
Abstract
A case of pancreatic acinar cell tumor (ACC) is presented in a 10-year-old boy. The tumor manifested clinically with Cushing’s syndrome, high serum adrenocorticotropic hormone (ACTH) and cortisol concentrations. In addition, excessive serum levels of alpha-fetoprotein (AFP) were detected. Surgical resection was not possible due to retroperitoneal invasion. Biopsy of the mass showed a solid, poorly differentiated ACC of the pancreas. Periodic acid Schiff positive cytoplasmic granules, trypsinogen, keratins, alpha-1-antitrypsin, and AFP were identified in the tumor cells. Electron microscopy demonstrated zymogen granules as well as isolated dense core granules. Using immunochemiluminometric assay, a high quantity of ACTH was found in the fresh frozen tumor extract. ACTH, chromogranin A, and corticotropin-releasing factor were identified only in a few cells by immunohistochemistry. Combined radiochemotherapy was temporarily effective in reducing the tumor mass and serum AFP. Serum ACTH and cortisol levels dropped progressively and definitively to normal values after chemotherapy, and the Cushing’s syndrome subsided. Two years later, the patient died with metastatic disease. The presented case of ACC is interesting due to high serum AFP values and ectopic ACTH secretion resulting in Cushing’s syndrome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exocrine as well as endocrine tumors of the pancreas are rare in children. The most common types are solid pseudopapillary tumors and two tumors closely related with acinar differentiation, pancreatoblastoma (PB), and acinar cell carcinoma (ACC) [1]. In fact, ACC is more often seen in adults between the fifth and seventh decades of life; only a few cases have been reported in children [1, 2]. The distinction between PB and ACC may be difficult because of the many shared morphologic and immunohistochemical features [3, 4].
High levels of serum alpha-fetoprotein (AFP) are regarded as sensitive tumor markers for hepatocellular and germ cell tumors. In the case of pancreatic neoplasia, AFP is suggestive of PB or ACC, indicating the diagnostic usefulness of this marker in these acinary differentiated tumors [5–12].
Ectopic ACTH secretion was previously not described in pancreatic ACCs but it was presented and serologically proven in one case of recurrent PB [13].
In this case, we report a child with Cushing’s syndrome associated to an AFP and ACTH-secreting ACC diagnosed from a biopsy.
Case Report
A 10-year-old white boy presented with abdominal and bone pain, polyuria, and convulsions. On examination, there were signs of Cushing’s syndrome, and an abdominal mass was felt. The child’s blood pressure was 170/100 mmHg.
The initial laboratory findings displayed glycosuria, hyperglycemia, and hypokalemia. The 24-h urine measurement was negative for catecholamine excess, and vanilyl-mandelic acid (VMA) excretion was repeatedly normal. A [(123) I] metaiodobenzylguanidine scintigraphy scan was also negative.
Further investigations for Cushing’s disease showed no abnormality of the pituitary fossa. Serum ACTH was 300 pg/ml (normal, 20–70 pg/ml); serum and urinary cortisol levels were elevated up to 495 μg/day and 44 μg/dl, respectively, suggesting ectopic ACTH secretion. Serum tumor markers were normal (CEA, CA-19-9, CA-72-4, NSE, HCG), except for an excessively high AFP of 7,664 IU/ml (normal, 0–5.8 IU/ml).
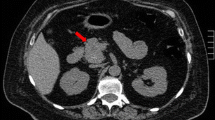
On abdominal ultrasound and computer tomography (CT) a retroperitoneal mass of 10 × 10 × 8 cm was seen adjacent the liver and the kidneys (Fig. 1). The tumor had well-circumscribed pushing borders, and it showed small areas of lucency. The tail of the pancreas and the adrenal glands were not involved. Associated mediastinal paraaortic and lower abdominal parailiac lymph node enlargement was also observed with nodes of up to 3 × 2 × 2 cm.

Abdominal CT scans showing a well-circumscribed, slightly inhomogeneous midline mass. The tumor appears to arise from pancreas, and it is in contact with the liver hilum (a), and kidneys and aorta (b)
The child underwent laparotomy that revealed the bulky tumor mass appearing to arise from the body of the pancreas with anterior displacement of the stomach. The tumor invaded around the hilum of the left kidney, and the portal and inferior caval veins. Due to the involvement of these vessels, it was considered inoperable. Tumor sample was collected for histology, and pieces of tissue were instantly frozen and stored at −80°C.
