
Abstract
Purpose
11β-hydroxylase deficiency accounts for 5% of congenital adrenal hyperplasia cases. Diagnosis suspiction is classically based on the association between abnormal virilization, precocious puberty, and hypertension in 46XX or 46XY subjects. We investigated two families with siblings presenting with opposed clinical features, and provided a review of the mechanisms involved in mineralocorticoid-dependent phenotypic heterogeneity.
Methods
The coding region of the CYP11B1 gene of 4 patients was sequenced and familial segregation was confirmed. Clinical characterization and blood steroid profile were performed.
Results
Family 1 comprised a female and a male siblings who presented in middle childhood with genital ambiguity (Prader II) and precocious puberty, respectively, associated with hypertension. In the second decade of life, the woman had three full-term pregnancies, and then evolved normotensive with no treatment over a 5-year follow up. On the other hand, her brother had hypertensive end-organ damage at age 24. In family 2, a 2.9 year-old boy presented with precocious puberty and hypertension, whereas his 21 days-old sister had genital ambiguity (Prader III) and salt wasting. A homozygous exon 4 splice site mutation was identified (IVS4ds-1G > A; c.799 G > A) in family 1, while a nonsense mutation in exon 6 (p. Q356X; c.1066 C > T) was found in family 2.
Conclusion
CYP11B1 mutations were associated with highly variable phenotypes, from mild to severe virilization, and early-onset hypertension or salt wasting. Further analysis of variants in other hypertension-related genes, steroid synthesis and metabolism compensatory pathways, and/or the investigation of chimeric CYP11B genes are needed to clarify the phenotypic heterogeneity in 11β-hydroxylase deficiency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Steroid 11β-hydroxylase deficiency (11β-OHD) is caused by CYP11B1 mutations and inherited as an autosomal recessive disorder. It is the second most common cause of congenital adrenal hyperplasia (CAH), accounting for 5–8% of all cases [1]. It occurs in ~1:100.000 live births in the general non-consanguineous population, although the incidence may be as high as 1:5000–1:7000 live births in Jews of Moroccan ancestry [1,2,3].
Steroid 11β-hydroxylase catalyzes the conversion of 11-deoxycortisol and 11-deoxycorticosterone (DOC) to cortisol and corticosterone, respectively. In 11β-OHD, chronic elevation of ACTH in response to low serum cortisol levels results in increased synthesis and secretion of steroid intermediates proximal to 11β-hydroxylase block. These abnormally increased precursors include DOC and 11-deoxycortisol produced in the zona glomerulosa and fasciculata, respectively. Although DOC and 11-deoxycortisol cannot be directly converted into androgens, there is a concomitant increase in their upstream precursors, progesterone and 17-hydroxyprogesterone, which in turn may be shunted into the androgen synthesis pathway in the zona reticularis, leading to hyperandrogenism. [1, 4, 5]. Classical 11β-OHD commonly results in virilization of the external genitalia in female newborns. After birth, hyperandrogenism leads to peripheral precocious puberty and advanced bone maturation, with premature epiphyseal closure. Since DOC exhibits mineralocorticoid activity, untreated patients may present severe hypertension, sometimes early in life [4,5,6]. The study of a large cohort from the International Consortium on Rare Steroid Disorders has shown that 11β-OHD is more associated with overt hypertension, significant virilization, and bone age advancement than 21-hydroxylase deficiency, but with a low genotype–phenotype correlation. Further analysis showed no correlation between protein structural changes due to disruptive CYP11B1 mutations and the severity of CAH [7].
The present study describes the clinical, biochemical, and molecular features of two Brazilian families with CAH due to 11β-OHD, whose phenotypic heterogenity ranged from a case with severe hypertension since infancy to a transient, but severe salt-wasting phenotype. In addition, a comprehensive review of potential mechanisms underlying phenotypic variability in 11β-OHD is provided.
Methods
Subjects
We studied 4 subjects from two unrelated Brazilian families, born from consanguineous marriages, who had a clinical diagnosis of 11β-OHD. Unaffected parents were included when available. The study was approved by the Research Ethics Committee of the School of Health Sciences, University of Brasilia, Brazil. All participants or their legal representatives signed the informed consent before inclusion.
