
Abstract
Gitelman syndrome is an autosomal recessive genetic disease caused by pathogenic variants in SLC12A3 resulting in the loss of function of the Na–Cl co-transporter (NCC) in the distal tubules. Hypokalemia and diuretic effects can cause secondary type 2 diabetes and renal function decline. Here, we present the case of a 49-year-old male patient with chronic persistent treatment-resistant hypokalemia for the past 13 years who had been receiving treatment for type 2 diabetes mellitus for 6 years. He was referred to our department due to the presence of urinary protein, impaired renal function, high renin activity, and hyperaldosteronism. Laboratory test results showed hypokalemia, hypomagnesemia, hypocalciuria, and metabolic alkalosis. Using next-generation and Sanger sequencing, we identified a novel stop-gain variant (NM_000339.3:c.137del [p.His47fs]) and a missense variant (NM_000339.3:c.2927C > T [p.Ser976Phe]) in the SLC12A3 gene. This novel pathogenic variant was located at the intracellular N-terminus of the NCC. Based on these findings, the patient was diagnosed with Gitelman syndrome. The use of next-generation sequencing facilitated the exclusion of diseases with similar clinical symptoms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gitelman syndrome (OMIM #263,800) is an autosomal recessive genetic disease caused by pathogenic variants in the SLC12A3 gene and is characterized by hypokalemia, hypocalciuria, hypomagnesemia, and metabolic alkalosis [1]. SLC12A3 encodes the Na–Cl co-transporter (NCC), which functions as the target site of thiazide diuretics, responsible for sodium reabsorption in the distal tubules. Pathogenic variants in SLC12A3 prevent the reabsorption of sodium in the distal tubules and cause a diuretic effect. To date, more than 350 pathogenic variants loci associated with Gitelman syndrome have been identified [1]. However, some patients with clinically suspected Gitelman syndrome do not carry variants in SLC12A3 [2]. For example, some patients who carry pathogenic variants in other genes, such as CLCNKB or HNF1B [1], present with a phenotype similar to that of Gitelman syndrome. Therefore, we performed a comprehensive genetic analysis using next-generation sequencing and confirmed the results using Sanger sequencing to diagnose Gitelman syndrome. Here, we report the case of a patient with Gitelman syndrome accompanied by chronic kidney disease (CKD) and diabetes who carried a novel frameshift variant in SLC12A3 gene.
Case Report
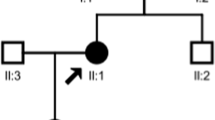
A 49-year-old male patient presenting with persistent treatment-resistant hypokalemia, high plasma renin activity, and high aldosterone levels was referred to our department. Thirteen years ago, he had visited a local hospital with complaints of lethargy and weakness in his lower limbs. He was found to have hypokalemia (2.1 mEq/L) and was administered potassium chloride and followed up. Serum potassium was below 3.0 mEq/L despite treatment, but he did not wish to see a specialist. He was diagnosed with diabetes at the age of 43 and was treated at a local hospital. He was recommended to visit a specialist several times, which brought him to our hospital. High plasma renin activity and aldosterone levels were observed, and the patient was referred to our department. His height was 170 cm, body weight was 69.6 kg, and blood pressure was 116/75 mmHg. No abnormalities were observed upon physical examination. On renal echocardiography, both kidneys showed no atrophy, and there were no findings suggestive of renal artery stenosis. No calcification was observed in the kidneys. There were no diabetes-associated changes in the fundus. The patient was taking glimepiride, febuxostat, potassium chloride, and enalapril maleate and was on insulin degludec. There was no use of drugs, such as diuretics and laxatives, other than those prescribed, and alcohol consumption was limited to occasional drinking. There was no family history of hypokalemia (Fig. 1).

Pedigree of the patient’s family. The black-filled square indicated by the arrow shows this case
As presented in Table 1, laboratory blood and urine tests showed hypokalemia, hypomagnesemia, hypercalcemia, and hypocalciuria. There was an increase in plasma renin activity and aldosterone levels. Venous blood gas analysis revealed alkalosis. The urinary protein level was 1.13 g/gCr, and eGFR was 84.5 mL/min/1.73m2. Hypercalcemia was observed; however, the intact parathyroid hormone level was normal (25 pg/mL), and parathyroid hormone-related protein levels were below the detection limit.