Chemotherapy was given to debulk the tumor before attempting total surgical resection. The protocol chosen for the treatment consisted of ifosfamide/vincristine and adriamycin/carboplatin/etoposide in three-week cycles to which the cortisol secretion inhibitor mitotane was added. After 3 months of treatment, the patient was normotensive, normokalemic, and euglycemic without need for mitotane or antihypertensive medication. The retroperitoneal mass decreased in size significantly but remained inoperable mainly due to its attachments to the great vessels. The lymphadenopathy regressed with chemotherapy. Radiotherapy was applied at this time; however, it resulted to no further benefit. Serum ACTH and serum cortisol levels definitively normalized (22 pg/ml and 5 μg/dl, respectively), whereas serum AFP values lowered significantly to 14,1 IU/ml. Half a year later, the general condition of the patient was stable; however, the AFP concentration increased again to 161 IU/ml, and several lung and liver metastases were detected without any biochemical or clinical evidence for ectopic hormone production. Two years after diagnosis, the child died and an autopsy was not performed.
Materials and Methods
Part of the excisional biopsy was fixed in 10% neutral buffered formalin, and blocks were embedded in paraffin. Sections were stained with hematoxylin-eosin (H&E) and periodic acid Schiff (PAS) with and without diastase digestion as well as mucicarmine.
For electron microscopy, tissue pieces were fixed in 2.5% glutaraldehyde and osmium tetroxide. Ultrathin sections were cut and double contrasted with uranyl acetate and lead citrate.
For immunohistochemistry, mouse monoclonal and rabbit polyclonal antibodies were used. The antibodies and working dilutions used are summarized in Table 1. The labeled streptavidin–biotin 2 (LASB 2) system was used (Dako Cytomation, Ghostrup, Denmark, DK) with chromogen diaminobenzidine tetrahydrochloride (DAB from View Ventana, Tucson, AZ, USA) or chromogen 3-amino-9-ethyl-carbazole (AEC from Biogenex substrate pack, Biogenex, San Ramon, CA, USA) for peroxidase reaction. A polyclonal rabbit anti-rat trypsinogen was used to demonstrate acinar differentiation in the tumor. This antibody was prepared and affinity-purified at the Department of Biochemistry, Eötvös University, Budapest [14], and it was known from Western-blot studies to cross react with human trypsinogen (unpublished data). Two anti-corticotropin releasing factor (CRF) antibodies were tried; one (Peninsula Laboratories, Inc., Belmont, CA, USA) for frozen sections; the other was prepared and characterized by Görcs TJ [14] for paraffin sections. For CRF and ACTH immunohistochemical reactions, antibodies were used in carefully experienced titres, and the assays were repeated on frozen sections using indirect immunofluorescence technique to avoid unspecific marking. After nuclear staining with hematoxylin, slides were mounted with either the Ultramount aqueous permanent mounting medium (Dako) or the GLC mounting medium (Sakura, Zoeterwoude, The Netherlands). Immunohistochemical reactions for secretin and vasoactive intestinal polypeptide (VIP) were prepared using the indirect immunofluorescence technique as described earlier [15, 16]. Antibody for claudin 4 was applied for the detection of tight junction components that are specific for pancreatic ductal or biliary origin.
For control reactions, the chronic pancreatitis and pancreatic ductal adenocarcinoma samples that were used were obtained from pancreatectomy specimens of adult patients. In the case of ACTH immunohistochemistry, positive control reactions were prepared on a hypophysis obtained at autopsy and on an ACTH-secreting carcinoid tumor of the lung from a 50-year-old patient.
For direct determination of ACTH content in the tumor, an immunochemiluminometric assay was also used. For this, pieces of frozen tumor tissue were homogenized in Krebs–Ringer bicarbonate buffer containing ethylenediaminetetraacetic acid (EDTA), and after centrifugation, supernates were used for the assay. For comparison, pieces of pancreatic ductal adenocarcinoma and chronic obstructive pancreatitis were similarly processed. The ACTH concentration in supernates was determined by a commercial kit according to the manufacturer’s instructions (Nichols Institute Ltd., San Juan, CA, USA).