Family 1
Patient 1 (P1) was a 46,XX girl who presented at 5 years old with precocious pubarche, genital ambiguity (Prader stage II), and high blood pressure. The diagnosis of 11β-OHD was made on a clinical basis, and glucocorticoid replacement therapy was initiated. Plasma steroids’ levels at the first medical evaluation were not accessible to the study. Lifelong adherence to therapy was poor and the patient remained hypertensive throughout adolescence; her final height was 141 cm. At the age of 17, she underwent genitoplasty and then had three uneventful pregnancies with full term cesarean deliveries. Since her third delivery, she remained off any specific treatment. Five years after her last delivery, at the age of 36, the patient was reassessed for this study, when she was found to be normotensive, with regular menstrual cycles and only mildly oily skin. A 24 h ambulatory blood pressure monitoring had shown averages of systolic and diastolic blood pressure within normal limits and a normal circadian variation of blood pressure.
Patient 2 (P2) was P1’s 46,XY sibling who presented at the age of 7 with GnRH-independent precocious puberty, hypertension, and asymptomatic hypokalemia. He also had poor adherence to glucoc n damage including left ventricular hypertrophy and bilateral hypertensive retinopathy.
Family 2
Patient 3 (P3) was a 35-month-old 46,XY boy, who was referred to the endocrinology clinic due to precocious pubarche. Pubertal stage was G1P3 (Tanner), with macro a blood pressure of 134/85 mmHg (above the 99th percentile for age, gender, and height). Bone age was 10, by Greulich–Pyle method. Hydrocortisone at 15 mg/m²/day was initiated and spironolactone was added for blood pressure control, after confirmation of a high 11-deoxycortisol plasma level. Poor adherence to treatment and irregular follow-up ensued, and the patient evolved with progressive bone age advancement and secondary central precocious puberty as a result of an activation of the hypothalamic GnRH pulse generator due to long-term exposure to sex steroids [9].
Patient 4 (P4) was P3’s 46,XX sister, who was born at 36 weeks and 6 days (gestational age); her weight was 2100 g and length 44 cm. She presented in the outpatient clinic at 21 days of life with genital ambiguity (Prader III) and deficient weight gain. She was admitted to hospital, normotensive, dehydrated, and persistent symptomatic hyponatremia was noted (Na = 128nmol/L (reference values: 135–148); K = 5.1nmol/L (reference values: 3.5–5.1)), associated with a high 11-deoxycortisol plasmatic level. Hydrocortisone was started, but fludrocortisone and oral sodium replacement were added for fluid and electrolyte balance. Mineralocorticoid withdrawal was possible only at the age of 2, and the infant remained normotensive thereafter.
Hormonal assays
Blood samples were collected in the morning. Both P1 and P2 had been off glucocorticoid therapy at the time of hormonal assessment. The woman (P1) had withdrawn medications since her last pregnancy, five years before, and her brother (P2), for 2 weeks before hormonal evaluation, with carefully follow-up, in order to compare their basal hormonal profile. In P3 and P4, hormonal levels were determined before treatment was started.
Serum cortisol, total testosterone, androstenedione, and ACTH were determined by chemiluminescence; 17-hydroxyprogesterone, 11-deoxycortisol, DOC, aldosterone, and plasmatic renin activity (PRA) were evaluated by radioimmunoassays.
Molecular analysis of CYP11B1 gene
Genomic DNA was isolated from peripheral blood leukocytes by the salting out method. The coding exons 1 to 8 of CYP11B1 gene were initially amplified by PCR, including exon–intron boundaries and avoiding simultaneous amplification of the homologous CYP11B2 sequence, as previously described [2]. Exon 9 sequencing was waived once potentially causative mutations were consistently determined.
PCR products were automatically sequenced by Sanger's method. The mutations were confirmed in a second independent reaction. Data were analyzed with SEQUENCHER 5.1 (Gene Codes Corporation) and compared with the CYP11B1 gene sequence deposited in ENSEMBL database (ENSG00000160882).
