
Abstract
Mutations in SLC4A1, encoding the chloride-bicarbonate exchanger AE1, cause distal renal tubular acidosis (dRTA), a disease of defective urinary acidification by the distal nephron. We searched for SLC4A1 gene mutations in six patients from a Chinese family with a severe phenotype of dRTA (growth impairment, severe metabolic acidosis, with/or without gross nephrocalcinosis and renal impairment). All coding regions of kidney isoform of AE1, including intron–exon boundaries, were analyzed using PCR followed by direct sequence analysis. A novel 1-bp duplication at nucleotide 2713 (c.2713dupG, band 3 Qingdao) in exon 20 of SLC4A1 in this family was identified by direct sequencing analysis. This duplication alters the encoded protein through codon 905, and results in a reading frame for 15 extra condons (instead of 8) before the new stop condon at position 919 (p.Asp905Glyfs15). We suggest that RTA should be considered as a diagnostic possibility in adult subjects with nephrocalcinosis and chronic renal insufficiency, and family survey should be carefully performed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Distal renal tubular acidosis (dRTA) results from the impaired secretion of hydrogen ions from the distal nephron, causing metabolic acidosis, often with hypokalemia, nephrocalcinosis, and/or nephrolithiasis. dRTA is mainly caused by autoimmune and/or systemic disease, or by drugs. Inversely, hereditary dRTA is rare. Both autosomal dominant and autosomal recessive (with deafness or preserved hearing) inheritance patterns have been reported in primary dRTA [1–17]. Recessive dRTA (rdRTA) was mainly reported in the studies of patients from Southeast Asia; however, dominant dRTA (ddRTA) was predominantly described in Caucasian. The spectrum of clinical severity is very wide, ranging from compensated mild acidosis, absence of symptoms, and the incidental finding of stones and/or renal tract calcification to major effects in infancy with severe acidosis, impaired growth, and early nephrocalcinosis causing eventual renal insufficiency. In general, patients with rdRTA display a severe phenotype than do those with dominantly inherited disease [18].
The physiology of distal tubular function is complex and remained somewhat elusive until recently. It is now clear that at least three molecules of the α-intercalated cell play an important role in the distal renal tubular acidification function [18]. Cytoplasmic carbonic anhydrase II (CA II) catalyzes the hydration of carbon dioxide to carbonic acid, which dissociates to form bicarbonate (HCO3 −) and hydrogen ions (H+), the latter being secreted into the tubule through the action of H+-ATPase on the apical membrane of the α-intercalated cells in the renal collecting duct. The HCO3 − generated by this process is transported across the basolateral membrane through the anion exchanger 1(AE1), a HCO3 −/Cl− AE. Defects of each of the three molecules (CA II, H+-ATPase, and AE1) will lead to destruction of normal distal tubular acidification function. Mutations in AE1 have been reported as the sole genetic cause of autosomal ddRTA (MIM#179800) and also rarely cause autosomal rdRTA [1–8, 12–17].
AE1 (also known as erythrocyte band 3) is a 911 amino acid membrane protein encoded by SLC4A1 (MIM 109270) located on chromosome 17q21–q22 presenting 20 exons [19–21]. AE1 comprises two main domains: a 40-kda NH2-terminal cytoplasmic domain, which anchors the membrane to the red cell skeleton, and a 55-kda COOH-terminal membrane-associated domain, which carries out anion exchange. The short cytoplasmic COOH-terminal tail binds carbonic anhydrase [22, 23]. A renal form of AE1 (kAE1), truncated at the NH2-terminus, is expressed in the basolateral membrane of the α-intercalated cells of kidney collecting ducts and is involved in acid secretion [21].
Several mutations in the SLC4A1 gene have been found associated to different diseases, e.g., hereditary spherocytosis, Southeast Asian ovalocytosis, chorea-acanthocytosis, and familial dRTA [24], thus demonstrating that the effects of SLC4A1 mutation are dependent on type and location. Three kinds of mutations have been reported in COOH-terminal of AE1, two of them (Walton and Dourados) were related with dRTA [12, 13], another (Vesuvio) was associated with hereditary spherocytosis but not with dRTA [24]. Herein, we report a novel COOH-terminal variant in a Chinese ddRTA family with a severe phenotype.