Results
The tumor consisted of well-demarcated compact sheets of epithelial cells surrounded by dense fibrous tissue stroma. The cells were arranged in places around small lumina. Nuclei were moderately pleomorphic with coarse marginal chromatin and prominent nucleoli that were centrally located (Fig. 2). At the periphery of the tumor sheets, the cells were small and condensed; centrally, they were large and pale. Small necrotic areas were present. Mitotic figures were extremely rare. A finely granular PAS staining resistant to digestion by diastase was seen mostly in the cytoplasm of peripherally located cells and in intercellular spaces. The mucicarmine stain was negative. “Squamoid nodules” or “a pancreas-like organoid structure” was not observed.

Tumor with solid (a) and acinar (b, arrows) pattern. Nuclei are moderately pleomorphic with prominent nucleoli. H&E, a 100× and b 800×
A summary of the immunohistochemical findings is presented in Table 1. Cells showed diffuse staining with cytokeratin cocktails. Cytokeratin 20 was negative, but cytokeratin 7 showed strong focal reactivity in irregular cohesive cell groups. A large proportion of tumor cells showed granular cytoplasmic positivity for trypsinogen (Fig. 3). Diffuse alpha-1-antitrypsin positivity was seen, whereas lysozyme was negative. An intensive AFP reaction of all tumor cells was in concordance with the high serum concentration (Fig. 4b). Claudin 4 was not expressed in the tumor. P53 proved to be negative (<5% nuclear staining). Beta-catenin showed normal membranous staining. A proliferation rate of up to 20% was seen by the expression of the nuclear Ki 67 antigen. Vimentin was negative.

a Expression of tripsinogen in the tumor. b Control human pancreas with the same reaction. Immunohistochemical reaction with anti-trypsinogen antibody; a, b 600×

a Paranuclear cytoplasmic chromogranin A reaction (arrow) in scattered tumor cells. b Widespread expression of AFP. c Immunoreactivity for ACTH in some tumor cells (arrows). d Focally accentuated CRF positivity (arrows). The strong granular staining in c and d represents immunoreactivity in macrophages (arrowheads). a 800×, b 800×, c and d 1,000×
Isolated endocrine cells were identified by chromogranin A reaction, but in most cells, only a paranuclear granular staining was seen (Fig. 4a), and there was no positivity with neuron-specific enolase or synaptophysin. Hormones including insulin, glucagon, somatostatin, gastrin, serotonin, secretin, and VIP were not detected. CRF was found in very few tumor cells and in macrophages infiltrating the tumor (Fig. 4d).
ACTH immunohistochemical staining using different methods was surprisingly weak and rare. Fine cytoplasmic staining of scattered tumor cells and macrophage granules was seen with both detection procedures (Fig. 4c).
Using the immunochemiluminometric assay, 1,790 pg ACTH/g wet tissue weight of tumor was detected. This represents an extremely high concentration as compared with the patient’s serum value (300 pg/ml), the control pancreatic ductal adenocarcinoma (49 pg/g), the control non-tumoral fibrotic pancreas (<8 pg/g), and the usual normal serum value of 20–70 pg/ml. This assay was conclusive for ACTH production by the ACC.
Electron microscopy revealed epithelial tumor cells containing dense zymogen granules of 300 to 500 nm in diameter and a few dense core granules of 100 nm in diameter. There were occasional tonofilaments and desmosomes. Rough endoplasmic reticulum and mitochondria were abundant (Fig. 5).

Zymogen granules (a) and dense core granules (b) in tumor cells shown by electron microscopy. The intracellular dense fibrilar structures are tonofilaments (T). Mitochondria (M). a 6,000×, b 10,000×
Diagnosis of ACC was made by taking into consideration the phenotypic appearance of the tumor, the presence of zymogen granules, and the marked AFP secretion. The tumor contained isolated endocrine cells, some positive for CRF and ACTH but negative for other hormones.
Discussion
In this report, a case of a child with ACC and high ACTH and AFP secretion is presented. The imaging techniques showed a large pancreatic tumor with features common for both PB and ACC [17–21].
The diagnosis, based on pure morphology, was hindered by the fact that the tumor was a solid type with hidden acinar structures. Nevertheless, ACC was proved by the presence of cytoplasmic PAS positivity after diastase digestion, the presence of immunoreactive trypsinogen, and of zymogen granules by electron microscopy. Alpha-1-antitrypsin and AFP secretion were also detected. Furthermore, focal endocrine and ductal differentiation seem to be also characteristic of acinar tumors [4, 17, 18, 22].
