
Abstract
MEN1 is the main gene responsible for tumorigenesis of syndromic and sporadic primary hyperparathyroidism (PHPT). Germline mutations of the CDKN1B/p27Kip gene have been associated with multiple endocrine tumors in rats and humans. To evaluate the involvement of the CDKN1B gene and its relationship with MEN1 in sporadic PHPT, we carried out sequencing and loss of heterozygosity analyses of the CDKN1B gene in 147 sporadic parathyroid adenomas. p27 immunohistochemistry and genetic screening of the MEN1 gene were performed in 50 cases. Three germline CDKN1B variants (c.-80C>T, c.-29_-26delAGAG, c.397C>A) were identified in 3/147 patients. Reduction of CDKN1B gene transcription rate was demonstrated in vitro for the novel c.-80C>T and the c.-29_-26delAGAG variants. Loss of p27 expression was detected in the tumor carrying the c.-29_-26delAGAG variant. Two tumors carrying the CDKN1B variants also harbored a MEN1 mutation. Fifty-four percent of 50 CDKN1B mutation-negative tumors had a reduction of p27 nuclear staining. Somatic MEN1 mutations, identified in 15/50 samples, significantly segregated in tumors negative for nuclear and cytoplasmic p27 staining. The germline nature of the CDKN1B mutations suggests that they might predispose to PHPT. The lack of somatic CDKN1B mutations in our samples points to a rare involvement in parathyroid adenomas, despite the frequent loss of nuclear p27 expression. MEN1 biallelic inactivation seems to be directly related to down-regulation of p27 expression through the inhibition of CDKN1B gene transcription.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The molecular pathogenesis of primary hyperparathyroidism (PHPT) is not completely understood. PHPT mostly occurs (90–95 % of cases) as a sporadic form, and rarely as part of hereditary syndromes. Cyclin D1 overexpression (20–40 %), loss of heterozygosity at chromosome 11q13 (25–40 %), and/or somatic mutations of Multiple Endocrine Neoplasia type 1 (MEN1) gene (20–35 %) represent the main known molecular defects of sporadic PHPT [1–3]. Mutations of Beta-Catenin (CTNNB1), Aryl hydrocarbon receptor Interacting Protein (AIP), and methyltransferase Enhancer of Zester Homolog 2 (EZH2) genes have been detected in a minority of cases of PHPT, while a role of other candidate genes has been excluded [4–7].
Dysregulation of cell cycle checkpoints is a key event in tumorigenesis. Cyclin D1 promotes the G1–S phase transition, and menin, the protein encoded by the MEN1 gene, is involved in the cell cycle control through the regulation of gene transcription. Menin, as a component of a SET1-Like Histone Methyltransferase Complex, directly regulates the expression of the Cyclin-Dependent Kinase Inhibitor 1B (CDKN1B/p27Kip1), by specifically binding to transcriptional regulatory elements of its promoter [8, 9].
CDKN1B gene, located on chromosome 12q13.1, encodes a protein of 198 amino acids, p27Kip1 (hereafter p27), a member of the CIP/KIP family of kinase inhibitors. In quiescent cells, p27 is mainly localized in the nucleus, where it negatively regulates the G1-phase cell cycle progression, by binding to and inhibiting cyclin A, E/cyclin-dependent kinase 2 (CDK2) complexes. Conversely, in proliferating cells, a fraction of p27 is phosphorylated on Ser10, promoting its nuclear export into the cytoplasm, removing the inhibitory function from its nuclear targets. The activity of p27 is dependent on its concentration (regulated at transcriptional, translational, and post-translational level), its interaction with different cyclin/CDK complexes, and its subcellular localization (mainly driven by p27 phosphorylation events) [10]. CDKN1B is a tumor suppressor gene, but somatic loss-of-function mutations have rarely been detected in endocrine and non-endocrine human cancers [11–15]. Biallelic inactivation is also an uncommon event, suggesting a haploinsufficient behavior [16, 17]. On the other hand, loss or decreased p27 nuclear expression has been commonly described in human cancers with a poor prognosis [18].
Heterozygous CDKN1B germline mutations have been reported in patients with MEN1-like conditions, namely patients with at least one of the main MEN1-associated tumors, but without MEN1 gene mutations. This condition is called MEN4, although affected patients do not show significant phenotypic differences in comparison with MEN1 mutation-positive cases [19]. The involvement of CDKN1B gene alterations in hereditary multiple endocrine tumors suggested its potential role in the pathogenesis of sporadic PHPT. Indeed, CDKN1B mutations, mostly germline, have been detected in about 3 % of sporadic parathyroid adenomas [20, 21].
In the present study, we further investigate the pathogenic role of the CDKN1B gene in a large series of sporadic parathyroid adenomas and the relationship between the expression of p27 and the MEN1 mutational status.
Materials and methods
Patients and tissue samples
All patients and control subjects gave their informed consent for genetic studies. Our internal Review Board approved the study.
One hundred forty-seven patients with histologically confirmed PHPT due to parathyroid adenoma were studied. No patient had a family history of PHPT or other endocrine tumors. The patients underwent an extensive clinical, biochemical, and instrumental evaluation to exclude manifestations of familial PHPT. Tissues were obtained at surgery, immediately snap frozen in liquid nitrogen, and stored at −80 °C. Blood samples were also obtained from all patients. One hundred Italian healthy subjects were included in the study as controls.
For immunohistochemistry (IHC), we used slices of normal parathyroid glands inadvertently removed from normocalcemic patients undergoing thyroid surgery for non-toxic multinodular goiter. For Real-time PCR, we collected small specimens of normal parathyroid glands that appeared ischemic during major neck surgery for laryngeal or thyroid diseases before their implantation in the sternocleidomastoid muscle.
CDKN1B and MEN1 sequencing
DNA variant numbering is based on reference cDNA sequence (+1 corresponds to the A of the ATG initiation codon), and mutations are reported according to the nomenclature recommendations of the Human Genome Variation Society.
Genetic screening of the CDKN1B gene was performed on the entire cohort of parathyroid adenomas, while sequencing of MEN1 gene was carried out in the three adenomas carrying CDKN1B mutations and in a subgroup of 50 consecutive CDKN1B mutation-negative parathyroid adenomas, which were also investigated for p27 expression by immunohistochemistry (IHC). DNA was extracted from parathyroid tissues and peripheral blood leukocytes with Maxwell™ 16 Instrument (Promega Corp., Madison, USA). The entire coding region and splicing junctions of CDKN1B and MEN1 were amplified and sequenced as previously reported [22, 23]. Both strands of the PCR fragments were separated on ABI 3130XL sequencer (Applied Biosystems, Foster City, USA). The region of interest was also examined in the germline DNA of all carriers of MEN1 and CDKN1B variants.