Results
The hormonal profile of all affected subjects is presented in Table 1. Siblings P1 and P2 had increased serum levels of ACTH, 11-deoxycortisol and DOC after glucocorticoid withdrawal, in addition to a suppressed PRA. Both siblings P3 and P4 had elevated 11-deoxycortisol levels, also compatible with the clinical diagnosis of 11β-OHD [1, 6].
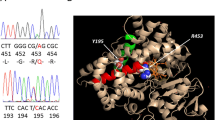
In family 1, we found that siblings P1 and P2 were homozygous for a splice site mutation in the last nucleotide of CYP11B1 exon 4 IVS4-1G > A (c.799 G > A). Their father was deceased, and the mother and other five apparently unaffected siblings in family 1 were not accessible to the study (Fig. 1a, c).

a Electropherograms of the exon–intron junction, showing the homozygous G > A change in the last nucleotide of exon 4, which generates the splice site IVS4-G > A mutation in patients 1 and 2. b Electropherograms of CYP11B1 exon 6 partial sequence, showing the p.Q356X (c.1066 C > T) mutation in homozygosity in siblings 3 and 4. c Family pedigrees, where circles represent women, and squares, men; black boxes represent mutant alleles; question marks denote relatives that were not available for the study. d. Schematic representation of the CYP11B1 gene showing coding exons (filled boxes) and the identified mutations positions (arrows) (Adapted from www.ensembl.org (version ENST00000292427.8, NM_000497)
In family 2, both siblings (P3 and P4) harbored a homozygous nonsense mutation in exon 6, p.Q356X (c.1066 C > T), while their parents were heterozygous. Of note, the first child in this family had deceased at the age of 1 month due to dehydration (Fig. 1b and d).
Discussion
We described the clinical features of four Brazilian patients with CAH due to 11β-OHD, from two unrelated families, who were born from consanguineous marriages. Patients from family 1 presented with of virilization and had hypertension at the time of diagnosis. Intriguingly, hypertension resolved during adulthood in the woman (P1), whereas her brother had persistent severe hypertension. Both harbored the homozygous IVS4-1G > A mutation. To our knowledge, this mutation has been uniquely described in in a 46XX child of Brazilian origin, which suggests the possibility of a founder effect [10]. Of note, the clinical evolution of P1 was unusual with regard to her mild hyperandrogenism, and to the complete resolution of hypertension after three uncomplicated pregnancies. This is in contrast to the clinical phenotype of previously reported case, who was assigned male gender due to the high degree of masculinization of the genitalia [10]. The IVS4-1G > A mutation impairs the splicing of CYP11B1 transcript by leading to the recognition of an alternative splice site within exon 4, which results in the loss of 45 nucleotides at the 3′ end. This partial exon deletion alters the reading frame from codon 251 and creates a premature stop codon resulting in a truncated protein with loss of enzymatic activity [10].
We identified a nonsense mutation in exon 6 (p.Q356X), which leads to a truncated and presumably nonfunctional protein in P3 and P4, from family 2. This mutation was first described by Curnow, et al. [11] in an African-American 46,XY boy, and then by De Carvalho, et al. [12] in a 46,XX Brazilian girl [11, 12], both with severe hyperandrogenism and hypertension in childhood and infancy, respectively. Thereafter, Kharrat et al identified this mutation in 4 of 15 unrelated Tunisian patients: a 46,XY boy with precocious pseudopuberty and hypertension and three affected 46,XX girls with ambiguous genitalia (Prader III and IV) [13]. In the current study, the 46,XX affected sibling had a salt-wasting phenotype, which persisted during a follow-up of ~24 months. To our knowledge, the p.Q356X mutation has not been previously associated with salt loss so far.
