
Abstract
Thymic carcinoid is an important component of the tumor spectrum causing Ectopic ACTH Syndrome (EAS) and usually carries a poor prognosis. Efforts have been focused on exploring the mechanism of the excessive ACTH production in non-pituitary tumors, whereas few studies have reported the molecular events underlying the tumor progression. In this study, seven patients with ACTH producing thymic carcinoids were enrolled. Of note is that five of them showed either lymph node metastasis, local invasion or distant metastasis. By using cDNA profiling approach, we evaluated the expression of cell adhesion pathway genes and found a remarkable overexpression of p21-activated kinase 3 (PAK3) in all thymic carcinoids which was further confirmed at both transcriptional and translational level. RAC1, an upstream activator of PAK3, was also overexpressed in thymic carcinoids. Overexpression of PAK3 in NIH3T3 cell enhanced cell migration and invasion. Importantly, we observed c-Jun NH2-terminal kinase (JNK) was activated in PAK3 transfected cells, and inhibition of JNK activity by SP600125, a JNK pathway inhibitor, abolished PAK3 mediated cell migration. Activation of JNK pathway was also detected in thymic carcinoid with high level of PAK3 expression. Our findings suggested a potential role of PAK3 in the progression of ACTH-producing thymic carcinoid.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ectopic ACTH Syndrome (EAS) occurs while excess ACTH is produced from non-pituitary tumors, especially neuroendocrine tumors [1]. EAS accounts for 12–17% of Cushing’s syndrome and has been associated with a broad spectrum of tumors, including small cell lung cancers (SCLC), bronchus carcinoid, thymic carcinoid, and other neuroendocrine tumors (NETs) such as medullary thyroid carcinomas and adrenal pheochromocytomas [2]. In our series, thymic carcinoid interestingly ranks the first cause of EAS: six out of the eight cases with removable tumors are pathologically diagnosed as thymic carcinoids [3, 4].
The biggest challenge to manage patients with EAS is to localize the ACTH producing tumors. Generally, a variety of tests and long-term follow-up has to be performed before the occult tumors are exposed. However, there are approximately 20% patients who remain as covert [5, 6]. In term of patient management, surgery is the most effective therapy but with only 65% patients cured. For patients with advanced disease, no effective systematic treatment is available for the control of tumor growth: somatostatin analogs can only ameliorate the symptoms related to excessive hormone production; the most recent clinical trials of tyrosine kinase inhibitor are too early to reach any conclusion. Thymic carcinoid in general has a poor prognosis, with most patients developing local recurrence or metastasis within 5 years after surgery and died within 10 years [7, 8].
The molecular mechanism of thymic carcinoid has been barely identified. Chromosomal imbalances including gains on 7, 11q, 12q, 19p, 20q, Xp and losses on 1p, 3, 4q, 6, 9p21-pter,10q, 11q, 13q [9–11] have been reported in thymic carcinoid. Although 25% of the reported thymic NETs are from MEN1 patients [10], LOH of the MEN1 locus on chromosome 11q13 has been found in only one thymic NET. High level of ACTH production in thymic carcinoid has been related to overexpression of corticotrophic transcription factor, such as Tpit and NeuroD1, as well as abnormal methylation of POMC promoter [4, 12].
In our previous study, we performed cDNA microarray to compare the expression profile between thymic carcinoids and noncancerous thymuses [13]. One of the most prominent features was the deregulation of cell adhesion pathway genes, particularly those involved in integrin-mediated cell adhesions. p21-activated kinase 3 (PAK3) was the most upregulated gene in this pathway. It is an evolutionarily conserved serine/threonine protein kinase and belongs to p21-activated kinase family, which is known as the downstream effectors of the small Rho GTPase, Cdc42, and Rac1 [14]. Consistently, Rac1 was also upregulated based upon our array data. Numerous studies have demonstrated PAKs not only regulate cell cytoskeleton organization and promote cell motility but also participate in cell proliferation, apoptosis, and mitosis [15]. Aberrant expression of PAK1 has been reported in various human tumors, particularly in breast cancers, correlated with breast cancer invasiveness as well as tumor cyclin D1 expression [16]. Overexpression of PAK1 in hepatocellular carcinoma could enhance cancer metastasis by activation of JNK pathway [17]. Unlike PAK1 and PAK2 which have a wide distribution over a number of tissues, PAK3 is specifically expressed in brain [18]. Therefore, studies are mainly focused on its role in neuronal system. PAK3 mutations lead to nonsyndromic mental retardation characterized by selective deficits in cognition [19]; PAK3 knock-out mice exhibited a significant abnormality in synaptic plasticity and deficiency in learning as well as memorizing [20]; in Xenopus, PAK3 mediated cell cycle arrest and participated in neurogenesis [21]. More recently, it was reported that PAK3 knockdown abrogated CRF-induced cell proliferation in AtT-20 cells [22].