Following approval for genetic analysis from the Ethics Committee of Osaka Minami Medical Center (31–15) and Tokyo Medical and Dental University (G2000-080–02), we obtained written informed consent from the patient. DNA was extracted from whole blood, and 166 genes associated with kidney disease were analyzed using next-generation sequencing [3]. We identified heterozygous variants in SLC12A3 gene (NM_000339.3:c.137del [p.His47fs] and NM_000339.3:c.2927C > T [p.Ser976Phe]). These variants were confirmed using Sanger sequencing (Fig. 2). Variant c.137del is novel variant and meets the criteria of very strong pathogenicity (PSV1), in addition to moderate (PM2) and supporting (PP4) evidence of pathogenicity, according to the American College of Medical Genetics and Genomics/the Association for Molecular Pathology (ACMG/AMP) standards guidelines, thereby leading to pathogenic based on the scoring rules [4]. Variant c.2927C > T has been previously reported as the cause of Gitelman syndrome [5, 6]. Based on these results, we diagnosed this patient with Gitelman syndrome. In addition, there were no pathogenic variants in CLCNKB or HNF1B, often responsible for clinical symptoms similar to those of Gitelman syndrome with hypokalemia [1].

Analysis of the SLC12A3 gene using Sanger sequencing in the patient The upper panel presents the features and sequencing chromatogram of the c.137del (p.His47fs) variant; the lower panel presents the features and sequencing chromatogram of the c.2927C > T (p.Ser976Phe) variant
Treatment for hypokalemia using eplerenone 100 mg and potassium chloride 2400 mg and for hypomagnesemia with magnesium oxide 330 mg was initiated. Subsequently, the serum potassium level improved and was effectively maintained above 3 mEq/L. Due to diarrhea caused by magnesium oxide administration, only 330 mg/day could be administered; serum magnesium level was still low at 1.2–1.5 mg/dL.
Discussion
Gitelman syndrome is an autosomal recessive genetic disorder caused by pathogenic variants in the SLC12A3 gene, resulting in the loss of function of the thiazide-sensitive NCC. The prevalence of this syndrome is 1–10 per 4,000 people, and it is common in Asia [1]. More than 350 variants in SLC12A3 have been identified to date [1], with 59% being missense variants and 14% insertions or deletions [7]. In this case, a novel stop-gain variant (NM_000339.3:c.137del) and a missense variant (NM_000339.3:c.2927C > T) [5] were found. We detected no variants in CLCNKB or HNF1B, which can lead to a phenotype similar to that of Gitelman syndrome, leading us to the diagnosis of Gitelman syndrome.
Codons downstream of the novel frameshift variant encode 65 amino acids. As the frameshift site is located at the N-terminal side of the transmembrane region, this protein variant does not contain the main body of the transporter, losing its function. Alternatively, this condition is thought to be degraded by the nonsense codon-dependent mRNA decay (NMD) mechanism, wherein mRNAs with an abnormal termination codon upstream of the original termination codon are degraded [8]. Previously, knock-in mice expressing the human p.Ser710X (c.2135C > A) variant in Gitelman syndrome presented a marked decrease in NCC mRNA levels and almost no NCC protein in the kidney, suggesting the involvement of NMD [9].
Variant c.137del is classified as PSV1 in ACMG/AMP guideline as it is a null variant [4]. In addition, this variant is not detected in the Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium, and there is no family history of a disease in this case, which is consistent with Gitelman syndrome. The patient had an autosomal recessive form of inheritance, thereby it is also classified as PM2 and PP4 [4]. Thus, this stop-gain variant is classified as pathogenic, because it satisfies all the criteria of PSV1, PM2, and PP4 [4]. The genomes of the parents in our present study were not analyzed; therefore, the possibility that the two variants exist in the same allele cannot be ruled out. This is a potential limitation. However, according to the Association for Clinical Genomic Science recommendations [10], when two mutations in an autosomal recessive disease are pathogenic, there is > 99% certainty that each variant is pathogenic, and the possibility of occurrence of a compound heterozygous condition is extremely high. Therefore, the variant in the patient could be a compound heterozygous. Furthermore, our next-generation sequencing panel-based analysis excluded any causal variants in both CLCNKB and HNF1B.