There is significant variability in biochemical parameters such as plasma levels of 11-deoxycortisol and DOC. As seen in P1, who was normotensive despite elevated DOC levels, there is poor correlation between blood pressure and DOC serum concentration. [1, 4, 7, 11, 14]. Serum 11-deoxycortisol, albeit elevated in all patients, were also variable, similar to reported by Khattab et al. [7]. The mechanisms underlying such a high variability among patients harboring the same CYP11B1 mutations are still elusive. Clinical manifestations may be modulated by inter-individual variations in steroid action, differences in metabolism and clearance of androgens, variability in end-organ responsiveness to circulating androgens and DOC [4]. In P1, specifically, the resolution of the hypertensive state in adulthood after multiple pregnancies is a remarkable finding, and, to our knowledge, previously unreported. Furthermore, there is only one report of a successful pregnancy in a woman with 11β-OHD, albeit no systematic observation is available regarding blood pressure behavior during pregnancy and thereafter in this case [15]. Nevertheless, it is tempting to speculate that the high levels of circulating progesterone secreted by the placenta may influence blood pressure control due to its antagonistic action at the mineralocorticoid receptor, which partially block aldosterone action as seen in a few pregnant women with primary aldosteronism [16, 17]. Thus, a modulatory antagonistic effect of progesterone deserves further investigation in mineralocorticoid-related hypertension in women, especially in high sodium intake conditions [18].
Hypertension has traditionally been described in approximately two thirds of 11β-OHD cases at diagnosis, but the time point when it arises along life is uncertain and may be associated with diverse clinical outcomes [5]. Various genetic and environmental factors may influence the prevalence and variable degree of severity of hypertension in this cohort. It is speculated whether it could reflect polymorphic variants in CYP11B2 or in others genes implicated in the pathogenesis of essential hypertension [19]. On the other hand, salt wasting is very uncommon in 11β-OHD due to mineralocorticoid receptor stimulation by its active precursors, mainly 11-deoxycortisol and DOC. The mechanisms involved in salt wasting in 11β-OHD are currently unknown and it has been attributed to the natriuretic activity of steroids such as progesterone, pregnenolone, and 16-hydroxylated compounds produced by the remaining fetal adrenal tissue [20, 21]. Another possible explanation is an insensitivity of mineralocorticoid receptors to DOC and aldosterone or even DOC suppression by excessive glucocorticoid treatment [22]. Salt-wasting during neonatal period was also reported in a boy carrying a homozygous hybrid CYP11B2-CYP11B1 deletion, probably as a result of aldosterone deficiency of the hybrid [23].
Aldosterone synthase (CYP11B2) catalyzes the final steps of aldosterone biosynthesis and several lines of argument designate CYP11B2 as a candidate gene for hypertension predisposition [19, 24]. The -344T/C polymorphism has been associated with aberrant transcriptional regulation and increased genetic predisposition to hypertension in several studies [24,25,26,27]. Nonetheless, considering that hypertension is classically a complex heterogeneous genetic trait, variants in many other genes beyond CYP11B2, may influence disease risk.
Finally, in the last decade, GWAS and other genomic based studies have addressed hypertension risk and development [28, 29]. Recently, a compilation of GWAS or other genomic-based studies have found more than 40 genetic variants associated with hypertension risk [30]. However, the clinical impact of these findings is currently uncertain and the vast majority of the genetic contribution to variation in blood pressure remains unexplained [28].
Conclusion
We present the unique case of a woman homozygous for the IVS4-1G > A mutation, who had resolution of hypertension during adulthood, after three spontaneous successful pregnancies. At the other end, a novel case of a 46,XX newborn homozygous for the CYP11B1 p.Q356X mutation with a salt-wasting phenotype is described. The molecular mechanisms underlying such a degree of phenotypic variability are incompletely understood, but may be related to individual variation in androgen, progesterone, or mineralocorticoid action on specific target organs, differences in metabolism, and clearance of androgens and DOC, or by co-expression of disease modifying alleles or environmental factors. Expanding the knowledge of the molecular basis of 11β-OHD by systematically analyzing a larger number of cases, together with the investigation of compensatory pathways of biosynthesis and metabolism of adrenal steroids might contribute to better understand phenotypic modulation of blood pressure control and to develop new treatment approaches.
