
Abstract
The review assesses immunohistochemical findings of somatostatin receptors and of metalloproteinases in different pituitary adenoma types and the significance of molecular genetic data. Current evidence does not support routine immunohistochemical assessment of somatostatin or dopamine receptor subtype expression on hormone-secreting or nonfunctioning pituitary adenomas. Further prospective studies are needed to define its role for clinical decision making. Until then we suggest to restrict membrane receptor profiling to individual cases or for study purposes. The problems of adenoma expansion and invasion are discussed. Despite partially contradictory publications, proteases clearly play a major role in permission of infiltrative growth of pituitary adenomas. Therefore, detection of at least MMP-2, MMP-9, TIMP-2, and uPA seems to be justified. Molecular characterization is important for familial adenomas, adenomas in MEN, Carney complex, and McCune-Albright syndrome and can gain insight into pathogenesis of sporadic adenomas.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The histopathological, immunocytochemical, and electron microscopical features of the different adenoma types according to the current WHO classification of pituitary adenomas [36] and their clinical significance are subjects of a recent review in this journal [53]. In this second review, further methods are discussed. Immunostainings of somatostatin receptors and dopamine receptors in the different adenoma types may be important for further treatment of patients with somatostatin analogs or dopamine agonists. Metalloproteinases influence the invasive character of adenomas. Finally, molecular genetic analyses deliver important results in different adenomas to account for familial adenomas, adenomas in MEN, McCune-Albright syndrome, Carney complex, gsp mutations, X-linked acrogigantism syndrome, and mutations of the deubiquitinase gene USP8. Details will be reviewed.
Expression of Membrane Receptors
Several somatostatin receptor (SSTR) and dopamine receptor (DR) subtypes are expressed in normal human pituitaries [41, 42]. These receptors are also present on a subset of hormone-secreting and nonfunctioning pituitary adenoma cells [10, 21, 41, 42] and can be pharmacologically targeted to control hormone over-secretion and tumor growth.
Information about the expression pattern of specific receptors on adenoma cells may predict the therapeutic outcome to receptor agonist treatment and may thus be helpful in therapeutic decision making. In this respect, the immunohistochemical expression pattern of SSTRs on somatotroph adenoma cells is the best studied membrane receptor system so far. Somatostatin analogs like octreotide and lanreotide are widely used in the medical treatment of acromegaly and exert their therapeutic response predominantly through activation of SSTR2A. Several studies reported a significant correlation between the in vitro immunohistochemical SSTR2A expression on somatotroph adenoma tissue with the clinical in vivo response [2, 11, 17, 21, 33]. There are, however, potential confounders that must be considered, e.g., material representativeness, heterogeneous receptor expression, antibody specificity, scoring system, pretreatment with somatostatin analogs, etc. This may well limit in the individual patient the applicability of routine immunohistochemical assessment of SSTR expression for therapeutic decision making. Given that disease activity and the therapeutic response to somatostatin analogs can be easily and timely monitored by measuring GH and/or IGF-1, there appears to be no added clinical value by immunohistochemical receptor profiling. To date, there is no evidence that routine immunohistochemical assessment of SSTR subtype or DR2 expression patterns may distinctly affect therapeutic decision making in patients with hormone-secreting pituitary adenomas.
In nonfunctioning adenomas, the clinical situation is somewhat different. Growth control is the prime therapeutic aim, but there are no markers that could easily and promptly predict the efficacy of receptor agonists like dopamine agonists or somatostatin analogs on controlling adenoma growth. Unfortunately, existing data do not support a predictive value of SSTR subtype or DR2 expression profiles on growth control by the respective agonists that would justify their routine immunohistochemical assessment [7, 22].
Expression of certain membrane receptors may be associated with adenoma phenotypes with distinct biological behavior [33, 38, 60] which could be of prognostic value and thus may influence therapeutic decisions or follow-up care. This, however, has not been prospectively studied in any adenoma subtype so far.
At present immunohistochemical evaluation of SSTR subtype or DR2 expression on hormone-secreting or nonfunctioning pituitary adenomas does not seem to impact on clinical decision making that would warrant routine assessment and the extra costs.
Perisellar Extension of Pituitary Adenomas
Extension of pituitary adenomas into the perisellar compartments is the main cause of incomplete tumor resection in transsphenoidal surgery. Especially the involvement of the parasellar space (synonymous: cavernous sinus, CS) frequently limits surgical success {17055}. However, the assessment of “parasellar infiltration” varies significantly in different investigations dealing with properties of “infiltrative” vs. “non-infiltrative” pituitary adenomas: radiological (preoperative) vs. surgical (intraoperative) vs. histological (in case the anterior sellar envelope has been investigated) vs. postoperative imaging [1, 9, 27, 39, 48]. Therefore, the comparability of case allocation in different series may not be given and explain some of the contradictory data published so far. Moreover, insufficient surgery may be a cause for primarily accessible tumor remnants [37].
Expansion vs. Invasion
Intrasellar tumor growth may be directed by anatomical variations, e.g., a strong diaphragma sellae [6, 32]. Therefore, it still is a matter of debate, whether extension of pituitary adenomas into CS is a consequence of their biological properties only or whether it may be facilitated by low anatomical resistance of the medial wall of CS against chronic tumor growth [25, 31, 65]. On the other hand different markers have been identified, which may correlate to the aggressiveness of adenomas [15, 16, 28, 40, 54]. Parasellar extension of pituitary adenoma may therefore be a result of expansion and/or invasion.