Multiplex ligation-dependent probe amplification (MLPA) assay
MLPA was used to detect potential large deletions in MEN1 and/or CDKN1B genes, which might escape detection by conventional PCR-based techniques, and to detect loss of heterozygosity (LOH). Experiments were performed on tumor samples of the 50 patients studied for MEN1 mutations and of 51 patients who were wild-type or homozygous for two common CDKN1B polymorphisms, using the SALSA MLPA probemix kit P244-C1, containing probes for the MEN1 and CDKN1B exons (MRC-Holland, Amsterdam, The Netherlands). MLPA assay was performed as previously described [4]. Coffalyser.Net (MRC-Holland) was used to identify copy number variations. Experiments were repeated at least twice. Three reference blood DNAs from healthy subjects and a negative control (a sample without DNA) were included in all experiments.
In silico analysis
Bioinformatics tools, including Align-GVGD (http://agvgd.iarc.fr), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), and SIFT (http://sift.jcvi.org), were used to predict the impact of the CDKN1B missense variant on protein structure and function.
In vitro study
Plasmid construct
The p27_-80T construct was generated by introducing the detected nucleotide changes into the wild-type (WT) human CDKN1B gene promoter (spanning nucleotides -821/-1 to the start site of translation), cloned in the pGL4-p27WT construct, by Quikchange II site-directed mutagenesis kit (Stratagene, Germany). The construct containing the c.-29_-26delAGAG deletion was previously reported [24]. The constructs were verified by sequencing.
Cell culture, transfections, and luciferase assay
GH3 cells (purchased from ATCC) were grown in F12 medium supplemented with 15 % horse serum, 2.5 % FCS, 20 mM l-glutamine, 100 U/ml penicillin G sodium, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, USA).
Transient transfection with the above-mentioned plasmid DNAs was performed using the Amaxa 4D-Nucleofector, following the manufacturer’s instructions (Lonza, Cologne, Germany). The pRL-TK plasmid (Promega, Madison, USA), a wild-type Renilla luciferase control reporter vector, was co-transfected with the p27 reporter construct and used to normalize the transfection efficiency. Empty pGL4 vector was used as control. Following electroporation, GH3 cells were plated into 96-wells plates. After 36 h, the relative luciferase activity was measured using the Dual-Luciferase Assay System and a GloMax 20/20 luminometer, according to the manufacturer’s instructions (Promega).
Real-time quantitative RT-PCR
Total RNA extraction was performed using the Maxwell® LEV Simply RNA Tissue kit on a Promega’s robotics platform. Parathyroid samples from patients #87 and #216 and three fragments of normal parathyroid glands were homogenized and loaded in the cartridge. Total RNA concentration and the A260:A280 ratio were determined with NanoDrop 2000 (Thermo Fisher Scientific, Waltham, USA). Total RNA from each sample and from a pool of the three normal parathyroids was reverse transcribed into cDNA with Superscript IV reverse transcriptase (Life Technologies, Carlsbad, USA). Quantitative gene expression study was performed by real-time PCR using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, USA). PCR reaction was carried out in CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, USA) by cDNA equivalent to 25 ng of total RNA and TaqMan Gene Expression Mix according to the manufacturer’s instructions (Applied Biosystems). The analysis of relative gene expression data was performed with the ΔΔC T method. The results were expressed as the amount of target gene normalized to the endogenous reference gene, Ribosomal Protein L13 (RPL13).
Immunohistochemistry (IHC)
IHC was performed using the Ventana Benchmark immunostaining system (Ventana Medical System, Tucson, USA). A monoclonal anti-p27 antibody (clone SX53G, Ventana) was used to detect p27 expression. Antibody binding was visualized employing 3,3′-diaminobenzidine as chromogen and nuclei were counterstained with hematoxylin. A negative control was included in each experiment by omitting the primary antibody. The rim of adjacent not adenomatous parathyroid parenchyma, infiltrating lymphocytes, and/or endothelial cells served as internal positive control. Three specimens of normal parathyroid gland were used as controls.
IHC scoring assessment
For each slide, we evaluated the percentage of positive cells in at least 4 randomly selected fields under high-power magnification (40×). The cytoplasmic stains were scored separately. The cut-off value for p27 expression was selected generating receiver operating characteristic (ROC) curves (MedCalc Software bvba, Belgium). The sensitivity and specificity of different IHC scores were plotted for the outcome under study (MEN1 genetic status, dichotomized as negative or positive). Tumors were defined as p27 negative, both at nuclear and cytoplasmic level, when the IHC score was below that threshold.
Statistics
The relationship between the nuclear and cytoplasmic p27 expression, either separately or combined, and the genetic status of MEN1 gene was evaluated using the Fisher’s exact test. All p values were two tailed, and statistical significance was set at p < 0.05.
Results
CDKN1B gene
Three CDKN1B variants in 3 out of 147 parathyroid adenomas were identified (Table 1).
A heterozygous missense variant c.397C>A in exon 1 (p.Pro133Thr) was identified in tumor #216. This substitution was previously identified in two sporadic parathyroid adenomas [20]. Multiple alignment query revealed that proline 133 is not a conserved residue across species.
An already reported heterozygous four nucleotides’ deletion at 5′ UTR (c.-29_-26delAGAG) was detected in tumor #87 [24, 25].
An unreported heterozygous substitution at 5′ UTR c.-80C>T was found in tumor #57.
Unexpectedly, all three variants were germline. The c.-80C>T variant was not detected in any of 100 healthy subjects, suggesting that it could not be a common polymorphism. All the three mutated tumors showed retention of heterozygosity at 12p13 (Table 1).
The presence of large deletions of the CDKN1B gene could be excluded in 96 out of 147 tumors carrying one or two among the common CDKN1B polymorphisms (i.e., c.-79C>T and p.Val109Gly) in the heterozygous state. In the remaining 51 tumors, which were wild-type or homozygous for both polymorphisms, MLPA analysis did not identify any CDKN1B large deletion.