References
S. Nimkarn, M.I. New, Steroid 11beta- hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol. Metab. 19(3), 96–99 (2008). https://doi.org/10.1016/j.tem.2008.01.002
N. Krone, W. Arlt, Genetics of congenital adrenal hyperplasia. Best Pract. Res. Clin. Endocrinol. Metab. 23(2), 181–192 (2009). https://doi.org/10.1016/j.beem.2008.10.014
D.P. Merke, T. Tajima, A. Chhabra, K. Barnes, E. Mancilla, J. Baron, G.B. Cutler, Novel CYP11B1 mutations in congenital adrenal hyperplasia due to steroid 11 beta-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 83(1), 270–273 (1998). https://doi.org/10.1210/jcem.83.1.4513
W.L. Miller, R.J. Auchus, The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32(1), 81–151 (2011). https://doi.org/10.1210/er.2010-0013
K. Bulsari, H. Falhammar, Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Endocrine 55(1), 19–36 (2017). https://doi.org/10.1007/s12020-016-1189-x
M.P. Mello, J.Y. Penachioni, F.C. Amaral, M. Castro, Deficiência da 11 -Hidroxilase. Arq. Bras. Endocrinol. Metabol. 48(5), 713–723 (2004)
A. Khattab, S. Haider, A. Kumar, S. Dhawan, D. Alam, R. Romero, J. Burns, D. Li, J. Estatico, S. Rahi, S. Fatima, A. Alzahrani, M. Hafez, N. Musa, M. Razzghy Azar, N. Khaloul, M. Gribaa, A. Saad, I.Ben Charfeddine, B. Bilharinho de Mendonça, A. Belgorosky, K. Dumic, M. Dumic, J. Aisenberg, N. Kandemir, A. Alikasifoglu, A. Ozon, N. Gonc, T. Cheng, U. Kuhnle-Krahl, M. Cappa, P.-M. Holterhus, M.A. Nour, D. Pacaud, A. Holtzman, S. Li, M. Zaidi, T. Yuen, M.I. New, Clinical, genetic, and structural basis of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Proc. Natl Acad. Sci. USA 114(10), E1933–E1940 (2017). https://doi.org/10.1073/pnas.1621082114
P.K. Whelton, R.M. Carey, W.S. Aronow, B. Ovbiagele, D.E. Casey, S.C. Smith, K.J. Collins, C.C. Spencer, C.D. Himmelfarb, R.S. Stafford, S.M. Depalma, S.J. Taler, S. Gidding, R.J. Thomas, K.A. Jamerson, K.A. Williams, D.W. Jones, J.D. Williamson, E.J. Maclaughlin, J.T. Wright, P. Muntner, G.N. Levine, P.T.O. Gara, J.L. Halperin, I. Past, S.M. Al-khatib, F. Gentile, J.A. Beckman, S. Gidding, K.K. Birtcher, Z.D. Goldberger, M.A. Hlatky, R.G. Brindis, J. Ikonomidis, J.A. Joglar, L. Mauri, S.J. Pressler, High blood pressure clinical practice guideline: executive summary 2017 ACC / AHA / AAPA / ABC / ACPM / AGS /APhA / ASH / ASPC / NMA / PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 71, 1269–1324 (2017)
A.C. Latronico, V.N. Brito, J.C. Carel, Causes, diagnosis and treatment of central precocious puberty. Lancet Diabetes Endocrinol. 8587(15), 1–10 (2016). https://doi.org/10.1016/S2213-8587(15)00380-0
F.C. Soardi, J.Y. Penachioni, G.Z. Justo, T.A.S.S. Bachega, M. Inácio, B.B. Mendonça, M. de Castro, M.P. de Mello, Novel mutations in CYP11B1 gene leading to 11 beta-hydroxylase deficiency in Brazilian patients. J. Clin. Endocrinol. Metab. 94(9), 3481–3485 (2009). https://doi.org/10.1210/jc.2008-2521
K.M. Curnow, L. Slutsker, J. Vitek, T. Cole, P.W. Speiser, M.I. New, P.C. White, L. Pascoe, Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7, and 8. Proc. Natl Acad. Sci. USA 90(10), 4552–4556 (1993)