Variant c.2927C > T has been reported as a causative mutation for Gitelman syndrome [5, 6]. The Japanese database, jMorp (Japanese Multi Omics Reference Panel) indicates an occurrence rate of 0.042%. However, it was not included in other major databases, such as Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium, suggesting that it is a rare mutation. This variant is located at the C-terminus of the NCC protein. Many genetic abnormalities have been reported at the C-terminus, and it is assumed to be involved in NCC trafficking and functional regulation [11]. This variant was predicted as pathogenic using SIFT, PolyPhen-2, and mutation taster. Based on the above, according to the ACMG/AMP guidelines, strong (PS4), moderate (PM1, PM3), and supporting (PP1, PP3, and PP4) evidence of pathogenicity are met and classified as pathogenic.
The patient had hypokalemia, hypomagnesemia, and hypocalciuria, which are typical laboratory findings of Gitelman syndrome. Impaired renal function, proteinuria, and diabetes mellitus—relatively common complications of Gitelman syndrome [12,13,14,15,16,17,18,19]—were also observed. In a study from Taiwan, seven out of forty patients with Gitelman syndrome had CKD, and five out of forty patients had diabetes [16]. Urinary protein was not detected among the seven patients with CKD [16]. Five of these seven patients underwent renal biopsy, and the results showed chronic tubular damage, assumed to be caused by hypokalemia [16]. However, the renin–angiotensin system is activated in Gitelman syndrome, and angiotensin II and aldosterone exacerbate CKD through oxidative stress, inflammation, fibrosis, and hemodynamics [20, 21]. Therefore, some reports attribute the disease to the effects of the activation of the renin–angiotensin system and renal hemodynamics on renal function, rather than hypokalemia [15, 17, 22]. In this case, the patient was on enalapril maleate, which increases plasma renin activity. However, aldosterone levels were also high, suggesting that the activity of the renin–angiotensin system was augmented, which may have resulted in impaired renal function and proteinuria. When a renal biopsy was performed on patients with Gitelman syndrome with proteinuria, glomerular basement membrane thickening was observed, which may also be due to the activation of the renin–angiotensin system [17]. Contrastingly, patients with Gitelman syndrome with a urinary protein level of 3.98 g/day displayed a minor lesion in renal biopsy [15]. Further studies are needed to determine the exact mechanism underlying renal function decline and the presence of proteinuria in Gitelman syndrome.
As noted above, Gitelman syndrome is often associated with diabetes mellitus [14, 16, 18, 19]. Abnormal glucose tolerance may be caused by increased insulin resistance and decreased insulin secretion due to hypomagnesemia and hypokalemia [14, 18, 23]. In this case, Gitelman syndrome may be involved in the development of diabetes.
In conclusion, we examined a case of Gitelman syndrome complicated by CKD and diabetes mellitus. Next-generation and Sanger sequencing revealed that the patient had a novel stop-gain variant, NM_000339.3:c.137del in SLC12A3, located on the intracellular N-terminal of the NCC protein, and a previously reported pathogenic variant, NM_000339.3:c.2927C > T. The use of next-generation sequencing facilitated the exclusion of other diseases with similar clinical symptoms.
References
Blanchard A, Bockenhauer D, Bolignano D, Calò LA, Cosyns E, Devuyst O, et al. Gitelman syndrome: consensus and guidance from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. 2017;91:24–33.
Mori T, Chiga M, Fujimaru T, Kawamoto R, Mandai S, Nanamatsu A, et al. Phenotypic differences of mutation-negative cases in Gitelman syndrome clinically diagnosed in adulthood. Hum Mutat. 2021;42:300–9.
Mori T, Hosomichi K, Chiga M, Mandai S, Nakaoka H, Sohara E, et al. Comprehensive genetic testing approach for major inherited kidney diseases, using next-generation sequencing with a custom panel. Clin Exp Nephrol. 2017;21:63–75.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Lee JW, Lee J, Heo NJ, Cheong HI, Han JS. Mutations in SLC12A3 and CLCNKB and their correlation with clinical phenotype in patients with Gitelman and Gitelman-like syndrome. J Korean Med Sci. 2016;31:47–54.
Nozu K, Nozu Y, Nakanishi K, Konomoto T, Horinouchi T, Shono A, et al. Cryptic exon activation in SLC12A3 in Gitelman syndrome. J Hum Genet. 2017;62:335–7.