The molecular pathogenesis of ACC and PB could involve the adenomatous polyposis coli (APC)/beta-catenin pathway [23, 24]. In adult pancreatic ductal adenocarcinomas, loss of Dpc4 protein expression and alterations in the expression of K-ras and p53 genes are evident. However, the nuclear expressions of beta-catenin and p53 were both negative in the present neoplasm.
In children, the biological behavior of PB is considered less aggressive than that of ACC [21]. The unfavorable clinical outcome of the presented case is concordant with the diagnosis of ACC.
At first, PB was considered in this case because of the young age of the patient. The immunohistochemical profile and endocrine features of PB and ACC are much the same, although PB should have a pancreas-like organoid structure by definition [3–5, 7, 25, 26]. ACC could be undistinguishable from PB in small biopsy samples [5].
ACC should also be distinguished from solid pseudopapillary neoplasms that are usually benign tumors, composed of monomorphic cells forming solid and pseudopapillary structures that express vimentin and endocrine markers [4, 27].
Beside germ-cell neoplasms, hepatoblastomas, and hepatocellular carcinomas, most PBs and many ACCs express and secrete AFP [5–12]. In pancreatic tumors, the phenomenon could have a diagnostic benefit, as in the present case when our reasoning was influenced by knowing the high serum AFP values at the biopsy examination. AFP was detected subsequently also in the tumor by immunohistochemistry.
We identified cells containing electron-dense granules on electron microscopy, but immunohistochemically, the endocrine differentiation was not totally conclusive due to the fact that, chromogranin A-positive cells were repeatedly negative for synaptophysin and neuron-specific enolase. Moreover, the number of these partially differentiated endocrine cells was low; therefore, mixed acinar–endocrine carcinoma was not diagnosed. Usually, tumors that contain a more significant proportion (greater than 30%) of endocrine components are diagnosed as mixed acinar–endocrine carcinomas [4, 17, 19, 28]. Nevertheless, scattered endocrine cells were usually found in tumors with acinar differentiation, and they reportedly contained hormones like insulin, glucagon, somatostatin, pancreatic polypeptide, thyroid-stimulating hormone, follicle-stimulating hormone, and neurotensin [25]. We could not identify the above-mentioned hormones in the present tumor, nor could we identify VIP, secretin, gastrin, and serotonin.
It seemed most likely that the overproduction of ACTH in our case originated from the pancreatic tumor, as the elevated serum ACTH and the Cushing’s syndrome disappeared after chemotherapy. To our knowledge, there is no previous publication to date that demonstrates ACTH at tissue level in a non-endocrine pancreatic tumor with ectopic ACTH syndrome. Passmore et al. [13] reported a recurrent PB with raised serum ACTH concentration. Histology diagnosis was obtained in their case 3 1/2 years earlier from the resected tissue, at which time the tumor was negative by immunoperoxidase reaction for ACTH, and there were no clinical signs of ectopic hormone secretion. It was concluded that an endocrine differentiation took place in the recurrent neoplasm. Most of our investigations focused on identifying ACTH in the presented tumor. Initially, we were uncertain on our immunohistochemical reactions because only very few ACTH-positive cells were evidentiated, less than endocrine differentiated cells. Later, we ingeniously implemented a tissue-extract-based analysis. By immunochemiluminescent assay (ICMA), a sensitive detection method used for serum analyses, the expected high concentration of ACTH was proved in the tumor tissue. Literary data on comparable results from extractions in ectopic ACTH-secreting tumors are available only for adrenomedullin [29]. Furthermore, in some neuroendocrine tumors with corticotropic differentiation, like small-cell lung carcinomas, proopiomelanocortin could be processed in an aberrant way, and high concentrations of ACTH precursors and few intact ACTH molecules are elaborated [30]. Similarly, in our case, an aberrant antigen structure may explain the difficulty of hormone detection by immunohistochemistry.
It is known that ACTH can also be produced by macrophages (and monocytes) in response to CRF stimulation [31–33]. In the tumor presented, there were many ACTH-positive macrophages. We speculated that these cells could elaborate only small quantities of ACTH or could merely store ACTH released from nearby-located parenchymal cells. It is highly improbable that the Cushing’s syndrome was caused by them. It seems more likely that ACTH was secreted in the present tumor by non-endocrine tumor cells or by dispersed cells with endocrine differentiation.
In addition to ACTH, we have also shown the presence of CRF in some of the tumor cells, as well as in macrophages. Due to the low expression of this neuropeptide, it is likely to have exerted paracrine effects.