The medical records of these patients were carefully reviewed. There was no history of familial PHPT or other endocrine tumors; hormonal data were not available in any of the relatives. Patient #216 had asymptomatic PHPT diagnosed at 49 years, autoimmune hypothyroidism, and fibrocystic breast disease. Patient #87 had severe PHPT diagnosed at 61 years and no other relevant diseases. Patient #57 had PHPT with nephrolithiasis diagnosed at 38 years and no other relevant diseases. A single clear cell and chief cell adenoma was excised from patients #216 and #87 and a double chief cell adenoma from patient #57. All patients remained normocalcemic after parathyroidectomy for 16, 14, and 8 years, respectively. At the latest follow-up visit after the identification of the CDKN1B variants, all three patients refused further evaluation, but no history of endocrine or non-endocrine tumors was reported. Three well-known benign single nucleotide polymorphisms (SNPs), a c.-79C>T nucleotide replacement in the 5′ UTR (rs34330), a valine for glycine substitution at position 109 (rs2066827:T>G, p.Val109Gly), and a synonymous c.426G>A change (p.Thr142Thr) were detected in our cohort. The SNP rs34330 was found in heterozygous state in patients #87 and #216 and the SNP rs2066827 in all three CDKN1B-mutated samples, in homozygosity in patient #87.
MEN1 gene
MEN1 mutations were found in two of the three CDKN1B mutated tumors.
The already described variant c.1621G>A (p.Ala541Thr) in exon 10 was detected in tumor #216 and in peripheral leukocytes. A somatic MEN1 deletion, c.152delA, in exon 2, causing a shift in the reading frame and a premature termination of menin at codon 117 (p.Asn51ThrfsX68), was identified in tumor #87. Both MEN1 mutations were in hemizygous state at the somatic level for the concurrent presence of allelic loss at the 11q13 locus, leading to a biallelic inactivation of menin. No MEN1 mutation, large deletion, or LOH was detected in tumor #57 (Table 1).
MEN1 mutational screening and copy number variation detection were also performed in the 50 CDKN1B mutation-negative adenomas that were investigated for the expression of p27 by IHC. Somatic MEN1 mutations were found in 15 tumors (Table 2). Allelic loss at 11q13 locus was detected in all but one mutated adenoma samples (Table 2). MEN1 large deletions were not detected in any of the 50 parathyroid tumors.
In silico analysis
The three different tools used, based on different algorithms, gave comparable results and all indicate that the p.Pro133Thr has a neutral effect on protein function.
In vitro study
We cloned the 5′-UTR region of the CDKN1B promoter into a luciferase reporter gene vector and introduced the c.-80C>T change by site-directed mutagenesis. The c.-29_-26delAGAG construct was also included as a positive control. GH3 cells (devoid of endogenous p27) were transfected with constructs bearing the WT or the mutated (p27_-80T and p27_-29_-26del) promoter region, and luciferase activity was measured. Cells transfected with the p27_-80T and p27_-29_-26delAGAG showed a highly significant reduction (p < 0.0001) in luciferase activity (threefold and 5.5-fold reduction, respectively) when compared with cells transfected with the WT construct (Fig. 1).

Relative luciferase activity (vs empty-vector-control) was measured in GH3 cells transfected with p27_WT, p27_-80T (genetic condition of the patient #57) and p27_-29_-26delAGAG deletion (genetic condition of the patient #87) (p27_-29_-26del) constructs. Luciferase activity of the c.-80T construct is significantly lower than that of the WT construct. The deletion-containing construct shows a luciferase activity lower than the other ones. p values were calculated using a two-tailed t test. ****p < 0.0001. Values were expressed relative to those generated as mean of three replicates. Error bars indicate standard deviation
Real-time quantitative RT-PCR
The comparative quantitative PCR showed that parathyroid adenomas from patients #87 and #216 had a lower level of p27 expression with respect to the pool of normal parathyroid glands (p < 0.001 and <0.01, respectively), which was on average 16 and 3.6-fold less for samples bearing the c.-29_-26delAGAG and c.397C>A variant, respectively (Fig. 2).

Bar graph representing real-time PCR results. The mRNA expression fold change of the CDKN1B gene by the ΔΔC t method relative to the internal control gene (RPL13) in a pool of three normal parathyroids (Pool PN) and in parathyroid adenomas carrying c.-29_-26delAGAG (sample #87) and c.397C>A (samples #216) CDKN1B variants. The fold difference was calculated as \(2^{{ - \Delta \Delta {C}_{\text{t}} }}\), where ΔΔC t = ΔC t sample- ΔC t calibrator and ΔC t = Ct value of CDKN1B - Ct value of RPL13. The average Ct value of a pool of three normal parathyroids (PN) was used as the calibrator in the analysis. Statistical significance was determined by Student’s t test. **p < 0.01; ***p < 0.001. Values were expressed relative to those generated as mean of three replicates. Error bars indicate standard deviation
Immunohistochemistry
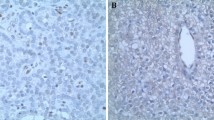
Immunostaining for the p27 protein was performed on one (#87) of the three CDKN1B mutated tumors (specimens #216 and #57 were not available) and on 50 CDKN1B mutation-negative parathyroid adenomas. Three specimens of normal parathyroid gland were also studied. Normal parathyroid gland showed a moderate to strong nuclear staining intensity, with a percentage of positive cells ranging between 60 and 90. No staining was detected in the cytoplasm. Neither nuclear nor cytoplasmic staining was observed in the tumor specimen #87 (Fig. 3).

Representative immunohistochemical p27 staining (original magnification ×40). a Normal parathyroid gland with nearly all nuclei showing p27 staining. b Parathyroid adenoma carrying the c.-29_-26delAGAG CDKN1B mutation (patient #87), showing a complete absence of p27 nuclear and cytoplasmic staining. Endothelial cells (arrows) show the typical strong nuclear positivity and serve as internal control to check for the adequacy of the staining. c A CDKN1B mutation-negative parathyroid adenoma scored as negative in the nucleus and in the cytoplasm (arrows indicate the rim of normal parathyroid tissue showing nuclear and weak cytoplasmic immunoreactivity for p27). d A CDKN1B mutation-negative parathyroid adenoma with a strong nuclear immunoreactivity for p27 and no cytoplasmic staining. e A CDKN1B mutation-negative parathyroid adenoma showing the loss of p27 expression in the nucleus and moderate cytoplasmic staining. f A CDKN1B mutation-negative parathyroid adenoma showing both nuclear and cytoplasmic staining with moderate intensity. Scale bar 100 μm
The cut-off values generated by the ROC curves (Fig. 4) were used to categorize the parathyroid specimens into two groups, namely positive and negative. We presented the following results according to the threshold values of 20 and 10 % of positive stained cells, respectively, relative to the nuclear and cytoplasmic staining (Table 3).