Vargas-Poussou R, Dahan K, Kahila D, Venisse A, Riveira-Munoz E, Debaix H, et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22:693–703.
Kurosaki T, Maquat LE. Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci. 2016;129:461–7.
Yang SS, Lo YF, Yu IS, Lin SW, Chang TH, Hsu YJ, et al. Generation and analysis of the thiazide-sensitive Na+-Cl− cotransporter (Ncc/Slc12a3) Ser707X knockin mouse as a model of Gitelman syndrome. Hum Mut. 2010;31:1304–15.
Ellard S, Baple EL, Berry I, Forrester N, Turnbull C, Owens M, et al. ACGS best practice guidelines for variant classification 2020: association for clinical genetics science (ACGS), 2020. Available at: https://www.acgs.uk.com/quality/best-practice-guidelines/#VariantGuidelines.
Glaudemans B, Yntema HG, San-Cristobal P, Schoots J, Pfundt R, Kamsteeg EJ, et al. Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome. Eur J Hum Genet. 2012;20:263–70.
Berry MR, Robinson C, Karet Frankl FE. Unexpected clinical sequelae of Gitelman syndrome: hypertension in adulthood is common and females have higher potassium requirements. Nephrol Dial Transplant. 2013;28:1533–42.
Walsh PR, Tse Y, Ashton E, Iancu D, Jenkins L, Bienias M, et al. Clinical and diagnostic features of Bartter and Gitelman syndromes. Clin Kidney J. 2018;11:302–9.
Blanchard A, Vallet M, Dubourg L, Hureaux M, Allard J, Haymann JP, et al. Resistance to insulin in patients with Gitelman syndrome and a subtle intermediate phenotype in heterozygous carriers: a cross-sectional study. J Am Soc Nephrol. 2019;30:1534–45.
Chen Q, Wu Y, Zhao J, Jia Y, Wang W. A case of hypokalemia and proteinuria with a new mutation in the SLC12A3 gene. BMC Nephrol. 2018;19:275.
Tseng MH, Yang SS, Hsu YJ, Fang YW, Wu CJ, Tsai JD, et al. Genotype, phenotype, and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation. J Clin Endocrinol Metab. 2012;97:E1478–82.
Demoulin N, Aydin S, Cosyns JP, Dahan K, Cornet G, Auberger I, et al. Gitelman syndrome and glomerular proteinuria: a link between loss of sodium-chloride cotransporter and podocyte dysfunction? Nephrol Dial Transplant. 2014; 29 Suppl 4:iv117–20.
He G, Gang X, Sun Z, Wang P, Wang G, Guo W. Type 2 diabetes mellitus caused by Gitelman syndrome-related hypokalemia: A case report. Medicine (Baltimore). 2020; 99:e21123.
Ren H, Qin L, Wang W, Ma J, Zhang W, Shen PY, et al. Abnormal glucose metabolism and insulin sensitivity in Chinese patients with Gitelman syndrome. Am J Nephrol. 2013;37:152–7.
Barrera-Chimal J, Girerd S, Jaisser F. Mineralocorticoid receptor antagonists and kidney diseases: pathophysiological basis. Kidney Int. 2019;96:302–19.
Georgianos PI, Agarwal R. Revisiting RAAS blockade in CKD with newer potassium-binding drugs. Kidney Int. 2018;93:325–34.
Walsh SB, Unwin E, Vargas-Poussou R, Houillier P, Unwin R. Does hypokalaemia cause nephropathy? An observational study of renal function in patients with Bartter or Gitelman syndrome. QJM. 2011;104:939–44.
Zillich AJ, Garg J, Basu S, Bakris GL, Carter BL. Thiazide diuretics, potassium, and the development of diabetes: a quantitative review. Hypertension. 2006;48:219–24.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Research involving Human Participants and/or Animals
This article does not describe any studies with animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Iio, K., Mori, T., Bessho, S. et al. Gitelman syndrome with a novel frameshift variant in SLC12A3 gene accompanied by chronic kidney disease and type 2 diabetes mellitus. CEN Case Rep 11, 191–195 (2022). https://doi.org/10.1007/s13730-021-00652-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-021-00652-4