Invasion and Proteases
“True” invasion of tumors into their surroundings is a three-step process starting first with receptor-mediated adhesion of tumors cells, which involves cell-adhesion molecules. Secondly, the neighboring extracellular matrix (ECM) is degraded by proteases (synonymous: proteinase, endopeptidase). Lastly, the tumor cells migrate into the intercellular space [13]. Endopeptidases are distinguished by their active site into four main classes: serine, cysteine, aspartic, and metallo-endopeptidases and play a physiological role in tissue remodelling [20]. Serine-proteases (urokinase-type plasminogen activator, uPA, and tissue type plasminogen activator, tPA) gain their proteolytic activity by transforming plasminogen to plasmin, which bears a broad substrate-specificity for glykoproteins and precursors of metalloproteinases. The activity of uPA is mediated by a cellular surface receptor (uPAR) and modulated by plasminogen activator inhibitors PAI-1 and PAI-2 [13, 46]. Metalloproteinases (MMP) are zinc-containing proteases specified by their substrate-specificity: e.g., collagenases (MMP-1 and MMP-9), gelatinase (MMP-2), and stromelysine (MMP-3). They are regulated by tissue inhibitors of metalloproteinases (TIMP); physiologically, they also play a permissive role in angiogenesis [46].
Interaction with ECM and Sampling Artefact
Since proteases may mainly be overexpressed adjacent to their substrate (ECM) [15, 63] in pituitary surgery, a sampling artefact may result with sampling of the accessible sellar tumor areas only, whereas the lateral parts of the tumors are rather sucked away.
Type III collagen is the main component of fetal CS walls [31]; the lateral wall of the sella turcica (synonymous: medial wall of CS) in adults mainly is composed of type IV collagen [23, 44]. Therefore, MMP-9 which is said to specifically ferment type IV collagen is a candidate to be a biological marker of laterally infiltrative pituitary adenomas [14].
Expression of Proteases and Invasion of Pituitary Adenomas
Overexpression of MMP-9 in invasive pituitary adenomas compared to non-invasive adenomas has been reported by different groups [14, 18, 19, 46, 61], as well as its mRNA [50]. Other groups could not confirm these findings [23, 64, 65]. Using Western blot analysis, MMP-1, MMP-2, and MMP-3 could not be correlated to invasiveness of 28 pituitary adenomas [4]; for MMP-2, this was confirmed immunohistochemically in 75 tumors [24]. Contrarily, one group reported an overexpression of MMP-2 in 12 invasive tumors compared to 42 non-invasive adenomas and of its mRNA [35]. MMP-1 was overexpressed in 50 non-secreting adenomas compared to 19 silent ACTH cell adenomas, which are supposed to be more aggressive. A correlation to invasiveness was not given in this report [40]. Others reported that in cases with pituitary adenomas, those with a homozygous 2G allele of the promoter region of the MMP-1 gene tend to be invasive more often [3]. Sophisticated analysis of different secreting and non-secreting adenoma types with respect to their infiltrative behavior uncovers peculiarities of the adenoma subgroups concerning their pattern of expression of proteases, e.g., tendency for overpression of MMP-2 and MMP-9 in ACTH-secreting tumors, which often are infiltrative although of small size [26] compared to their non-infiltrative counterparts and, likewise, of uPA in infiltrative non-secreting adenomas [24]. A protective role of TIMP-2 and TIMP-3 against infiltrative behavior of pituitary adenomas seems to be elucidated by the relative overexpression of both factors in non-invasive adenomas [4], which for TIMP-2 is supported by others [24].
Determination of MMP-2, MMP-9, TIMP-2, and uPA in a large scale of pituitary adenomas, like in tumor registries, may be helpful, when modifications of tumor classification as proposed in part I of the manuscript are implemented. By this way, the limitations of the previous data concerning (a) definition of infiltration, (b) sampling artefact, (c) underestimation of tumor type, and (d) surgical technique may be overcome, and thus the correlation of proteases to infiltrative adenoma growth could be further elucidated. However, a systemic therapeutic approach for modulation of proteases is not yet available and principally would also act on their physiologic role in tissue remodelling.
Genetic Analysis of Pituitary Tumor Samples
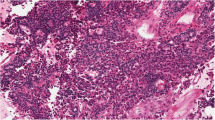
Pituitary adenomas arise in a sporadic setting or rarely as part of the hereditary syndromes. Clear structural especially histopathological differences between sporadic and familial or MEN-associated adenomas do not exist (Figs.. 1 and. 2). Molecular characterization has allowed for a deeper understanding of the pathogenesis both in familial and isolated pituitary adenomas. Such information may be clinically important with respect to the choice of treatment and the prognosis. Furthermore, it allows for early detection of other manifestations in neoplastic syndromes as well as screening of family members. Recent publications have summarized suggestions whom to screen for germline mutations [29, 56], whereas molecular analysis of tumor samples in routine use is less established. This part aims to summarize the molecular pathology from a pathologist’s perspective, trying to answer the questions, whether if any genetic investigation of tumor samples is required in routine care.

Sparsely granulated GH cell adenoma in MEN: diffusely arranged medium-sized cell with slightly eosinophilic cytoplasm and fibrous bodies. Hematoxylin-eosin stain, magnification ×440

Sporadic sparsely granulated GH cell adenoma: same features as Fig. 1. Hematoxylin-eosin stain, magnification ×300
Germline Syndromes
To raise suspicion for germline mutations, a thorough history of the patient with a pituitary adenoma is necessary, with special emphasis on other affected family members and further clinical manifestations related to the known syndromes.