In summary, the presented case of ACC in a child is remarkable due to its appearance with high serum AFP values and Cushing’s syndrome caused by ectopic ACTH secretion. Imaging and laboratory data could guide medical reasoning to achieve diagnosis in such rare cases.
References
Shorter NA, Glick RD, Klimstra DS, Brennan MF, Laquaglia MP. Malignant pancreatic tumors in childhood and adolescence: the Memorial Sloan-Kettering experience, 1967 to present. J Pediatr Surg 37:887–92, 2002.
Huang Y, Cao YF, Lin JL, Gao F, Li F. Acinar cell cystadenocarcinoma of the pancreas in a 4-year-old child. Pancreas 33(3):311–2, 2006.
Hoorens A, Gebhard F, Kraft K, Lemoine NR, Kloppel G. Pancreatoblastoma in an adult: its separation from acinar cell carcinoma. Virchows Arch 424:485–90, 1994.
Hamilton SR, Aaltonen LA, editors. Pathology and genetics of tumours of the digestive system. Lyon: IARC Press, 2000.
Cingolani N, Shaco-Levy R, Farruggio A, Klimstra DS, Rosai J. Alpha-fetoprotein production by pancreatic tumors exhibiting acinar cell differentiation: study of five cases, one arising in a mediastinal teratoma. Human Pathol 31(8):938–44, 2000.
Balian A, Hammel P, Terris B, Sauvanet A, Belghiti J, Ruszniewski P, Bernades P. Increase of serum alpha-fetoprotein in a patient with acinar cell carcinoma of the pancreas [French]. Gastroenterologie Clinique et Biologique 21:231–32, 1997.
Defachelles AS, De Lassalle M, Boutard P, Nelken B, Schneider P, Patte C. Pancreatoblastoma in childhood: clinical course and therapeutic management of seven patients. Med Pediatr Oncol 37:47–52, 2001.
Eriguchi N, Aoyagi S, Hara M, Okuda K, Saito N, Fukuda S, Akashi H, Kutami R, Jimi A. Large acinar cell carcinoma of the pancreas in a patient with elevated serum AFP level. J Hepatobiliary Pancreat Surg 7(2):222–5, 2000.
Kawamoto S, Hiraoka T, Kanemitsu K, Kimura M, Miyauchi Y, Takeya M. Alpha-fetoprotein-producing pancreatic cancer-a case report and review of 28 cases. Hepato-Gastroenterology 39(3):282–6, 1992.
Morohoshi T, Sagawa F, Mitsuya T. Pancreatoblastoma with marked elevation of serum alpha-fetoprotein. An autopsy case report with immunocytochemical study. Virchows Arch A Path Anat Histopathol 416:265–70, 1990.
Nojima T, Kojima T, Kato H, Sato T, Koito K, Nagashima K. Alpha-fetoprotein-producing acinar cell carcinoma of the pancreas. Human Pathol 23(7):828–30, 1992.
Mueller SB, Micke O, Herbst H, Schaefer U, Willich N. Alpha-fetoprotein-positive carcinoma of the pancreas: a case report. Anticancer Res 25(3A):1671–4, 2005.
Passmore SJ, Berry PJ, Oakhill A. Recurrent pancreatoblastoma with inappropriate adrenocorticotrophic hormone secretion. Arch Dis Child 63:1494–96, 1988.
Nagy K, Palfia Z, Rez G. Characterisation of the progression of azaserine-induced rat pancreatic adenocarcinoma by proliferative cell nuclear antigen, basement membrane laminin and trypsinogen immunohistochemistry. Histochem Cell Biol 119:405–13, 2003.
Koves K, Kausz M, Reser D, Illyes G, Takacs J, Heinzlmann A, Gyenge E, Horvath K. Secretin and autism: a basic morphological study about the distribution of secretin in the nervous system. Regulatory Pept 123:209–16, 2004.
Gulyas AI, Gorcs TJ, Freund TF. Innervation of different peptide-containing neurons in the hippocampus by GABAergic septal afferents. Neuroscience 37:31–44, 1990.
Aqel B, Scolapio J, Nguyen J, Krishna M, Raimondo M. Recurrent pancreatitis due to a cystic pancreatic tumor: a rare presentation of acinar cell carcinoma. J Pancreas 5:151–54, 2004.
