopathy with tRNA(Leu(UUR)) mutation and complex I deficiency responsive to riboflavin. J Pediatr. 1997;130:138–45.
Opdal SH, Rognum TO, Torgersen H, Vege A. Mitochondrial DNA point mutations detected in four cases of sudden infant death syndrome. Acta Paediatr. 1999;88:957–60.
Opdal SH, Vege Å, Egeland T, Musse MA, Rognum TO. Possible role of mtDNA mutations in sudden infant death. Pediatr Neurol. 2002;27:23–9.
Arnestad M, Opdal SH, Musse MA, Vege A, Rognum TO. Are substitutions in the first hypervariable region of the mitochondrial DNA displacement-loop in sudden infant death syndrome due to maternal inheritance? Acta Paediatr. 2002;91:1060–4.
Divne AM, Råsten-Almqvist P, Rajs J, Gyllensten U, Allen M. Analysis of the mitochondrial genome in the sudden infant death syndrome. Acta Paediatr. 2003;92:386–8.
Arnestad M, Opdal SH, Vege A, Rognum TO. A mitochondrial DNA polymorphism associated with cardiac arrhythmia investigated in sudden infant death syndrome. Acta Paediatr. 2007;96:206–10.
Opdal SH, Vege A, Arnestad M, Musse MA, Rognum TO. Mitochondrial tRNA genes and flanking regions in sudden infant death syndrome. Acta Paediatr. 2007;96:211–4.
Stanton AN. Sudden infant death. Overheating and cot death. Lancet. 1984;2:1199–201.
Sawczenko A, Fleming PJ. Thermal stress, sleeping position, and the sudden infant death syndrome. Sleep. 1996;19:S267–70.
Rahim RA, Boyd PA, Ainslie Patrick WJ, Burdon RH. Human heat shock protein gene polymorphisms and sudden infant death syndrome. Arch Dis Child. 1996;75:451–2.
Fatemi A, Item C, Stockler-Ipsiroglu S, Ipsiroglu O, Sperl W, Patsch W, et al. Sudden infant death: no evidence for linkage to common polymorphisms in the uncoupling protein-1 and the beta3-adrenergic receptor genes. Eur J Pediatr. 2002;161:337–9.
Bross P, Li Z, Hansen J, Hansen JJ, Nielsen MN, Corydon TJ, et al. Single-nucleotide variations in the genes encoding the mitochondrial Hsp60/Hsp10 chaperone system and their disease-causing potential. J Hum Genet. 2007;52:56–65.
Ishibe N, Wiencke JK, Zuo ZF, McMillan A, Spitz M, Kelsey KT. Susceptibility to lung cancer in light smokers associated with CYP1A1 polymorphisms in Mexican- and African-Americans. Cancer Epidemiol Biomarkers Prev. 1997;6:1075–80.
Bartsch H, Nair U, Risch A, Rojas M, Wikman H, Alexandrov K. Genetic polymorphism of CYP genes, alone or in combination, as a risk modifier of tobacco-related cancers. Cancer Epidemiol Biomarkers Prev. 2000;9:3–28.
Wang X, Zuckerman B, Pearson C, Kaufman G, Chen C, Wang G, et al. Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. JAMA. 2002;287:195–202.
Rand CM, Weese-Mayer DE, Maher BS, Zhou L, Marazita ML, Berry-Kravis EM. Nicotine metabolizing genes GSTT1 and CYP1A1 in sudden infant death syndrome. Am J Med Genet A. 2006;140:1447–52.
Poetsch M, Czerwinski M, Wingenfeld L, Vennemann M, Bajanowski T. A common FMO3 polymorphism may amplify the effect of nicotine exposure in sudden infant death syndrome (SIDS). Int J Legal Med 2010; (in press).
Koukouritaki SB, Hines RN. Flavin-containing monooxygenase genetic polymorphism: impact on chemical metabolism and drug development. Pharmacogenomics. 2005;6:807–22.
Toruner GA, Kurvathi R, Sugalski R, Shulman L, Twersky S, Pearson PG, et al. Copy number variations in three children with sudden infant death. Clin Genet. 2009;76:63–8.
Opdal SH, Rognum TO. The sudden infant death syndrome gene: does it exist? Pediatrics. 2004;114:e506–12.
Hunt CE, Hauck FR. Sudden infant death syndrome. CMAJ. 2006;174:1861–9.
Weese-Mayer DE, Ackerman MJ, Marazita ML, Berry-Kravis EM. Sudden Infant Death Syndrome: review of implicated genetic factors. Am J Med Genet A. 2007;143A:771–88.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Opdal, S.H., Rognum, T.O. Gene variants predisposing to SIDS: current knowledge. Forensic Sci Med Pathol 7, 26–36 (2011). https://doi.org/10.1007/s12024-010-9182-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12024-010-9182-9