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal-dominant disorder with a mutation in the menin gene. Affected patients are especially prone to develop parathyroid hyperplasia (>90 %) and pancreatic neuroendocrine neoplasia (30–40 %), next to pituitary adenomas (25–40 %). Comparison of MEN1-positive pituitary tumors with an unselected group of non-MEN1 sporadic pituitary adenomas revealed that MEN1 tumors were significantly larger and more often invasive by histology [59]. Whereas no predominance of any hormone-producing subtype was found, plurihormonal and multiple adenomas were more frequent in MEN1. Therefore, in such rare conditions, either germline or tumor sample investigations for MEN1 mutations may be justified. Somatic MEN1 mutations are very rare [49].
More recently, a germline nonsense mutation in the cyclin-dependent kinase inhibitor 1B (CDKN1B or p27/KIP1) was found in a family with MEN1-like condition that was negative for menin mutations [45]. MEN1-related tumors in this family included acromegaly, hyperparathyreoidism, and renal angiomyolipoma. The mutation resulted in the loss of functional p27, which plays an important role as a tumor-suppressor. The condition was subsequently termed MEN4, and also includes the development of neuroendocrine neoplasia. However, mutations of CDKN1B appear to be exceptionally rare in families with familial isolated pituitary adenomas, as only 2 germline changes were found among 88 AIP-negative families with familial isolated pituitary adenomas [57]. Screening for MEN4 should therefore be limited to patients with MEN1-like syndrome, found to be negative for menin mutations.
The syndrome of familial isolated pituitary adenomas is defined as familial presentation of any type of pituitary adenoma in the absence of clinical and genetic evidence for syndromes like MEN 1 and Carney complex [62]. It is inherited in an autosomal-dominant pattern with variable penetrance. Affected family members may either experience homogenous demonstration of the same type of pituitary adenoma or demonstrate heterogenous expression of different pituitary tumor types in the same family. PRL-secreting (40 %) and GH-secreting (30 %) adenomas are the most common types, with somatoprolactinomas (7 %), gonadotropinomas (4 %), ACTH-secreting (4 %) adenomas, and TSH-secreting adenomas much rarer [62]. Inactivating mutations in the gene for the aryl-hydrocarbon receptor interacting protein (AIP) are found in approximately 25 % of patients with familial isolated pituitary adenomas, whereas the genetic cause for the remaining familial isolated pituitary adenoma patients is currently unclear. Over 50 different mutations are spread over the entire length of the gene, without any genotype-phenotype correlation. Although the exact molecular mechanisms remain to be elucidated, loss of heterozygosity in tumor tissues suggests the function as a tumor-suppressor gene. AIP mutations in familial isolated pituitary adenoma patients are associated with a younger onset of disease and a more aggressive tumor behavior, as well as difficulties in therapeutic control, e.g., more frequent resistance to medical treatment with dopamine agonists and somatostatin analogs in prolactinomas and acromegaly, respectively. In addition to patients with a pituitary adenoma and a positive family history, screening in apparently sporadic tumors may be justified in young patients <30 years of age, with mutations present in approximately 12 % (20 % in pediatric patients) [8].
Carney complex is a rare, dominantly inherited disease characterized by multiple manifestations including acromegaly. Diagnostic criteria include spotty skin pigmentation (lips, conjunctiva and inner or outer canthi, vaginal and penil mucosa), myxomas (cutaneous, mucosal, heart, breast), schwannomas of the peripheral nerves, large cell calcifying Sertoli cell tumors, (epitheloid) blue nevus, breast ductal adenomas, osteochondromyxomas, and various endocrine tumors [8]. The latter contain GH-secreting pituitary adenomas, thyroid carcinoma or nodules, and primary pigmented nodular adrenocortical disease (PPNAD). It is the PRKAR1A gene coding for the type I-A regulatory subunit (R-Ia) of protein kinase A (PKA) (situated at chromosome 17q and referred to as CNC1 locus) that is mutated in more than 70 % of patients with Carney syndrome. A second genetic locus associated with Carney complex in PRKAR1A-negative patients is a 10MB region at chromosome 2p16 (termed CNC2 locus). The genes in this region responsible for the phenotype are currently unknown.
Several lines of evidence suggest a role of PKA as a tumor-suppressor gene; in cells carrying mutations of PKA, its activity is irregularly activated by cAMP. In patients with GH-secreting pituitary adenomas, any combination with other Carney manifestations should trigger suspicion of the syndrome and subsequent genetic investigation.
McCune-Albright syndrome is caused by mosaicism for the gsp mutation (a mutation of the Gsalpha gene), resulting in constitutive activation of the cAMP pathway. The disorder is characterized by polyostotic fibrous dysplasia, pigmented skin lesions, and over-activity of endocrine organs [12]. Autonomous GH secretion is found in up to 20 % of patients, but defined pituitary adenomas are rarely detected. Screening for gsp mutations is therefore rarely required in this syndrome.
Sporadic Tumors
The gsp mutation found in McCune-Albright syndrome has also been demonstrated in a relevant proportion of sporadic pituitary tumors, occurring in approximately 30–40 % of GH-secreting pituitary adenomas. Tumors with this mutation tend to be smaller and have a higher GH secretion and are more sensitive to dopamine- or somatostatin-induced GH inhibition [55]. As there are no clear cutoffs to determine treatment response, investigation of gsp mutations in sporadic tumors is not part of the routine procedures.