Receiver operating characteristic (ROC) curves for the selection of optimal IHC cut-off values (criterion) relative to p27 expression. Each percentage scores a cells with p27 positive staining in the nucleus, and b cells with p27 positive staining in the cytoplasm—were plotted against the sensitivity and specificity for the presence or absence of MEN1 mutations. The area under the ROC curve (AUROC) was calculated to estimate the discriminatory power of p27 over the entire range of scores
Of the 50 CDKN1B mutation-negative adenomas, 23 were scored as nuclear positive (range of positive cells, 30–90 %) and 27 as nuclear negative. Five of the 23 positive and 7 of the 27 negative samples showed a cytoplasmic staining (Table 3).
Cytoplasmic/nuclear p27 expression and mutation of the MEN1 gene
We examined whether p27 expression (nuclear, cytoplasmic, or combined) was associated or not with MEN1 gene mutational status. We found a statistically significant association between both negative nuclear and cytoplasmic staining and the presence of MEN1 mutation (Table 3). Similar results were observed when the MEN1 mutational status was correlated with the combined nuclear and cytoplasmic p27 staining (Table 3). In particular, among p27 nuclear-negative tumors, MEN1 mutations were exclusively found in those scored as cytoplasmic negative (p = 0.008). On the other hand, among p27 cytoplasmic-negative tumors, MEN1 mutations were significantly more common in those scored as nuclear negative (60 %), as compared with those scored as nuclear positive (16.7 %, p = 0.009; Table 3).
Discussion
Up to date, only three studies examined the possible role of CDKN1B mutations in patients with sporadic PHPT, with discrepant results [20, 21, 26]. The overall prevalence of germline CDKN1B mutations in sporadic PHPT cases is 1.8 % (3/166) and decreases to 0.8 % (1/128) if we consider only somatic mutations.
In this study, we detected three different germline CDKN1B variants in 147 (2.0 %) apparently sporadic parathyroid adenomas, updating the current prevalence of germline variants to 1.9 %, also including variants of uncertain pathogenic significance (VUS) (6/313). The apparent sporadic nature of PHPT in our CDKN1B-mutated patients is in contrast with the finding of germline mutations. Two patients had a single parathyroid adenoma and the third—the youngest patient—a double adenoma; all were cured by surgery. The rather advanced age at diagnosis in the sporadic cases carrying CDKN1B germline mutations so far identified, with the exception of one case (15 years) [n = 6, mean (±SD) age 56.0 ± 11.6 years, range 38–68 years], is very similar to that reported in MEN4 cases (55.6 ± 11.8 years, 41–79 years) [19, 20, 27, 28]. These patients might represent a variant of MEN4 with incomplete phenotypic expression and might have a risk of developing other MEN4-associated tumors.
The c.-80C>T variant (#57) has not been previously reported and was shown to affect in vitro CDKN1B transcription rates. The lack of LOH at the p27 locus in this tumor is not surprising, since it has been detected in a few MEN4-related tumors. Thus, we cannot, at present, rule out that CDKN1B may act as a haploinsufficient gene also in human endocrine tissues, as previously demonstrated in animal models [16]. Unfortunately, the tumor tissue #57 was not available for p27 mRNA nor immunohistochemical studies. This variant is located next to a known polymorphism at −79, which is considered as a putative genetic risk factor for some cancers [29–31] and has been associated with a significant lower transcription rate [32]. Indeed, our patient did not carry this variant.
The 5′-UTR deletion c.-29_-26delAGAG (#87), previously reported as c.-32_-29delGAGA by Malanga et al. in a sporadic MEN4, and by Sambugaro et al. in a acromegalic patient, was associated with a complete loss of p27 expression, which, in the absence of LOH in the patient’s tumor, suggests the presence of an alternative somatic (genetic or epigenetic) second hit and excludes an haploinsufficient behavior [24, 25, 33]. The loss of p27 in our patient’s tumor is in keeping with previous and our functional studies showing a down-regulation of CDKN1B mRNA expression [24, 25]. Our mRNA expression data are in line with the previous observation by Malanga et al. in the blood cells of their patient. Interestingly, the transcription rate in our tissue sample was affected to a greater extent with respect to the peripheral blood leukocytes (16-fold vs. threefold reduction), suggesting that an additive somatic defect (i.e., menin alteration) could further affect transcription in the tumor [25]. These data further confirm that the 5′-UTR region including the GAGAGA element has a primary role in CDKN1B transcription [24, 25, 34].
The p.Pro133Thr variant has previously been reported in two sporadic parathyroid adenomas, both in germline, and in a novel MEN4 case presenting parathyroid adenoma in association with cerebral meningioma and papillary thyroid carcinoma [2, 20, 35]. LOH at CDKN1B locus was detected neither by us nor by Costa-Guda et al., arguing against a biallelic inactivation. Functional studies revealed the lack of abnormal protein degradation [20]. Conversely, the lack of p.Pro133Thr in any of the 240 alleles from healthy subjects nor in > 2000 alleles reported in the databases led the authors to conclude that this change might have a role in parathyroid tumorigenesis [20]. On the other hand, the ExAC database reported this variant (rs137985549) in three subjects in homozygosity and the in silico prediction indicates a neutral effect of the p.Pro133Thr on protein function. Unfortunately, no IHC studies could be performed either by us or by Costa-Guda et al. because of unavailability of tissue samples. Based on these findings, the p.Pro133Thr is considered as a VUS and its role as susceptibility factor has to be further investigated [36].
A reduced nuclear p27 staining was detected in 27 of the 50 (54 %) CDKN1B mutation-negative adenomas, suggesting that defects in the regulation of CDKN1B transcription or post-transcriptional, post-translational mechanisms may account for this finding.