Chiou YY, Chiang JH, Hwang JI, Yen CH, Tsay SH, Chang CY. Acinar cell carcinoma of the pancreas: clinical and computed tomography manifestations. J Comput Assist Tomogr 28:180–86, 2004.
Ogawa T, Isaji S, Yabana T. A case of mixed acinar-endocrine carcinoma of the pancreas discovered in an asymptomatic subject. Int J Pancreatol 27:249–57, 2000.
Takeda K, Goto H, Hirooka Y, Itoh A, Hashimoto S, Niwa K, Hayakawa T. Contrast-enhanced transabdominal ultrasonography in the diagnosis of pancreatic mass lesions. Acta Radiol 44:103–06, 2003.
Matsuura S, Fukuya T, Miyajima K, Koga M, Suga N, Seo Y, Fukuda T. Imaging features of pancreatoblastoma: a case report. Radiat Med17:365–68, 1999.
Ordonez NG, Mackay B. Acinar cell carcinoma of the pancreas. Ultrastruct Pathol 24:227–41, 2000.
Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee JH, Yeo CJ, Cameron JL, Hruban RH. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol 159:1619–27, 2001.
Abraham SC, Wu TT, Hruban RH, Lee JH, Yeo CJ, Conlon K, Brennan M, Cameron JL, Klimstra DS. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol 160:953–62, 2002.
Hua C, Shu XK, Lei C. Pancreatoblastoma: a histochemical and immunohistochemical analysis. J Clin Pathol 49:952–54, 1996.
Ordonez NG. Pancreatic acinar cell carcinoma. Adv Anat Pathol 8:144–59, 2001.
Zhou H, Cheng W, Lam KY, Chan GC, Khong PL, Tam PK. Solid-cystic papillary tumor of the pancreas in children. Pediatr Surg Int 17:614–20, 2001.
Kloppel G. Mixed exocrine-endocrine tumors of the pancreas. Sem Diagn Pathol 17:104–8, 2000.
Murakami O, Takahashi K, Satoh F, Totsune K, Sone M, Arihara Z, Andoh N, Mouri T. Expression of adrenomedullin and adrenomedullin mRNA in ectopic ACTH-secreting tumors. Eur J Endocrinol 138(4):436–9, 1998.
Terzolo M, Reimondo G, Ali A, Bovio S, Daffara F, Paccotti P, Angeli A. Ectopic ACTH syndrome: molecular bases and clinical heterogeneity. Ann Oncol 12(Suppl 2):S83–7, 2001.
Agelaki S, Tsatsanis C, Gravanis A, Margioris AN. Corticotropin-releasing hormone augments proinflammatory cytokine production from macrophages in vitro and in lipopolysaccharide-induced endotoxin shock in mice. Infect Immun 70:6068-74, 2002.
Hendricks GL, Mashaly MM. Effects of corticotropin releasing factor on the production of adrenocorticotropic hormone by leukocyte populations. Br Poult Sci 39:123–7, 1998.
Kawahito Y, Sano H, Mukai S, Asai K, Kimura S, YamamuraY, Kato H, Chrousos GP, Wilder RL, Kondo M. Corticotropin releasing hormone in colonic mucosa in patients with ulcerative colitis. Gut 37:544–51, 1995.
Acknowledgment
The authors wish to express their gratitude to Dr. Görcs TJ (Department of Human Morphology and Developmental Biology, Semmelweis University, Budapest) and Dr. Ildikó Miklós (Institute of Experimental Medicine of the Hungarian Academy of Sciences) for providing CRF antibodies and control tissues. Trypsinogen antibody was kindly given by Gábor Réz (Department of General Zoology, Eötvös University, Budapest). Many thanks to Ms. Ágnes Szík, Ms. Sándorné Tóth, and Ms Sklánitzné Somodai Erika for their devoted efforts to solve the technical problems arising in the laboratory work. The study was sponsored by the grants OTKA T 049559 and ETT-049/2006 by the Hungarian Ministry of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Andrea Luczay is working now at the Second Department of Pediatrics, Semmelweis University, Budapest, Hungary.
Rights and permissions
About this article
Cite this article
Illyés, G., Luczay, A., Benyó, G. et al. Cushing’s Syndrome in a Child with Pancreatic Acinar Cell Carcinoma. Endocr Pathol 18, 95–102 (2007). https://doi.org/10.1007/s12022-007-0018-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12022-007-0018-4