X-linked acrogigantism is a new syndrome of gigantism due to GH excess, caused by microduplications on chromosome Xq26.3 [58]. It was initially observed in 13 patients with disease onset during early childhood, including 4 patients from two unrelated kindreds and 9 patients with sporadic disease. In a subsequent study including a total of 18 patients (13 sporadic), rapid growth started as early as 2 months (median 12 months), with a median height and weight standard deviation scores (SDS) of >+3.9 SDS at diagnosis after a median of 27 months [5]. The inheritance pattern in the two families was dominant, and all patients demonstrated marked hypersecretion of GH and usually PRL due to a pituitary macroadenoma or hyperplasia. Disease control was challenging due to tumor size and frequent partial resistance to somatostatin analogs. In another study of 208 patients with gigantism, X-LAG was found in 2 familial isolated pituitary adenoma kindreds and in 10 sporadic patients [52]. Further molecular characterization of the duplicated genomic region shared by all affected patients did not reveal any single-nucleotide variants of likely pathogenicity but considerably upregulation of the GPR101 gene which encodes an orphan G protein-coupled receptor [58]. Investigation of 248 patients with sporadic acromegaly revealed an absence of microduplications on chromosome Xq26.3 but a c.924G → C substitution (p.E308D) in GPR101 in 4.4 % of DNA in tumor samples and in 1.9 % of peripheral blood cells (with at least one patients demonstrating a clear de novo somatic mutation). Overexpression of the mutation in GH3 cells significantly increased GH secretion and cell proliferation. Although the X-LAG syndrome awaits further characterization, screening for microduplications on Xq26.3 and/or GPR101 mutations may improve early diagnosis and treatment in selected cases.
Except for the rare manifestation as part of one of the germline syndromes described above, the pathogenesis of corticotropic pituitary adenomas was largely unclear. However, two recent studies applying genome sequencing identified somatic mutations of the USP8 deubiquitinase gene in 4/10 and 8/12 adenomas, respectively [30, 51]. Further evaluation of larger cohorts confirmed USP8 variants in 48/145 (33 %) and 67/108 (62 %) of tumor samples, respectively [30, 47]. Of interest, the former cohort included 11 silent corticotropic adenomas, of which none demonstrated an USP8 mutation. USP8 mutants diminished epidermal growth factor receptor ubiquitination with subsequent activation of EGF receptor signaling and induced POMC promoter activity in a cell model. With respect to the clinical presentation, mutations were associated with a younger age at diagnosis. Biochemical activity was lower in patients with USP8 mutations, at least with respect to preoperative cortisol levels after suppression by 8 mg dexamethasone, potentially explaining a lower rate of postoperative adrenal insufficiency. In contrast, maximum adenoma size was not significantly different between USP8 mutant and wild-type adenomas. The prevalence of USP8 mutants was higher in adults than in pediatric cases (41 vs 17 %) and in females than in males. As clarification of the underlying molecular changes may allow the development of new therapeutic strategies for these adenomas, screening for these mutations may become clinically important in the future.
Alterations in other well-known tumor suppressor genes (e.g., P53, RB) or oncogenes (e.g., Ras) are rarely involved in pituitary adenoma development and are therefore not routinely analyzed in sporadic adenomas [12]. However, cell signaling abnormalities may play some role for the induction of invasive behavior and recurrence. In this line, 9 % of invasive (vs. 0 % of non-invasive) pituitary adenomas harbored mutations of the phosphoinositol-3-kinase (PI3K) gene [34] (18), which could lead to phosphorylation of Akt and subsequently other proteins that affect cell growth. Of note, these mutations were also associated with a higher frequency of recurrence. In another study, tumors with a high level of expression of phosphorylated Akt, phosphorylated mitogen activated protein kinase (MAPK), and pituitary tumor transforming gene PTTG1 were associated with early recurrence [43]. In contrast, in the same study high levels of expression of phosphorylated cyclic AMP response element binding protein (CREB) and the zinc-finger protein ZAC1 were inversely correlated with recurrence. It remains to be seen, whether these observations have significant prognostic implications or lead to detection of new therapeutic targets, and may therefore gain relevance as part of routine investigation of pituitary adenomas.
In conclusion, there has been significant progress in the understanding of the pathogenetics of pituitary adenomas as part of germline syndromes, leading to the potential of screening for the molecular abnormalities in patients and relatives to detect and treat disease manifestations at an early stage. In contrast, the pathogenesis of sporadic pituitary adenomas is much less characterized, with the exception of recent advances for corticotropic adenomas and patients with gigantism. Although a number of molecular alterations have been published, the association with prognosis and tumor response to the various treatment options is weak, precluding any routine use during follow-up of these tumors so far. Therefore, further efforts are necessary to establish clear parameters to help us caring for patients with pituitary adenomas.