It is worth noting that one of the tumor-suppressor mechanisms of menin is mediated by the transcriptional activation of CDKN1B and CDKN2C (encoding p18) genes, through the recruitment of the Mixed Lineage Leukemia (MLL) family of proteins [8, 9]. These studies demonstrated the binding of menin to MLL in a histone methyltransferase (HMT) complex and their co-localization at target promoters. Moreover, a direct link between MLL or menin loss of function and down-regulation of p27 and p18 expression was also shown [8, 9]. In particular, Milne et al. [9] also showed a reduction of p27 expression in parathyroid and pancreatic MEN1-related tumors, confirming a relationship between the lack of a functional menin and the loss of p27 in human endocrine tumors. Menin-MLL-p27 pathway down-regulation was also demonstrated in sporadic pituitary adenomas [37].
Based on these premises, we evaluated MEN1 gene abnormalities in CDKN1B-mutated and non-mutated tumors. A MEN1 germline substitution (p.Ala541Thr) was found in CDKN1B-mutated tumor #216. This variant was firstly classified as a benign polymorphism, being present in 6.2–16.5 % of control population in ExAc and dbSNP databases, respectively, and in 2–7.6 % in different reports [3, 38]. However, functional studies have shown that, in contrast to wild-type, the overexpression of the p.Ala541Thr mutant protein does not inhibit cell growth, suggesting that p.Ala541Thr might be a pathogenic mutation or, at least, a pathogenic non-synonymous polymorphism [39–41]. This conclusion is supported by the identification of this variant in a somatic setting in sporadic parathyroid adenomas [39]. LOH at 11q13 region detected in tumor #216 further supports a role for the MEN1 gene in tumor growth. Based on these considerations, only functional experiments and clinical evaluation of carriers of variants initially classified as polymorphisms might establish its pathogenic role.
Interestingly, patient #87 carries the c.-29_-26delAGAG germline CDKN1B mutation and a biallelic inactivation of the MEN1 gene [i.e., somatic frameshift mutation (c.152delA) causing a probably deleterious menin protein and LOH at 11q13]. Although we could hypothesized that the loss of p27 expression could only be due to the MEN1 somatic defects, causing the inhibition of p27 transcription through menin-MLL deregulation, we cannot rule out a role of the germline CDKN1B c.-29_-26delAGAG in parathyroid tumorigenesis predisposition. Indeed, the combined loss of menin and p27 in the double p27/Men1 knockout mice does not seem to have synergistic effects on pancreatic tumor development [42]. However, the coexistence of germline CDKN1B and somatic MEN1 mutations might suggest that they could be sequential events leading to tumor development. Finally, we could hypothesize that the germline CDKN1B mutation, affecting a single allele, might cause a partial reduction in gene transcription and the somatic inactivation of menin might have the greatest effect on inhibiting the transcription of the wild-type CDKN1B allele, causing the complete loss of p27 expression, as shown by IHC analysis.
The choice of p27 IHC cut-off values is variable in the literature (0–62.4 %) [43]. Different studies have evaluated p27 nuclear expression on parathyroid tumors by IHC or tissue microarray [20, 44–51]. All studies found a progressive decrease, compared with normal parathyroid, in the percentage of p27 positive cells from hyperplasia–adenoma–carcinoma, suggesting that the reduction of p27 expression could be directly related to malignancy. Empiric cut-off values of p27 positivity were only used in 4/9 studies as shown in the table (Online Resource 1) [20, 47–49]. Therefore, to avoid the problem of misclassifying tumors regarding their p27 status, we used ROC curves to select the optimal cut-off value. We found a higher percentage (54 %) of adenomas with negative nuclear p27 expression with respect to the literature. MEN1 mutations were found in 30 % of tumors, in agreement with the literature [4, 5, 52]. Eighty percent of MEN1-mutated adenomas showed a decreased p27 nuclear staining, in agreement with the finding of a reduced p27 expression in 77 % of the insulinomas developed in conditional organ-specific Men1 knockout mice, suggesting a loss of menin-MLL-dependent transcription of the Cdkn1b target gene [53]. Our results suggest a link between menin mutations and p27 nuclear loss consistent with those obtained in MEN1-related pancreatic and parathyroid tumors and in sporadic pituitary adenomas [9, 37]. In the three MEN1 mutation-positive adenomas with positive nuclear p27 expression (20 %), other pathways and/or gene targets regulated by menin (SMAD proteins, Runx2, JunD, nuclear factor κB) might be altered in parathyroid tumorigenesis.
Forty-three percent of the tumors that did not carry MEN1 mutations and/or LOH at 11q13 had a reduced p27 nuclear staining. Unfortunately, in these cases, we were not able to assess CDKN1B transcription levels for unavailability of tissue samples. However, we can hypothesize that the down-regulation of p27 expression might be due to inhibition of p27 translation, post-transcriptional or post-translational events, and/or enhanced p27 proteolysis by MEN1-independent mechanisms.
Herein we also evaluated p27 cytoplasmic expression. It is well known that the regulatory function of p27 on cell proliferation depends upon its subcellular localization. The cytoplasmic displacement of p27, as well as its nuclear loss, has been associated with an adverse clinical outcome in various cancers, suggesting that p27 might exert dual functions in carcinogenesis, being either a nuclear tumor suppressor or a cytoplasmic oncoprotein [43]. To our knowledge, no data about the biological meaning of cytoplasmic p27 in benign neoplasia are available. In contrast with the findings in colorectal cancer, we found adenomas showing p27 cytoplasmic sequestration together with nuclear loss or retention, indicating that different mechanisms of p27 inactivation may coexist in parathyroid tumorigenesis [54]. All adenomas with negative nuclear p27 expression and MEN1 mutation-positive had low p27 cytoplasmic levels, supporting a menin-dependent loss of p27 transcription, while all adenomas with positive cytoplasmic p27 staining were MEN1 wild-type, suggesting that in these tumors, the p27 deregulation might be due to menin-independent mechanisms, i.e., accelerated p27 degradation and/or mislocalization, miRNA-mediated inhibition of translation, or post-translational events [55–57].
In summary, we did not identified CDKN1B somatic variants in any of the 147 sporadic PHPT, confirming that CDKN1B mutations are extremely rare in parathyroid tumors. However, we found three germline CDKN1B variants, and the in vitro study carried out for two of them suggested their potential role in parathyroid tumorigenesis. Of note, reduction of p27 nuclear staining in CDKN1B mutation-negative adenomas was statistically correlated with the presence of MEN1 somatic mutations. Further studies will be required to better understand the additional mechanisms of p27 function impairment and the dual role that p27 might have on parathyroid tumorigenesis depending on its cellular localization.