In the present study, we investigated the potential roles of PAK3 in the development and progression of thymic carcinoid with EAS.
Results
Alteration of cell adhesion pathway genes in thymic carcinoids
In total, we found 10 adhesion related genes were upregulated consistently and significantly in thymic carcinoid, as red highlighted in the heat map, and five genes downregulated as blue highlighted (Fig. 1a). By using pathway resources obtained from KEGG, GenMAPP, and Biocarta, we architected a potential deregulated network of cell adhesion and cell migration in thymic carcinoid (Fig. 1b). PAK3 was the most upregulated gene.

Altered expression of cell adhesion related genes, as revealed by gene expression profile between five ACTH-producing thymic carcinoids (AC1-AC5) and six noncancerous thymuses (NC1-NC6). a Hierarchical visualization of the expression patterns of those genes functionally involved in cell adhesions. Right panel lists the names of the genes, as highlighted by those annotated in integrin-mediated cell adhesion (red for upregulation, blue for downregulation, black for moderate expression). b A typical representation of integrin-mediated cell adhesion, potentially deregulated in ACTH producing thymic carcinoids. (Color figure online)
Overexpression of PAK3 and RAC1 in ACTH producing thymic carcinoids
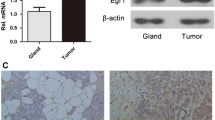
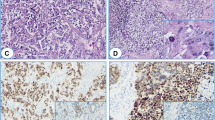
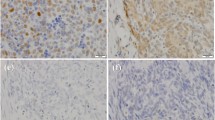
First, we confirmed the array data by qRT-PCR. PAK3 expression level in thymic carcinoids was an average of about 100-fold greater than in noncancerous thymuses. RAC1 was also upregulated in thymic carcinoids. In addition, there was no change for CDC42, another member of the small GTPase family (Fig. 2a). At the protein level, PAK3 was detected in all the thymic carcinoids, while in the three noncancerous thymuses no expression was found (Fig. 2b). Consistent with these results, by immunohistochemistry, we observed that the tumor samples showed strong staining of PAK3, while noncancerous tissues showed negative staining. The temporal lobe of brain was stained as a positive control of PAK3 (Fig. 2c).

Overexpression of PAK3 and RAC1 in ACTH producing thymic carcinoids. a qRT-PCR of PAK3 and its upstream regulators, RAC1 and CDC42. *** P < 0.001, t-test. b Western blot. Extracts from ACTH producing thymic carcinoids (AC), and noncancerous thymuses (NC) were immunoblotted using PAK3 and β-actin antibodies. c Representative immunostaining of PAK3 in NC and AC. Upper panel ×200; lower panel ×400. The temporal lobe of brain was used as a positive control of PAK3 staining. Magnification bar = 25 μm
PAK3 enhanced cell migration and invasion
We overexpressed PAK3 gene in NIH3T3 cell and this exogenous PAK3 showed kinase activity in promoting the phosphorylation of MBP in the presence of [γ-32P]ATP (Fig. 3a). Then we analyzed cell mobility and invasion. In wound healing assay, cells with PAK3 overexpression showed enhanced ability to migrate to close the wound (Fig. 3b). Meanwhile, in matrigel invasion chamber assay, we detected more cells with high PAK3 level invaded through the matrigel membrane compared with the non-PAK3 expressed cells (Fig. 3c).