References
Abe, T., Lüdecke, D.K.: Effects of preoperative octreotide treatment on different subtypes of 90 GH-secreting pituitary adenomas and outcome in one surgical centre. Eur J Endocrin 145: 137–145 (2001)
Albiger, N.M., Ceccato, F., Zilio, M., Barbot, M., Occhi, G., Rizzati, S., Fassina, A., Mantero, F., Boscaro, M., Iacobone, M., Scaroni, C.: An analysis of different therapeutic options in patients with Cushing’s syndrome due to bilateral macronodular adrenal hyperplasia: a single-centre experience. Clin Endorin 82: 808–815 (2015)
Altas, M., Bayrak, O.F., Ayan, E., Bolukbasi, F., Silav, G., Coskun, K.K., Sahin, F., Elmaci, I.: The effect of polymorphisms in the promoter region of the MMP-1 gene on the occurrence and invasiveness of hypophyseal adenoma. acta neurochir (Wien) 152: 1611–1617 (2010)
Beaulieu, E., Kachra, Z., Mousseau, N., Delbecchi, L., Hardy, J., Beliveau, R.: Matrix metalloproteinases and their inhibitors in human pituitary tumors. Neurosurgery 45: 1432–1440 (1999)
Beckers, A., Lodish, M., Trivellin, G., Rostomyan, L., Lee, M., Faucz, F.R., Yuan, B., Choong, C.S., Caberg, J.H., Verrua, E., Naves, L.A., Cheetham, T.D., Young, J., Lysy, P.A., Petrossians, P., Cotterill, A., Shah, N.S., Metzger, D., Castermans, E., Ambrosio, M.R., Villa, C., Strebkova, N., Mazerkina, N., Gaillard, S., Barra, G.B., Casulari, L.A., Neggers, S.J., Salvatori, R., Jaffrain-Rea, M.L., Zacharin, M., Santamaria, B.L., Zacharieva, S., Lim, E.M., Mantovani, G., Zatelli, M.C., Collins, M.T., Bonneville, J.F., Quezado, M., Chittiboina, P., Oldfield, E.H., Bours, V., Liu, P.W., Pellegata, N.S., Lupski, J.R., Daly, A.F., Stratakis, C.A.: X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer 22: 353–367 (2015)
Campero, A., Martins, C., Yasuda, A., Rhoton, A.L., Jr.: Microsurgical anatomy of the diaphragma sellae and its role in directing the pattern of growth of pituitary adenomas. Neurosurgery 62: 717–723 (2008)
Colao, A., Filippella, M., Di Somma, C., Manzi, S., Rota, F., Pivonello, R., Gaccione, M., De Rosa, M., Lombardi, G.: Somatostatin analogs in treatment of non-growth hormone-secreting pituitary adenomas. Endocrine 20: 279–283 (2003)
Correa, R., Salpea, P., Stratakis, C.A.: Carney complex: an update. Eur J Endocrin 173: M85-M97(2015)
Cottier, J.P., Destrieux, C., Brunereau, L., Bertrand, P., Moreau, L., Jan, M., Herbreteau, D.: Cavernous sinus invasion by pituitary adenoma: MR imaging. Radiology 215: 463–469 (2000)
de Bruin, C., Feelders, R.A., Lamberts, S.W.J., Hofland, L.J.: Somatostatin and dopamine receptors as targets for medical treatment of Cushing’s Syndrome. Reviews in Endocrine & Metabolic Disorders 10: 91–102 (2009)
Demirci, S., Unubol, M., Demirci, K.: Galactorrhea With Normal Prolactin Levels Associated With Duloxetine. Journal of Clinical Psychopharmacology 35: 346–347 (2015)
Dworakoska, D., Korbonits, M., Aylwin, S., McGregor, A., Grossman, A.B.: The pathology of pituitary adenomas from a clinical perspective. Front Biosci (Schol Ed) 3: 105–116 (2011)
Giese, A., Westphal, M.: Glioma invasion in the central nervous system. Neurosurgery 39: 235–250 (1996)
Gong, J., Zhao, Y., Abdel-Fattah, R., Amos, S., Xiao, A., Lopes, M.B.S., Hussaini, I.M., Laws, E.R.: Matrix metalloproteinase-9, a potential biological marker in invasive pituitary adenomas. Pituitary 11: 37–48 (2008)
Gurlek, A., Karavitaki, N., Ansorge, O., Wass, J.A.: What are the markers of aggressiveness in prolactinomas? Changes in cell biology, extracellular matrix components, angiogenesis and genetics Eur J Endocr 156: 143–153 (2007)
Honegger, J., Prettin, C., Feuerhake, F., Petrick, M., Schulte-Monting, J., Reincke, M.: Expression of Ki-67 antigen in nonfunctioning pituitary adenomas: correlation with growth velocity and invasiveness. J Neurosurg 99: 674–679 (2003)
Ju, H.J., Pan, Y., Lv, J., Mao, X.W., Zhang, Y.F.: Pituitary Involvement of Langerhans Cell Histiocytosis in an Adult Unveiled by FDG PET/CT. Clinical Nuclear Medicine 40: 509–511 (2015)
Kawamoto, H., Kawamoto, K., Mizoue, T., Uozumi, T., Arita, K., Kurisu, K.: Matrix metalloproteinase-9 secretion by human pituitary adenomas detected by cell immunoblot analysis. acta neurochir (Wien) 138: 1442–1448 (1996)
Kawamoto, H., Uozumi, T., Kawamoto, K., Arita, K., Yano, T., Hirohata, T.: Type IV collagenase activity and cavernous sinus invasion in human pituitary adenomas. Acta Neurochir.Wien 138: 390–395 (1996)
Kenny, A.J.: Introduction: Nomenclature and classes of peptidases. In: Edited by Sterchi, E.E. Stoecker, W.: Springer Lab Manual.Proteolytic enzymes. Tools and targets., pp.1-10. Springer-Verlag, Berlin, Heidelberg, New York: 1999.