References
F. Cetani, E. Pardi, S. Borsari, C. Marcocci, Molecular pathogenesis of primary hyperparathyroidism. J. Endocrinol. Investig. 34, 35–39 (2011)
J. Costa-Guda, A. Arnold, Genetic and epigenetic changes in sporadic endocrine tumors: parathyroid tumors. Mol. Cell. Endocrinol. 386, 46–54 (2014)
M.C. Lemos, R.V. Thakker, Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum. Mutat. 29, 22–32 (2008)
E. Pardi, C. Marcocci, S. Borsari, F. Saponaro, L. Torregrossa, M. Tancredi, B. Raspini, F. Basolo, F. Cetani, Aryl hydrocarbon receptor interacting protein (AIP) mutations occur rarely in sporadic parathyroid adenomas. J. Clin. Endocrinol. Metab. 98, 2800–2810 (2013)
M.K. Cromer, L.F. Starker, M. Choi, R. Udelsman, C. Nelson-Williams, R.P. Lifton, T. Carling, Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J. Clin. Endocrinol. Metab. 97, E1774–E1781 (2012)
L.F. Starker, A.L. Fonseca, G. Akerstrom, P. Bjorklund, G. Westin, T. Carling, Evidence of a stabilizing mutation of beta-catenin encoded by CTNNB1 exon 3 in a large series of sporadic parathyroid adenomas. Endocrine 42, 612–615 (2012)
L.F. Starker, A. Delgado-Verdugo, R. Udelsman, P. Bjorklund, T. Carling, Expression and somatic mutations of SDHAF2 (SDH5), a novel endocrine tumor suppressor gene in parathyroid tumors of primary hyperparathyroidism. Endocrine 38, 397–401 (2010)
S.K. Karnik, C.M. Hughes, X. Gu, O. Rozenblatt-Rosen, G.W. McLean, Y. Xiong, M. Meyerson, S.K. Kim, Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. U.S.A. 102, 14659–14664 (2005)
T.A. Milne, C.M. Hughes, R. Lloyd, Z. Yang, O. Rozenblatt-Rosen, Y. Dou, R.W. Schnepp, C. Krankel, V.A. Livolsi, D. Gibbs, X. Hua, R.G. Roeder, M. Meyerson, J.L. Hess, Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 102, 749–754 (2005)
G. Viglietto, M.L. Motti, A. Fusco, Understanding p27(kip1) deregulation in cancer: down-regulation or mislocalization. Cell Cycle 1, 394–400 (2002)
B. Belletti, G. Baldassarre, New light on p27(kip1) in breast cancer. Cell Cycle 11, 3701–3702 (2012)
S. Dietrich, J. Hullein, S.C. Lee, B. Hutter, D. Gonzalez, S. Jayne, M.J. Dyer, M. Oles, M. Else, X. Liu, M. Slabicki, B. Wu, X. Troussard, J. Durig, M. Andrulis, C. Dearden, C. von Kalle, M. Granzow, A. Jauch, S. Frohling, W. Huber, M. Meggendorfer, T. Haferlach, A.D. Ho, D. Richter, B. Brors, H. Glimm, E. Matutes, O. Abdel, T. Zenz, Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood 126, 1005–1008 (2015)
J.E. Maxwell, S.K. Sherman, G. Li, A.B. Choi, A.M. Bellizzi, T.M. O’Dorisio, J.R. Howe, Somatic alterations of CDKN1B are associated with small bowel neuroendocrine tumors. Cancer Genet. 208(11), 564–570 (2015)
R. Morosetti, N. Kawamata, A.F. Gombart, C.W. Miller, Y. Hatta, T. Hirama, J.W. Said, M. Tomonaga, H.P. Koeffler, Alterations of the p27KIP1 gene in non-Hodgkin’s lymphomas and adult T-cell leukemia/lymphoma. Blood 86, 1924–1930 (1995)
J. Crona, T. Gustavsson, O. Norlen, K. Edfeldt, T. Akerstrom, G. Westin, P. Hellman, P. Bjorklund, P. Stalberg, Somatic Mutations and Genetic Heterogeneity at the CDKN1B Locus in Small Intestinal Neuroendocrine Tumors. Ann. Surg. Oncol. 45(12), 1483–1486 (2015)
M.L. Fero, E. Randel, K.E. Gurley, J.M. Roberts, C.J. Kemp, The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 396, 177–180 (1998)
J. Philipp-Staheli, S.R. Payne, C.J. Kemp, p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp. Cell Res. 264, 148–168 (2001)
J. Slingerland, M. Pagano, Regulation of the Cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 183, 10–17 (2000)
M. Lee, N.S. Pellegata, Multiple endocrine neoplasia syndromes associated with mutation of p27. J. Endocrinol. Investig. 36, 781–787 (2013)
J. Costa-Guda, I. Marinoni, S. Molatore, N.S. Pellegata, A. Arnold, Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J. Clin. Endocrinol. Metab. 96, E701–E706 (2011)
T. Gluick, Z. Yuan, S.K. Libutti, S.J. Marx, Mutations in CDKN2C (p18) and CDKN2D (p19) may cause sporadic parathyroid adenoma. Endocr. Relat. Cancer 20, L27–L29 (2013)
F. Cetani, E. Pardi, A. Giovannetti, P. Cerrai, S. Borsari, E. Vignali, A. Picone, L. Cianferotti, P. Miccoli, A. Pinchera, C. Marcocci, Six novel MEN1 gene mutations in sporadic parathyroid tumors. Hum. Mutat. 16, 445 (2000)
N.S. Pellegata, L. Quintanilla-Martinez, H. Siggelkow, E. Samson, K. Bink, H. Hofler, F. Fend, J. Graw, M.J. Atkinson, Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. U.S.A. 103, 15558–15563 (2006)
S. Sambugaro, M. Di Ruvo, M.R. Ambrosio, N.S. Pellegata, M. Bellio, A. Guerra, M. Buratto, M.P. Foschini, F. Tagliati, E. Degli Uberti, M.C. Zatelli, Early onset acromegaly associated with a novel deletion in CDKN1B 5′UTR region. Endocrine 49, 58–64 (2015)
D. Malanga, S. De Gisi, M. Riccardi, M. Scrima, C. De Marco, M. Robledo, G. Viglietto, Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur. J. Endocrinol. 166, 551–560 (2012)