C.E. De Carvalho, J.Y. Penachioni, M. Castro, A.C. Moreira, M.P.De Mello, CYP11B1 intragenic polymorphisms give evidence for a different Q356X allele in an African-Brazilian patient. J. Endocr. Genet. 1(2), 79–86 (1999)
M. Kharrat, S. Trabelsi, M. Chaabouni, F. Maazoul, L. Kraoua, L. Ben Jemaa, N. Gandoura, S. Barsaoui, Y. Morel, R. M’rad, H. Chaabouni, Only two mutations detected in 15 Tunisian patients with 11β-hydroxylase deficiency: the p.Q356X and the novel p.G379V. Clin. Genet. 78(4), 398–401 (2010). https://doi.org/10.1111/j.13990004.2010.01403.x
Y.S. Zhu, J.J. Cordero, S. Can, L.Q. Cai, X. You, C. Herrera, M. DeFilo-Ricart, C. Shackleton, J. Imperato-McGinley., Mutations in CYP11B1 gene: phenotype - genotype correlations. Am. J. Med. Genet. 122A(15), 193–200 (2003)
P.J. Simm, M.R. Zacharin, Successful pregnancy in a patient with severe 11- beta-hydroxylase deficiency and novel mutations in CYP11B1 gene. Horm. Res. 68(6), 294–297 (2007). https://doi.org/10.1159/000107651
C. Campino, P. Trejo, C.A. Carvajal, A. Vecchiola, C. Valdivia, C.A. Fuentes, J.F. Delgado, C.F. Lagos, M. Aglony, C. Carrasco, A. Martinez-Aguayo, H. Garciá, C. Loureiro, C.E. Fardella, Pregnancy normalized familialhyperaldosteronism type I: a novel role for progesterone. J. Hum. Hypertens. 29(2), 138–139 (2015). https://doi.org/10.1038/jhh.2014.49
V. Ronconi, F. Turchi, M.C. Zennaro, M. Boscaro, G. Giacchetti, Progesterone increase counteracts aldosterone action in a pregnant woman with primary aldosteronism. Clin. Endocrinol. 74(2), 278–279 (2011). https://doi.org/10.1111/j.1365-2265.2010.03901.x
J.W. Funder, Aldosterone and mineralocorticoid receptors—physiology and pathophysiology. Int. J. Mol. Sci. 18(5), 1–9 (2017). https://doi.org/10.3390/ijms18051032
J.M.C. Connell, R. Fraser, S.M. MacKenzie, E.C. Friel, M.C. Ingram, C.D. Holloway, E. Davies, The impact of polymorphisms in the gene encoding aldosterone synthase (CYP11B2) on steroid synthesis and blood pressure regulation. Mol. Cell. Endocrinol. 217(1–2), 243–247 (2004). https://doi.org/10.1016/j.mce.2003.10.025
M. Zachmann, D. Tassinari, A. Prader, Clinical and biochemical variability of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. A study of 25 patients. J. Clin. Endocrinol. Metab. 56(2), 222–229 (1983)
Z. Zadik, L. Kahana, H. Kaufman, A. Benderli, Z. Hochberg, Salt loss in hypertensive form of congenital adrenal hyperplasia (11-β-hydroxylase deficiency). J. Clin. Endocrinol. Metab. 58(2), 384–387 (1984). https://doi.org/10.1210/jcem-58-2-384
Z. Hochberg, A. Benderly, Z. Zadik, Salt loss in congenital adrenal hyperplasia due to 11 β-hydroxylase deficiency. Arch. Dis. Child. 59, 1092–1094 (1984)
B. Ezquieta, C. Luzuriaga, Neonatal salt-wasting and 11 β-hydroxylase deficiency in a child carrying a homozygous deletion hybrid CYP11B2 (aldosterone synthase)-CYP11B1 (11 β-hydroxylase). Clin. Genet. 66(3), 229–235 (2004). https://doi.org/10.1111/j.1399-0004.2004.00291.x