Overexpression of PAK3 in NIH3T3 cell enhanced cell migration and invasion. a Western blot and PAK3 kinase activity assay. Cell lysates from NIH3T3 cell transfected with pcDNA3.1+ vector (control) and mouse PAK3 (PAK3) were immunoblotted using PAK3 and β-actin antibodies (middle and lower band). The cell extracts were immunoprecipitated by PAK3 antibody, and PAK3 kinase activity was measured using [γ-32P]ATP and MBP as substrates. The reaction was terminated and the γ-32P-labeled MBP was visualized by autoradiography (upper band). b Wound healing test. Control and PAK3 overexpressing Cells were plated 24 h before wounding. Immediately after scraping the cell monolayers with a pipette tip, the cell culture were exchanged with fresh medium. 20 h later, images of the wound size were taken in an Olympus microscope. c Transwell assay. Left, Upper panel are the migrated cells in the control wells. Bottom panels are those cells invading through the matrigel insert membranes. Right quantification of the rate of cell invasion, which was calculated by dividing the number of cells invading through matrigel insert membrane by the number of cells migrating through control insert membrane. ** P < 0.01, t-test
PAK3 induced cell migration was partially mediated by JNK activation
We detected the activation of JNK as well as other MAPK pathways to explore the possible mechanism involved in PAK3 induced cell migration. We observed increased phosphorylation of JNK and c-Jun after PAK3 overexpression, while no significant difference for phospho-ERK and phosphor-p38 (Fig. 4a). Inhibition of JNK pathway by SP600125 could substantially abrogate cell migration induced by PAK3 overexpression in NIH 3T3 cells (Fig. 4c). We did not observe any change of cell mobility for ERK inhibitor, U0126 (data not shown).

PAK3 induced cell migration was partially mediated by JNK activation. a Western blot results of the phosphorylation of Erk, p38 and JNK in PAK3 transfected NIH3T3 cells. b Relative quantification of indicated genes in control and PAK3 transfected NIH3T3 cells are shown as percentage of phosphorylation level to its total level which is already divided by β-actin level. The data are shown as mean ± SEM of three independent experiments. c Wound healing test. Transfected NIH3T3 cells were wounded and the cell culture was then replaced with fresh medium in the presence or absence of JNK pathway inhibitor, SP600125, at the concentration of 40 μmol/l, DMSO as a negative control. At 20 h, the wound size was visualized by microscopy
JNK activation was detected in ACTH producing thymic carcinoids
Consistent with the data obtained from cell line, we also detected elevated phosphorylation of both JNK and c-Jun in the thymic carcinoid tissues obtained from patients with EAS, parallel to overexpression of PAK3 (Fig. 5). No significant difference was observed for phospho-ERK and p38 between thymic carcinoids and noncancerous thymuses (data not shown).

JNK was activated in the thymic carcinoids. Western blot results of phospho-JNK and c-Jun as well as PAK3 expression in thymic carcinoids and noncancerous thymuses, human β-actin was used as an internal control
PAK3 overexpression correlated with the aggressive feature of thymic carcinoids
We further examined PAK3 expression level in a total of seven patients with ACTH producing thymic carcinoid, which included the five patients we did the profiling assay. All tumors showed high level of PAK3 expression. Interestingly, 71% (five out of the seven) of patients showed signs of tumor progression: local invasion, lymph node metastasis, or distant metastasis (Table 1); PAK3 level was higher in tumors with progressive disease than tumors without (Fig. 6). However, the two patients with localized tumor require long term follow-up to exclude any metastatic disease (Table 1).

Thymic carcinoids with tumor progression have a higher level of PAK3 expression than localized tumors. a Representative western blot of PAK3 in the seven thymic carcinoids. Number 1–7 is the same patient in Table 1. b The relative level of PAK3 is indicated as the ratio to GAPDH level. The band densities were calculated by densitometry analysis. The data are shown as mean ± SEM of three independent experiments. * P < 0.05, t-test
Discussion
In this study, we investigated the possible role of PAK3 in the development and progression of ACTH producing thymic carcinoids. We found that PAK3 overexpressing NIH3T3 cell showed higher level of JNK phosphorylation and increased cell migration as well as invasion. More importantly, 71% (five out of seven) of thymic carcinoids with overexpression of PAK3 showed tumor invasion or metastasis.
As a member of PAK family, PAK3 was first identified as a target protein for Rho family of small GTP-binding protein in 1995 [14]. Later, PAK3 mutations have been linked to nonsyndromic X-linked mental retardation [19]. Its role in the development of nervous system such as synaptic plasticity and spine morphogenesis has been well established [23, 24]. However, little is known for its roles in cancer development and progression. Our findings indicated PAK3 might participate in the progression of thymic carcinoid.