S.K. Agarwal, C.M. Mateo, S.J. Marx, Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J. Clin. Endocrinol. Metab. 94, 1826–1834 (2009)
E. Pardi, S. Mariotti, N.S. Pellegata, K. Benfini, S. Borsari, F. Saponaro, L. Torregrossa, A. Cappai, C. Satta, M. Mastinu, C. Marcocci, F. Cetani, Functional characterization of a CDKN1B mutation in a Sardinian kindred with multiple endocrine neoplasia type 4 (MEN4). Endocr. Connect. 4, 1–8 (2015)
M.S. Elston, G.Y. Meyer-Rochow, M. Dray, M. Swarbrick, J.V. Conaglen, Early onset primary hyperparathyroidism associated with a novel germline mutation in CDKN1B. Case Rep. Endocrinol. 2015, 510985 (2015)
B.L. Chang, S.L. Zheng, S.D. Isaacs, K.E. Wiley, A. Turner, G. Li, P.C. Walsh, D.A. Meyers, W.B. Isaacs, J. Xu, A polymorphism in the CDKN1B gene is associated with increased risk of hereditary prostate cancer. Cancer Res. 64, 1997–1999 (2004)
W. Wang, M.R. Spitz, H. Yang, C. Lu, D.J. Stewart, X. Wu, Genetic variants in cell cycle control pathway confer susceptibility to lung cancer. Clin. Cancer Res. 13, 5974–5981 (2007)
H. Xiang, H. Li, W. Ge, W. Wu, M. Gao, W. Wang, L. Hong, D. Jiang, C. Zhang, Association of CDKN1B gene polymorphisms with susceptibility to breast cancer: a meta-analysis. Mol. Biol. Rep. 40, 6371–6377 (2013)
I. Landa, C. Montero-Conde, D. Malanga, S. De Gisi, G. Pita, L.J. Leandro-Garcia, L. Inglada-Perez, R. Leton, C. De Marco, C. Rodriguez-Antona, G. Viglietto, M. Robledo, Allelic variant at −79 (C > T) in CDKN1B (p27Kip1) confers an increased risk of thyroid cancer and alters mRNA levels. Endocr. Relat. Cancer 17, 317–328 (2010)
C. Verdelli, I. Forno, V. Vaira, S. Corbetta, Epigenetic alterations in human parathyroid tumors. Endocrine 49, 324–332 (2015)
J. Coleman, W.K. Miskimins, Structure and activity of the internal ribosome entry site within the human p27 Kip1 5′-untranslated region. RNA Biol. 6, 84–89 (2009)
M.J. Bugalho, R. Domingues, Uncommon association of cerebral meningioma, parathyroid adenoma and papillary thyroid carcinoma in a patient harbouring a rare germline variant in the CDKN1B gene. BMJ Case Rep. (2016). doi:10.1136/bcr-2015-213934
N.S. Pellegata, MENX and MEN4. Clin. (Sao Paulo) 67(Suppl 1), 13–18 (2012)
K. Horiguchi, M. Yamada, T. Satoh, K. Hashimoto, J. Hirato, M. Tosaka, S. Yamada, M. Mori, Transcriptional activation of the mixed lineage leukemia-p27Kip1 pathway by a somatostatin analogue. Clin. Cancer Res. 15, 2620–2629 (2009)
S.K. Agarwal, M.B. Kester, L.V. Debelenko, C. Heppner, M.R. Emmert-Buck, M.C. Skarulis, J.L. Doppman, Y.S. Kim, I.A. Lubensky, Z. Zhuang, J.S. Green, S.C. Guru, P. Manickam, S.E. Olufemi, L.A. Liotta, S.C. Chandrasekharappa, F.S. Collins, A.M. Spiegel, A.L. Burns, S.J. Marx, Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum. Mol. Genet. 6, 1169–1175 (1997)
L. Shan, Y. Nakamura, M. Nakamura, T. Yokoi, M. Tsujimoto, R. Arima, T. Kameya, K. Kakudo, Somatic mutations of multiple endocrine neoplasia type 1 gene in the sporadic endocrine tumors. Lab. Investig. 78, 471–475 (1998)
W. Bazzi, M. Renon, C. Vercherat, Z. Hamze, A. Lacheretz-Bernigaud, H. Wang, M. Blanc, C. Roche, A. Calender, J.A. Chayvialle, J.Y. Scoazec, M. Cordier-Bussat, MEN1 missense mutations impair sensitization to apoptosis induced by wild-type menin in endocrine pancreatic tumor cells. Gastroenterology 135, 1698–1709 (2008)
C. Nozieres, C.X. Zhang, A. Buffet, S. Dupasquier, R. Vargas-Poussou, M. Guillaud-Bataille, M. Cordier-Bussat, P. Ruszniewski, S. Christin-Maitre, A. Murat, L. Groussin, D. Vezzosi, C. Cardot-Bauters, V. Hervieu, M.O. Joly, S. Giraud, M.F. Odou, A.P. Gimenez-Roqueplo, P. Goudet, F. Borson-Chazot, A. Calender, e. Groupe francais des tumeurs, p.Ala541Thr variant of MEN1 gene: a non deleterious polymorphism or a pathogenic mutation? Ann. Endocrinol. (Paris) 75, 133–140 (2014)
F. Bai, X.H. Pei, T. Nishikawa, M.D. Smith, Y. Xiong, p18Ink4c, but not p27Kip1, collaborates with Men1 to suppress neuroendocrine organ tumors. Mol. Cell. Biol. 27, 1495–1504 (2007)
I.M. Chu, L. Hengst, J.M. Slingerland, The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 8, 253–267 (2008)
R.V. Lloyd, L. Jin, X. Qian, E. Kulig, Aberrant p27kip1 expression in endocrine and other tumors. Am. J. Pathol. 150, 401–407 (1997)
L.A. Erickson, L. Jin, P. Wollan, G.B. Thompson, J.A. van Heerden, R.V. Lloyd, Parathyroid hyperplasia, adenomas, and carcinomas: differential expression of p27Kip1 protein. Am. J. Surg. Pathol. 23, 288–295 (1999)
M. Tokumoto, K. Tsuruya, K. Fukuda, H. Kanai, S. Kuroki, H. Hirakata, Reduced p21, p27 and vitamin D receptor in the nodular hyperplasia in patients with advanced secondary hyperparathyroidism. Kidney Int. 62, 1196–1207 (2002)
A. Stojadinovic, A. Hoos, A. Nissan, M.E. Dudas, C. Cordon-Cardo, A.R. Shaha, M.F. Brennan, B. Singh, R.A. Ghossein, Parathyroid neoplasms: clinical, histopathological, and tissue microarray-based molecular analysis. Hum. Pathol. 34, 54–64 (2003)