E. Brand, N. Chatelain, P. Mulatero, I. Fery, K. Curnow, X. Jeunemaitre, P. Corvol, L. Pascoe, F. Soubrier, Structural analysis and evaluation of the aldosterone synthase gene in. Hypertension Hypertension 32(2), 198–204 (1998). https://doi.org/10.1161/01.HYP.32.2.198
S. Alvarez-Madrazo, S.M. Mackenzie, E. Davies, R. Fraser, W.-K. Lee, M. Brown, M.J. Caulfield, A.F. Dominiczak, M. Farrall, M. Lathrop, T. Hedner, O. Melander, P.B. Munroe, N. Samani, P.M. Stewart, B. Wahlstrand, J. Webster, CN. Palmer, S. Padmanabhan, JM. Connell, Common polymorphisms in the CYP11B1 and CYP11B2 genes: evidence for a digenic influence on hypertension. Hypertension 61(1), 232–239 (2013). https://doi.org/10.1161/HYPERTENSIONAHA.112.200741
K. Tsukada, T. Ishimitsu, M. Teranishi, M. Saitoh, M. Yoshii, H. Inada, S. Ohta, M. Akashi, J. Minami, H. Ono, M. Ohrui, H. Matsuoka, Positive association of CYP11B2 gene polymorphism with genetic predisposition to essential hypertension. J. Hum. Hypertens. 16(11), 789–793 (2002). https://doi.org/10.1038/sj.jhh.1001484
M. Lisurek, R. Bernhardt, Modulation of aldosterone and cortisol synthesis on the molecular level. Mol. Cell. Endocrinol. 215(1–2), 149–159 (2004). https://doi.org/10.1016/j.mce.2003.11.008
S. Rafiq, S. Anand, R. Roberts, Genome-wide association studies of hypertension: have they been fruitful? J. Cardiovasc. Transl. Res. 3(3), 189–196 (2010). https://doi.org/10.1007/s12265-010-9183-9
D. Levy, G.B. Ehret, K. Rice, G.C. Verwoert, L.J. Launer, A. Dehghan, N.L. Glazer, A.C. Morrison, A.D. Johnson, T. Aspelund, Y. Aulchenko, T. Lumley, A. Köttgen, R.S. Vasan, F. Rivadeneira, G. Eiriksdottir, X. Guo, D.E. Arking, G.F. Mitchell, F.U.S. Mattace-Raso, A.V. Smith, K. Taylor, R.B. Scharpf, S.-J. Hwang, E.J.G. Sijbrands, J. Bis, T.B. Harris, S.K. Ganesh, C.J. O’Donnell, A. Hofman, J.I. Rotter, J. Coresh, E.J. Benjamin, A.G. Uitterlinden, G. Heiss, C.S. Fox, J.C.M. Witteman, E. Boerwinkle, T.J. Wang, V. Gudnason, M.G. Larson, A. Chakravarti, B.M. Psaty, C.M. van Duijn, Genome-wide association study of blood pressure and hypertension. Nat. Genet. 41(6), 677–687 (2009). https://doi.org/10.1038/ng.384.Genome-wide
G.B. Ehret, M.J. Caulfield, Genes for blood pressure: an opportunity to understand hypertension. Eur. Heart J. 34(13), 951–961 (2013). https://doi.org/10.1093/eurheartj/ehs455
Funding
This study was funded by CNPq (Conselho Nacional de Desenvolvimento Cientifico e Tecnológico), research grant number 462346/2014-15 to ALP
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study was approved by the Ethics Committee of the School of Health Sciences, University of Brasilia, Brazil (protocol number 112/11). All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standard.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Valadares, L.P., Pfeilsticker, A.C.V., de Brito Sousa, S.M. et al. Insights on the phenotypic heterogenity of 11β-hydroxylase deficiency: clinical and genetic studies in two novel families. Endocrine 62, 326–332 (2018). https://doi.org/10.1007/s12020-018-1691-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-018-1691-4