Subjects and methods
Patients
Kindred-RTA, A 68-year-old man (Proband, Fig. 1, Ia) was admitted to our hospital with the diagnosis of urinary system infection 5 years ago. He has a remarkable medical history of persistent hypokalemia (serum potassium: 2.3–3.5 mM) and calculus voiding. The biological workup performed confirmed mild hypokalemia, with a potassium level of 3.3 mM, in association with acidosis (plasma bicarbonate 18.2 mM), and inappropriate alkaline urine (pH 6.5) after hospitalization. His elevated serum creatinine levels (228 μM) demonstrated the existence of marked renal impairment and ultrasonography-revealed evidence of nephrocalcinosis in both kidneys. He received hemodialysis treatment as a result of renal failure 3 years later.

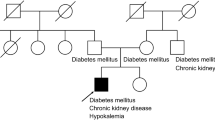
Pedigree of the kindred wth dRTA. Males and females are indicated by squares and circles, respectively. An affected individual is indicated by filled symbols
A son of the proband (Fig. 1, IId) visited our department of nephrology to seek medical help for poor appetite and weakness 3 years ago. He suffered recurrent problems with renal stones and chronic hypokalemia in previous medical history. Severe decrease in renal function and nephrocalcinosis was found in further investigation (Fig. 2). Despite partial recovery of his renal function by treatment, he finally required dialysis half of a year later. Two years ago, he received an allograft from a donor. He remained well since transplantation and his electrolyte disturbances completely restored (serum creatinine 89 μM, K+ 4.2 mM, CO2CP 26 mM).

Plain abdominal radiograph of patient IId. Nephrocalcinosis and enlarged volume of both kidneys
In view of the similar medical history and clinical features shared by the father and one of his descendants, a possible hereditary renal disease was considered and a search for a familial disease was started. Written informed consent was obtained from all individuals of the family participating in the study, which was approved by the Ethics Committee of the affiliated hospital of Qingdao University School of medicine. Complete metabolic evaluation and study of renal tubular acidification function was performed in the other first-degree relatives of the patient IId (Fig. 1). Their erythrocytes morphology was examined by peripheral blood films (Wright stain). Brainstem electric-response audiometry was used to assess the auditory function.
Mutation analysis
Genomic DNA was extracted from peripheral blood of the proband and their family members by the GenElute blood genomic DNA kit (Sigma, NA2010). Twelve pairs of oligonucleotide primers were generated to amplify all coding exons (from exons 4 to 20) and flanking intronic regions of the renal isoform of AE1 (Table 1). PCRs were performed in 50 μl of solution containing 0.2 mM dNTP, 0.03 U/μl Taq polymerase (Takara EX Taq Hot start version, DRR006B), 2.0 mM MgCl2, 2.5 μl 10× PCR Mg2+-free buffer (Takara), approximately 50 ng genomic DNA and 1 mM of each primer. PCR was performed with an initial denaturation step at 95°C for 5 min, subsequently followed by 33 cycles with denaturation at 95°C for 45 s, annealing at 58°C for 45 s and elongation at 72°C for 45 s. PCR samples were subjected to bidirectional sequencing. The sequence reactions were run on an ABI Prism 3730 DNA Analyzer (Applied Biosystems).
Results
As outlined in Fig. 1 and Table 2, four other members (IIa, IIe, IIId, and IIIe) were found to have abnormality of acid-base metabolism and renal tubular acidification function besides patients Ia and IId. Among of them, both cousins (IIId and IIIe) had inappropriately alkaline urine (pH > 6.4) in the presence of systemic metabolic acidosis (CO2CP < 18 mM) with normal renal function, and their urinary excretion of NH4 +, titratable acid, and bicarbonate was low. All the metabolic features of both cousins suggested the diagnosis of dRTA. In the meanwhile, other two cases in the kindred (the sister IIa and brother IIe) also presented classic features of dRTA, e.g., metabolic acidosis, hypokalemia, nephrocalcinosis, and inappropriate alkaline urine; meanwhile, both of them had moderate chronic renal insufficiency. By reanalysis, it became clear that the primary disease of patients Ia and IId should be dRTA, which led to nephrocalcinosis and progressive renal impairment, eventually requiring dialysis. With the combination of pedigree analysis (Fig. 1), the diagnosis of autosomal ddRTA in the kindred established. The prominent features in this family were growth impairment (short stature in four cases and thrive failure in two cases), gross nephrocalcinosis, and renal insufficiency (four of six patients), but no bone disease. All subjects did not have hearing loss, and red cell morphology was normal by Wright stain.