Keskin, F.E., Yetkin, D.O., Ozkaya, H.M., Haliloglu, O., Sadri, S., Gazioglu, N., Tanriover, N., Ak, H., Hatipoglu, E., Kadioglu, P.: The problem of unrecognized acromegaly: surgeries patients undergo prior to diagnosis of acromegaly. Journal of Endocrinological Investigation 38: 695–700 (2015)
Kitayama, K., Kashiwagi, S., Amano, R., Noda, S., Ohira, G., Yamazoe, S., Kimura, K., Hamamoto, K., Hamuro, A., Ohsawa, M., Onoda, N., Hirakawa, K., Pawlikowski, M.: Immunohistochemical detection of dopamine D2 receptors in human pituitary adenomas Folia Histochem. Cytobiol 48: 394–397 (2010)
Knappe, U.J., Fink, T., Fisseler-Eckhoff, A., Schoenmayr, R.: Expression of extracellular matrix-proteins in perisellar connective tissue and dura mater. acta neurochir (Wien) 152: 345–355 (2010)
Knappe, U.J., Hagel, C., Lisboa, B.W., Wilczak, W., Lüdecke, D.K., Saeger, W.: Expression of serine proteases and metalloproteinases in human pituitary adenomas and anterior pituitary lobe. acta neuropath.(Wien) 106: 471–478 (2003)
Knappe, U.J., Konerding, M.A., Schoenmayr, R.: Medial wall of the cavernous sinus: microanatomical diaphanoscopic and episcopic investigation. acta neurochir (Wien) 15: 961–967 (2009)
Knappe, U.J., Lüdecke, D.K.: Persistent and recurrent hypercortisolism after transsphenoidal surgery for Cushing’s disease. Acta Neurochir.Suppl.Wien 65: 31–34 (1996)
Knosp, E., Steiner, E., Kitz, K., Matula, C.: Pituitary Adenomas with Invasion of the Cavernous Sinus Space - A Magnetic Resonance Imaging Classification Compared with Surgical Findings. Neurosurgery 33: 610–618 (1993)
Kontogeorgos, G.: Predictive markers of pituitary adenoma behavior. Neuroendocrinology 83: 179–188 (2006)
Korbonits, M., Storr, H., Kumar, A.V.: Familial pituitary adenomas - who should be tested for AIP mutations? Clin Endorin 77: 351–356 (2012)
Kunz, A.S., Wirth, C., Ernestus, K., Veldhoen, S.: The childhood cancer of the Adrenal Cortex A rare and interdisciplinary Diagnosis. Rofo-Fortschritte Auf dem Gebiet der Rontgenstrahlen und der Bildgebenden Verfahren 187: 293–296 (2015)
Kural, C., Simsek, G.G., Guresci, S., Arslan, E., Kilic, C., Tehli, O., Izci, Y.: Histological structure of the medial and lateral walls of cavernous sinus in human fetuses 17. Childs Nerv Syst 31: 699–703 (2015)
Kursat, E., Yilmazlar, S., Aker, S., Aksoy, K., Oygucu, H.: Comparison of lateral and superior walls of the pituitary fossa with clinical emphasis on pituitary adenoma extension: cadaveric-anatomic study. Neurosurg Rev 31: 91–98 (2008)
Leporati, P., Fonte, R., de Martinis, L., Zambelli, A., Magri, F., Pavesi, L., Rotondi, M., Chiovato, L.: A male patient with acromegaly and breast cancer: treating acromegaly to control tumor progression. Bmc Cancer 15: 397 (2015)
Lin, Y., Jiang, X.F., Shen, Y., Li, M., Ma, H.L., Xing, M.Z., Lu, Y.: Frequent mutations and amplifications of the PIK3CA gene in pituitary tumors. Endocrine-Related Cancer 16: 301–310 (2009)
Liu, W., Kunishio, K., Matsumoto, Y., Okada, M., Nagao, S.: Matrix metalloproteinase-2 expression correlates with cavernous sinus invasion in pituitary adenomas. J clin Neurosci 12: 791–794 (2005)
Lloyd, R.V., Kovacs, K., Young, W.F., Jr., Farrell, W.E., Asa, S.L., Trouillas, J., Kontogeorgos, G., Sano, T., Scheithauer, B.W., Horvath, E., Watson, R.E., Jr., Lindell, E.P., Barkan, A.L., Saeger, W., Nosé, V., Osamura, R.Y., Ezzat, S., Yamada, S., Roncaroli, F., Lopes, M.B.S., Vidal Ruibal, S.: Tumours of the pituitary. In: Edited by DeLellis, R.A., Lloyd, R.V., Heitz, P.U.: Pathology and Genetics. Tumours of Endocrine Tumours, Ed.1: pp.9-48. International Agency for Research and Cancer (IARC), Lyon: 2004.
Mattozo, C.A., Dusick, J.R., Esposito, F., Mora, H., Cohan, P., Malkasian, D., Kelly, D.F.: Suboptimal sphenoid and sellar exposure: a consistent finding in patients treated with repeat transsphenoidal surgery for residual endocrine-inactive macroadenomas. Neurosurgery 58: 857–865 (2006)
Mayr, B., Buslei, R., Theodoropoulou, M., Stalla, G.K., Buchfelder, M., Schofl, C.: Molecular and functional properties of densely and sparsely granulated GH-producing pituitary adenomas. Eur J Endocrin 169: 391–400 (2013)
Meij, B.P., Lopes, M.B.S., Ellegala, D.B., Alden, T.D., Laws, E.R.: The long-term significance of microscopic dural invasion in 354 patients with pituitary adenomas treated with transsphenoidal surgery. J Neurosurg 96: 195–208 (2002)
Mete, O., Hayhurst, C., Alamadi, H., Monsalves, E., Gucer, H., Gentili, F., Ezzat, S., Asa, S.L., Zadeh, G.: The role of mediators of cell invasiveness, motility, and migration in the pathogenesis of silent corticotroph adenomas. Endocr Pathol 24: 191–198 (2013)