N. Bergero, R. De Pompa, C. Sacerdote, G. Gasparri, M. Volante, G. Bussolati, M. Papotti, Galectin-3 expression in parathyroid carcinoma: immunohistochemical study of 26 cases. Hum. Pathol. 36, 908–914 (2005)
G.G. Fernandez-Ranvier, E. Khanafshar, D. Tacha, M. Wong, E. Kebebew, Q.Y. Duh, O.H. Clark, Defining a molecular phenotype for benign and malignant parathyroid tumors. Cancer 115, 334–344 (2009)
B.M. Erovic, L. Harris, M. Jamali, D.P. Goldstein, J.C. Irish, S.L. Asa, O. Mete, Biomarkers of parathyroid carcinoma. Endocr. Pathol. 23, 221–231 (2012)
M.I. Alvelos, J. Vinagre, E. Fonseca, E. Barbosa, J. Teixeira-Gomes, M. Sobrinho-Simoes, P. Soares, MEN1 intragenic deletions may represent the most prevalent somatic event in sporadic primary hyperparathyroidism. Eur. J. Endocrinol. 168, 119–128 (2013)
P.J. Newey, M.A. Nesbit, A.J. Rimmer, R.A. Head, C.M. Gorvin, M. Attar, L. Gregory, J.A. Wass, D. Buck, N. Karavitaki, A.B. Grossman, G. McVean, O. Ansorge, R.V. Thakker, Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J. Clin. Endocrinol. Metab. 98, E796–E800 (2013)
S. Fontaniere, H. Casse, P. Bertolino, C.X. Zhang, Analysis of p27(Kip1) expression in insulinomas developed in pancreatic beta-cell specific Men1 mutant mice. Fam. Cancer 5, 49–54 (2006)
S. Ogino, T. Kawasaki, A. Ogawa, G.J. Kirkner, M. Loda, C.S. Fuchs, Cytoplasmic localization of p27 (cyclin-dependent kinase inhibitor 1B/KIP1) in colorectal cancer: inverse correlations with nuclear p27 loss, microsatellite instability, and CpG island methylator phenotype. Hum. Pathol. 38, 585–592 (2007)
S. Corbetta, V. Vaira, V. Guarnieri, A. Scillitani, C. Eller-Vainicher, S. Ferrero, L. Vicentini, I. Chiodini, M. Bisceglia, P. Beck-Peccoz, S. Bosari, A. Spada, Differential expression of microRNAs in human parathyroid carcinomas compared with normal parathyroid tissue. Endocr. Relat. Cancer 17, 135–146 (2010)
C. le Sage, R. Nagel, D.A. Egan, M. Schrier, E. Mesman, A. Mangiola, C. Anile, G. Maira, N. Mercatelli, S.A. Ciafre, M.G. Farace, R. Agami, Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 26, 3699–3708 (2007)
J. Lee, S.S. Kim, The function of p27 KIP1 during tumor development. Exp. Mol. Med. 41, 765–771 (2009)
J.H.D. Bassett, S.A. Forbes, A.A.J. Pannett, S.E. Lloyd, P.T. Christie, C. Wooding, B. Harding, G.M. Besser, C.R. Edwards, J.P. Monson, J. Sampson, J.A.H. Wass, M.H. Wheeler, R.V. Thakker, Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am. J. Hum. Genet. 62, 232–244 (1998)
C. Tanaka, K. Yoshimoto, S. Yamada, H. Nishioka, S. Li, M. Moritani, T. Yamaoka, M. Itakura, Absence of germ-line mutations of the multiple endocrine neoplasia type 1 (MEN1) gene in familial pituitary adenoma in contrast to MEN1 in Japanese. J. Clin. Endocr. Metab. 83(3), 960–965 (1998)
I. Lemmens, W.J.M. Van de Ven, K. Kas, C.X. Zhang, S. Giraud, V. Wautot, N. Buisson, K. De Witte, J. Salandre, G. Lenoir, M. Pugeat, A. Calender, F. Parente, D. Quincey, P. Gaudray, M.J. De Wit, C.J.M. Lips, J.W.M. Höppener, S. Khodaei, A.L. Grant, G. Weber, S. Kytölä, B.T. Teh, F. Farnebo, C. Phelan, N. Hayward, C. Larsson, A.A.J. Pannett, S.A. Forbes, J.H. Duncan Bassett, R.V. Thakker, Identification of the multiple endocrine neoplasia type1 (MEN1) gene. The European Consortium on MEN1. Hum. Mol. Genet. 6(7), 1177–1183 (1997)
S. Giraud, C.X. Zhang, O. Serova-Sinilnikova, V. Wautot, J. Salandre, N. Buisson, C. Waterlot, C. Bauters, N. Porchet, J.-P. Aubert, P. Emy, G. Cadiot, B. Delemer, O. Chabre, P. Niccoli, F. Leprat, F. Duron, B. Emperauger, P. Cougard, P. Goudet, E. Sarfati, J.-P. Riou, S. Guichard, M. Rodier, A. Meyrier, P. Caron, M.-C. Vantyghem, M. Assayag, J.-L. Peix, M. Pugeat, V. Rohmer, M. Vallotton, G. Lenoir, P. Gaudray, C. Proye, B. Conte Devolx, P. Chanson, Y.Y. Shugart, D. Goldgar, A. Murat, A. Calender, Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am. J. Hum. Genet. 63, 455–467 (1998)
Acknowledgments
We thank Drs. Antonella Picone, Antonella Meola, Silvia Chiavistelli for the help in the collection of parathyroid specimens. CDKN1B and MEN1 mutations were submitted to ClinVar database; accessions SCV000246271; SCV000246272; SCV000246273; SCV000246274; SCV000246275.
Funding
This work was supported by Grants from the University of Pisa (Fondi di Ateneo per la Ricerca) and Ministero dell’Istruzione, dell’Universita` e della Ricerca to C Marcocci (Grant Number 20094T89BR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Simona Borsari and Elena Pardi have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Borsari, S., Pardi, E., Pellegata, N.S. et al. Loss of p27 expression is associated with MEN1 gene mutations in sporadic parathyroid adenomas. Endocrine 55, 386–397 (2017). https://doi.org/10.1007/s12020-016-0941-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-016-0941-6