Administration of sodium citrate, potassium citrate, and citric acid corrected metabolic acidosis and hypokalemia in all patients, and restored normal growth in both cousins; the increase in their height (IIId and IIIe) was 18 and 20 cm, respectively, in 2 years. Additionally, the renal function and electrolyte metabolism of transplant recipient IId still kept normal without requirement of citrate therapy so far.
By direct sequencing analysis, a novel 1-bp duplication at nucleotide 2713 (c.2713dupG, Fig. 3) in exon 20 of SLC4A1 in the proband and all the other five patients was identified. This duplication alters the encoded protein through codon 905, and results in a reading frame for 15 extra condons (instead of eight) before the new stop condon at position 919 (p.Asp905Glyfs15, Fig. 3). Direct sequencing analysis failed to find this mutation in other 11 healthy members of this family and 100 unrelated healthy subjects. We named this mutation Qingdao (Band 3 Qingdao) in reference to the area of origin of the kindred of dRTA.

Sequencing chromatogram and schematic diagram of translation. a A novel SLC4A1 mutation (c.2713dupG, Qingdao) identified in patients with dRTA. The duplicated nucleotide of guanine is marked with asterisk. b Presumed translation of normal and mutant SLC4A1 cDNA sequence
Discussion
In this investigation, the similar clinical course observed in the father and one of his children urged us to seek a genetic cause of the disorder. Although the phenotype of the proband and his children was modified by metabolic disturbance of renal failure, both young cousins showed typical clinical features of dRTA, which facilitated the diagnosis of primary causes in this family. Complete dRTA with severe phenotype, such as growth impairment, severe metabolic acidosis, with/or without gross nephrocalcinosis and renal impairment, were detected in six members of the kindred. However, though not invariably, patients with ddRTA usually display a mild phenotype [3, 18]. The severe phenotype may contribute to the delay or neglect of treatment, and meanwhile may be partially related to the genotype. We suggest that dRTA should be considered as a diagnostic possibility in all cases with nephrocalcinosis and chronic renal insufficiency, and family survey should be further performed.
Even though there have been advances in understanding the etiology and pathogenesis of RTA in the recent years [1–18], the possibilities and strategies for its management remained almost the same. The main therapy to treat dRTA is still an oral administration of citrate and citric acid, which is usually sufficient to reverse most of the biochemical abnormalities, leading to the resumption of normal growth through repair of normal growth hormone-IGF axis’ action [25, 26]. Additionally, therapy with citrate, by complexing calcium in urine or in renal tissue, might diminish renal stone formation and deposition of insoluble calcium salts in the kidneys [27–29].
In this study, six members with dRTA from a family shared a novel 1-bp duplication (band 3 Qingdao) in exon 20 of AE1 gene, resulting in a reading frame from condon 905 to a new stop codon at position 919. Three mutations in the COOH-terminal tail, specifically in exon 20, were reported to be associated with dRTA so far. Karet et al. [3] were the first to identify a 13-bp duplication in tandem sequence extending from the second base in codon 896 to the second base in codon 900 in two brothers with dRTA and nephrocalcinosis. Toye et al. [12] reported the presence of dRTA and NC in two brothers who shared the same mutation described by Karet et al. and an additional deletion of 9-bp over the sequence that would have coded for amino acids Tyr904-Glu906 of normal AE1. Either above-mentioned mutations resulted in a premature termination codon at position 901, truncating the protein in the last 11 amino acids, and considered as band 3 Walton. More recently, Cheidde et al. reported two affected brothers with completed dRTA and NC and their father with incompleted dRTA and nephrolithiasis shared a 20-bp deletion (band 3 Dourados) in exon 20 of AE1, resulting in a premature termination codon at position 889, truncating the protein by 23 amino acids [13].