Miller, G.M., Alexander, J.M., Bikkal, H.A., Katznelson, L., Zervas, N.T., Klibanski, A.: Somatostatin receptor subtype gene expression in pituitary adenomas. J Clin Endocrinol Metab 80: 1386–1392 (1995)
Neto, L.V., Machado, E.D., Luque, R.M., Taboada, G.F., Marcondes, J.B., Chimelli, L.M.C., Quintella, L.P., Niemeyer, P., de Carvalho, D.P., Kineman, R.D., Gadelha, M.R.: Expression analysis of dopamine receptor subtypes in normal human pituitaries, nonfunctioning pituitary adenomas and somatotropinomas, and the association between dopamine and somatostatin receptors with clinical response to Octreotide-LAR in acromegaly. Journal of Clinical Endocrinology & Metabolism 94: 1931–1937 (2009)
Noh, T.W., Jeong, H.J., Lee, M.K., Kim, T.S., Kim, S.H., Lee, E.J.: Predicting Recurrence of Nonfunctioning Pituitary Adenomas. Journal of Clinical Endocrinology & Metabolism 94: 4406–4413 (2009)
Peker, S., Kurtkaya, Y., K l c, T., Pamir, M.N.: Microsurgical anatomy of the lateral walls of the pituitary fossa. Acta Neurochirurgica 147: 641–649 (2005)
Pellegata, N.S., Quintanilla-Martinez, L., Siggelkow, H., Samson, E., Bink, K., Hofler, H., Fend, F., Graw, J., Atkinson, M.J.: Germ-line mutations in p27(Kip1) cause a multiple endocrine neoplasia syndrome in rats and humans. Proceedings of the National Academy of Sciences of the United States of America 103: 15558–15563 (2006)
Pepper, M.S., Montesano, R.: Proteolytic balance and capillary morphogenesis. Cell Differ Dev 32: 319–327 (1990)
Perez-Rivas, L.G., Theodoropoulou, M., Ferrau, F., Nusser, C., Kawaguchi, K., Stratakis, C.A., Faucz, F.R., Wildemberg, L.E., Assi, G., Beschorner, R., Dimopoulou, C., Buchfelder, M., Popovic, V., Berr, C.M., Toth, M., Ardisasmita, A.L., Honegger, J., Bertherat, J., Gadelha, M.R., Beuschlein, F., Stalla, G., Komada, M., Korbonits, M., Reincke, M.: The ubiquitin-specific protease 8 gene is frequently mutated in adenomas causing Cushing disease. Endocrine Abstracts 37 OC12.2 | DOI:10.1530/endoabs.37.OC12.2 (2015)
Pinker, K., Ba-Ssalamah, A., Wolfsberger, S., Mlynarik, V., Knosp, E., Trattnig, S.: The value of high-field MRI (3T) in the assessment of sellar lesions. Eur J Radiol 54: 327–334 (2005)
Poncin, J., Stevenaert, A., Beckers, A.: Somatic MEN1 gene mutation does not contribute significantly to sporadic pituitary tumorigenesis. Eur J Endocrin 140: 573–576 (1999)
Qiu, L., He, D., Fan, X., Li, Z., Liao, C., Zhu, X., Wang, H.: The expression of interleukin (IL)-17 and Il-17 receptor and MMP-9 in human pituitary adenomas. Pituitary 14: 266–275 (2011)
Reincke, M., Sbiera, S., Hayakawa, A., Theodoropoulou, M., Osswald, A., Beuschlein, F., Meitinger, T., Mizuno-Yamasaki, E., Kawaguchi, K., Saeki, Y., Tanaka, K., Wieland, T., Graf, E., Saeger, W., Ronchi, C.L., Allolio, B., Buchfelder, M., Strom, T.M., Fassnacht, M., Komada, M.: Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nature Genetics 47: 31–40 (2015)
Rostomyan, L., Daly, A.F., Petrossians, P., Natchev, E., Lila, A.R., Lecoq, A.L., Lecumberri Santamaria, B., Trivellin, G., Salvatori, R., Moraitis, A., Holdaway, I., Kranenburg-van Klaveren, D., Zatelli, M.C., Palacios, N., Nozieres, C., Zacharin, M., Ebeling, T.M., Ojaniemi, M., Rozhinskaya, L., Verrua, E., Jaffrain-Rea, M.L., Filipponi, S., Guskova, D., Pronin, V., Bertherat, J., Belaya, Z., Ilovaiskaya, I., Sahnoun Fatallah, M., Sievers, C., Stalla, G.K., Castermans, E., Caberg, J.H., Sorkina, E., Auriemma, R., Mittal, S., Kareva, M., Lysy, P., Emy, P., de Menis, E., Choong, C.S., Mantovani, G., Bours, V., de Herder, W.W., Brue, T., Barlier, A., Neggers, S., Zacharieva, S., Chanson, P., Shah, N., Stratakis, C.A., Naves, L.A., Beckers, A.: Clinical and genetic characterization of pituitary gigantism: an international study of 208 patients. Endocr Relat Cancer 22: 745–757 (2015)
Saeger, W., Honegger, J., Theodoropoulou, M., Knappe, U.J., Schöfl, C., Petersenn, S., Buslei, R.: Clinical impact of the current WHO-classification of pituitary adenomas. Endocr Pathol (submitted):(2016)
Saeger, W., Lüdecke, B., Lüdecke, D.K.: Clinical tumor growth and comparison with proliferation markers in non-functioning (inactive) pituitary adenomas. Exper clin Endocr Metab 116: 80–85 (2007)
Spada, A., Arosio, M., Bochicchio, D., Bazzoni, N., Vallar, L., Bassetti, M., Faglia, G.: Clinical, biochemical, and morphological correlates in patients bearing growth hormone-secreting pituitary tumors with or without constitutively active adenylyl cyclase. J Clin Endocrinol Metab 71: 1421–1426 (1990)