The structure and function of the short COOH-terminal cytoplasmic tail of AE1 have not been well elucidated. Transport studies by Toye et al. demonstrated that band 3 Walton was able to reach the cell surface of erythrocytes and oocytes, but retained intracellularly within the kidney cells [12]. The results indicate that the COOH-terminal tail of AE1 is required to allow movement to the cell surface in kidney cells. We propose that band 3 Qingdao may, through the same mechanism as band 3 Walton, led to destruction of normal distal renal tubular acidification. Another disturbed mechanism caused by band 3 Qingdao may be the lack of proper CA II binding. Since the mutated region is close to the critical binding sites (D887ADD) [23] of AE1, we could not completely exclude the possibility that CA II binding is interrupted by conformation changes (such as by a new disulfide bond formation through Cys911) of AE1 caused by band 3 Qingdao, disturbing the cell capacity for bicarbonate transport. However, this presumption requires further study in vitro to confirm.
References
L.J. Bruce, D.L. Cope, G.K. Jones, A.E. Schofield, M. Burley, S. Povey, R.J. Unwin, O. Wrong, M.J. Tanner, J. Clin. Invest. 100, 1693–1707 (1997)
P. Jarolim, C. Shayakul, D. Prabakaran, L. Jiang, A. Stuart-Tilley, H.L. Rubin, S. Simova, J. Zavadil, J.T. Herrin, J. Brouillette, M.J. Somers, E. Seemanova, C. Brugnara, L.M. Guay-Woodford, S.L. Alper, J. Biol. Chem. 273, 6380–6388 (1998)
F.E. Karet, F.J. Gainza, A.Z. Györy, R.J. Unwin, O. Wrong, M.J. Tanner, A. Nayir, H. Alpay, F. Santos, S.A. Hulton, A. Bakkaloglu, S. Ozen, M.J. Cunningham, A. di Pietro, W.G. Walker, R.P. Lifton, Proc. Natl Acad. Sci. USA 95, 6337–6342 (1998)
V.S. Tanphaichitr, A. Sumboonnanonda, H. Ideguchi, C. Shayakul, C. Brugnara, M. Takao, G. Veerakul, S.L. Alper, J. Clin. Invest. 102, 2173–2179 (1998)
S. Vasuvattakul, P.T. Yenchitsomanus, P. Vachuanichsanong, P. Thuwajit, C. Kaitwatcharachai, V. Laosombat, P. Malasit, P. Wilairat, S. Nimmannit, Kidney Int. 56, 1674–1682 (1999)
L.J. Bruce, O. Wrong, A.M. Toye, M.T. Young, G. Ogle, Z. Ismail, A.K. Sinha, P. McMaster, I. Hwaihwanje, G.B. Nash, S. Hart, E. Lavu, R. Palmer, A. Othman, R.J. Unwin, M.J. Tanner, Biochem. J. 350, 41–51 (2000)
S. Sritippayawan, A. Sumboonnanonda, S. Vasuvattakul, T. Keskanokwong, N. Sawasdee, A. Paemanee, P. Thuwajit, P. Wilairat, S. Nimmannit, P. Malasit, P.T. Yenchitsomanus, Am. J. Kidney Dis. 44, 64–70 (2004)
P.T. Yenchitsomanus, S. Vasuvattakul, S. Kirdpon, S. Wasanawatana, W. Susaengrat, S. Sreethiphayawan, D. Chuawatana, S. Mingkum, N. Sawasdee, P. Thuwajit, P. Wilairat, P. Malasit, S. Nimmannit, Am. J. Kidney Dis. 40, 21–29 (2002)
F.E. Karet, K.E. Finberg, R.D. Nelson, A. Nayir, H. Mocan, S.A. Sanjad, J. Rodriguez-Soriano, F. Santos, C.W. Cremers, A. Di Pietro, B.I. Hoffbrand, J. Winiarski, A. Bakkaloglu, S. Ozen, R. Dusunsel, P. Goodyer, S.A. Hulton, D.K. Wu, A.B. Skvorak, C.C. Morton, M.J. Cunningham, V. Jha, R.P. Lifton, Nat. Genet. 21, 84–90 (1999)
F.E. Karet, K.E. Finberg, A. Nayir, A. Bakkaloglu, S. Ozen, S.A. Hulton, S.A. Sanjad, E.A. Al-Sabban, J.F. Medina, R.P. Lifton, Am. J. Hum. Genet. 65, 1656–1665 (1999)
A.N. Smith, J. Skaug, K.A. Choate, A. Nayir, A. Bakkaloglu, S. Ozen, S.A. Hulton, S.A. Sanjad, E.A. Al-Sabban, R.P. Lifton, S.W. Scherer, F.E. Karet, Nat. Genet. 26, 71–75 (2000)
A.M. Toye, L.J. Bruce, R.J. Unwin, O. Wrong, M.J. Tanner, Blood 99, 342–347 (2002)
L. Cheidde, T.C. Vieira, P.R. Lima, S.T. Saad, I.P. Heilberg, Pediatrics 112, 1361–1367 (2003)
N. Rungroj, M.A. Devonald, A.W. Cuthbert, F. Reimann, V. Akkarapatumwong, P.T. Yenchitsomanus, W.M. Bennett, F.E. Karet, J. Biol. Chem. 279, 13833–13838 (2004)
S. Kittanakom, E. Cordat, V. Akkarapatumwong, P.T. Yenchitsomanus, R.A. Reithmeier, J. Biol. Chem. 279, 40960–40971 (2004)
K.E. Choo, T.K. Nicoli, L.J. Bruce, M.J. Tanner, A. Ruiz-Linares, O.M. Wrong, Pediatr. Nephrol. 21, 212–217 (2006)
F.E. Anacleto, L.J. Bruce, P. Clayton, S. Hegde, L.P. Resontoc, O. Wrong, Nephron Physiol. 114, 19–24 (2010)
F.E. Karet, J. Am. Soc. Nephrol. 13, 2178–2184 (2002)
L.C. Showe, M. Ballantine, K. Huebner, Genomics 1, 71–76 (1987)
S.E. Lux, K.M. John, R.R. Kopito, H. Lodish, Proc. Natl Acad. Sci. USA 86, 9089–9093 (1993)
M.J.A. Tanner, Mol. Membr. Biol. 14, 155–165 (1997)
J.W. Vince, R.A. Reithmeier, J. Biol. Chem. 273, 28430–28437 (1998)
J.W. Vince, R.A. Reithmeier, Biochemistry 39, 5527–5533 (2000)
S. Perrotta, F. Polito, M.L. Cone, B. Nobili, S. Cutillo, V. Nigro, A. Iolascon, G. Amendola, Blood 93, 2131–2132 (1999)
A. Challa, W. Chan, R.J. Krieg Jr., M.A. Thabet, F. Liu, R.L. Hintz, J.C. Chan, Kidney Int. 44, 1224–1227 (1993)
A. Challa, R.J. Krieg Jr., M.A. Thabet, J.D. Veldhuis, J.C. Chan, Am. J. Physiol. 265, E547–E553 (1993)
G.M. Preminger, K. Sakhaee, C. Skurla, C.Y. Pak, J. Urol. 134, 20–23 (1985)
H.G. Tiselius, C. Berg, A.M. Fornander, M.A. Nilsson, Scan. Microsc. 7, 381–389 (1993)
G.A. Tanner, J. Am. Soc. Nephrol. 9, 1242–1248 (1998)
Acknowledgment
This study was supported by a grant from the prime foundation for scientific research of the Affiliated Hospital of Qingdao University School of Medicine.
Conflict of interest
All authors have no conflict of interest to report.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Shao, L., Xu, Y., Dong, Q. et al. A novel SLC4A1 variant in an autosomal dominant distal renal tubular acidosis family with a severe phenotype. Endocr 37, 473–478 (2010). https://doi.org/10.1007/s12020-010-9340-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-010-9340-6