Thakker, R.V., Newey, P.J., Walls, G.V., Bilezikian, J., Dralle, H., Ebeling, P.R., Melmed, S., Sakurai, A., Tonelli, F., Brandi, M.L.: Clinical Practice Guidelines for Multiple Endocrine Neoplasia Type 1 (MEN1). Journal of Clinical Endocrinology & Metabolism 97: 2990–3011 (2012)
Tichomirowa, M.A., Lee, M., Barlier, A., Daly, A.F., Marinoni, I., Jaffrain-Rea, M.L., Naves, L.A., Rodien, P., Rohmer, V., Faucz, F.R., Caron, P., Estour, B., Lecomte, P., Borson-Chazot, F., Penfornis, A., Yaneva, M., Guitelman, M., Castermans, E., Verhaege, C., Wemeau, J.L., Tabarin, A., Montanana, C.F., Delemer, B., Kerlan, V., Sadoul, J.L., Rudelli, C.C., Archambeaud, F., Zacharieva, S., Theodoropoulou, M., Brue, T., Enjalbert, A., Bours, V., Pellegata, N.S., Beckers, A.: Cyclin-dependent kinase inhibitor 1B(CDKN1B) gene variants in AIP mutation-negative familial isolated pituitary adenoma kindreds. Endocrine-Related Cancer 19: 233–241 (2012)
Trivellin, G., Daly, A.F., Faucz, F.R., Yuan, B., Rostomyan, L., Larco, D.O., Schernthaner-Reiter, M.H., Szarek, E., Leal, L.F., Caberg, J.H., Castermans, E., Villa, C., Dimopoulos, A., Chittiboina, P., Xekouki, P., Shah, N., Metzger, D., Lysy, P.A., Ferrante, E., Strebkova, N., Mazerkina, N., Zatelli, M.C., Lodish, M., Horvath, A., de Alexandre, R.B., Manning, A.D., Levy, I., Keil, M.F., Sierra, M.D., Palmeira, L., Coppieters, W., Georges, M., Naves, L.A., Jamar, M., Bours, V., Wu, T.J., Choong, C.S., Bertherat, J., Chanson, P., Kamenicky, P., Farrell, W.E., Barlier, A., Quezado, M., Bjelobaba, I., Stojilkovic, S.S., Wess, J., Costanzi, S., Liu, P., Lupski, J.R., Beckers, A., Stratakis, C.A.: Gigantism and Acromegaly Due to Xq26 Microduplications and GPR101 Mutation. New England Journal of Medicine 371: 2363–2374 (2014)
Trouillas, J., Labat-Moleur, F., Sturm, N., Kujas, M., Heymann, M.F., Figarella-Branger, D., Patey, M., Mazucca, M., Decullier, E., Verges, B., Chabre, O., Calender, A.: Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): A case–control study in a series of 77 patients versus 2509 non-MEN1 patients. Amer J Surg Pathol 32: 534–543 (2008)
Tuncer, B.B., Bavbek, N.C., Ozkan, C., Tuncer, C., Altinova, A.E., Gungor, K., Akturk, M., Toruner, F.B.: Craniofacial and pharyngeal airway morphology in patients with acromegaly. Acta Odontologica Scandinavica 73: 433–440 (2015)
Turner, H.E., Nagy, Z., Esiri, M.M., Harris, A.L., Wass, J.A.H.: Role of matrix metalloproteinase 9 in pituitary tumor behavior. J Clin Endocrinol Metab 85: 2931–2935 (2000)
Vasilev, V., Daly, A.F., Naves, L., acharieva, S., eckers, A.: Clinical and genetic aspects of familial isolated pituitary adenomas. Clinics (Sao Paulo) 67, Suppl 1: 37–41 (2012)
Weidle, U.H., Wollisch, E., Ronne, E., Ploug, M., Behrendt, N., de Vries, T.J., Quax, P.H., Verheijen, J.H., van Muijen, G.N., Ruiter, D.J.: Studies on functional and structural role of urokinase receptor and other components of the plasminogen activation system in malignancy. Ann Biol Clin (Paris) 52: 775–782 (1994)
Yamada, S., Taguchi, M., Takeshita, A., Morita, K., Takano, K., Sano, T.: A study of the correlation study of the correlation between morphological findings and biological activities in clinically nonfunctioning pituitary adenomas. Neurosurgery 61: 580–584 (2007)
Yokoyama, S., Hirano, H., Moroki, K., Goto, M., Imamura, S., Kuratsu, J.: Are nonfunctioning pituitary adenomas extending into the cavernous sinus aggressive and/or invasive? Neurosurgery 49: 857–862 (2001)
Acknowledgments
The funding for the German Pituitary Tumor Registry to WS from Novartis Pharma GmbH (Nuremberg, Germany), Novo Nordisk Pharma GmbH (Mainz, Germany), Pfizer Pharma GmbH (Karlsruhe, Germany), and Ipsen Pharma GmbH (Ettlingen, Germany) is gratefully acknowledged. We thank all colleagues for sending tumor material to the German Pituitary Tumor Registry.
Author Contributions
W. Saeger was responsible in the general pathology, conception, and plan; St. Petersen in the genetics; C. Schöfl for the receptors; U. J. Knappe for the invasion and proteases; M. Theodoropoulou for the immunocytochemistry; R. Buslei for immunocytochemistry and conception; and J. Honegger for atypical adenomas, conception, and plan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Due to the character and kind (review) of the theme of the manuscript, an approval on ethics appears to be not necessary.
Conflict of Interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Saeger, W., Petersenn, S., Schöfl, C. et al. Emerging Histopathological and Genetic Parameters of Pituitary Adenomas: Clinical Impact and Recommendation for Future WHO Classification. Endocr Pathol 27, 115–122 (2016). https://doi.org/10.1007/s12022-016-9419-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12022-016-9419-6