PAK1 is an important regulator of cell growth and metastatic phenotype in cancer [17, 25, 26]. PAK3 has 90% homology at the kinase domain with PAK1, and some active mutations in PAK3 gene have been identified in some tumors by genome sequencing [27], which strongly suggest that PAK3 may be implicated in some tumors’ etiology. We proposed that PAK3 may regulate cell growth and migration in cancer. Although we did not observe the cell proliferation effect of PAK3 overexpression in NIH3T3 cell (data not shown), we found PAK3 activation can promote cell motility in NIH3T3 cell. We noted that, very recently, Asano et al. has reported the depletion of PAK3 also polarized the cellular distribution of actin filaments, and is sufficient to induce nonmotile cells to migrate [28], and Cobos et al. disclosed the role of PAK3 in neuronal migration, that is PAK3 overexpression in the medial ganglionic eminences (MGE) was sufficient to arrest the tangential migration of interneurons to neocortex [29]. However, our result is consistent with the common well-known function of PAKs, i.e., to modulate cell skeleton organization and consequent cell movement [30]. In fibroblasts and epithelial cells, PAK is targeted with Paxillin and GIT1 to dramatically increase migration, protrusion, and adhesion turnover [31]. Overexpression of PAK3 in PC12 cells (derived from pheochromocytoma, a neuroendocrine tumor in adrenal medulla) could induce cell spreading, membrane ruffling, and increased lamellipodia formation [32]. In a panel of 25 pheochromocytoma, we found 84% (21/25) with PAK3 overexpression (data not shown). Altogether these observations might explain the relative aggressive feature of thymic carcinoid, with 5 year survival rate only 0–50% and 10 year less than 10% [33].
MAPKs play crucial roles in cell migration [34]. It has been reported that PAKs could stimulate the mitogen-activated protein kinase (p42/44 MAPK, p38 MAPK and JNK) [35]. Based on these findings, we investigated the activities of MAPKs in both the cell model and the tumor tissues. We found JNK pathway was positively associated with PAK3 expression and PAK3 mediated cell migration. A previous study described constitutively active PAK3 mutant could stimulate JNK activation in transfected COS-1 cells [36]. JNK has been implicated in the regulation of cell migration in a broad range of cell types and several developmental processes [34]. Several molecules, such as c-Jun and paxillin [37], may facilitate this regulation process. Ching et al. [17] reported that overexpression of PAK1 in hepatocellular carcinoma could promote cancer metastasis through c-Jun NH2-terminal kinase activation and its downstream paxillin phosphorylation. Activation of c-Jun was confirmed in our study, while there was no significant change for total and phosphorylated paxillin (Ser178) (data not shown). PAK3 may promote cell migration through a different mechanism.
In addition to cell migration, PAK3 may also play a role in neuroendocrine development, similar to its roles in brain development. It will be interesting to evaluate the expression of neuroendocrine markers, such as chromogranin A (CgA), neuron specific enolase (NSE), or other specific ones such as ACTH, catecholamine, or calcitonin after PAK3 knockdown or overexpression in the appropriate models. In summary, this study discovered a novel role of PAK3 in the development and progression of ectopic ACTH producing thymic carcinoid. Upregulation and activation of PAK3 may contribute to the metastatic behavior of the tumors through JNK mediated tumor cell migration and invasion.
Materials and methods
Samples and patients
A total of seven cases of thymic carcinoid with EAS (four males and three females, ages range from 29 to 58 years) were enrolled in this study. The diagnosis was based on clinical manifestations and histopathological examination [3]. Of the seven patients, five displayed lymph node metastasis, local invasion, or distant metastasis. All the patients were operated in between 2001 and 2008 and clinically monitored at the Shanghai Clinical Center for Endocrine and Metabolic Diseases in the Ruijin Hospital. Informed consent was obtained from every participant and this study was approved by the Ruijin Hospital Ethical Committee. Thymic carcinoid tissues obtained from patients with EAS (AC), and noncancerous thymic tissues (NC) from subjects with heart surgery were collected immediately after surgical excision and stored in liquid nitrogen.
Microarray analysis
Microarray and date management was described previously [13]. Gene Ontology (GO) categorizations were downloaded from NCBI database (ftp://ftp.ncbi.nih.gov/gene/DATA/gene2go.zip) and PANTHER database [38, 39]. Pathway resources were obtained from KEGG [40], GenMAPP [41], and Biocarta (http://www.biocarta.com/). These databases were subjected to hypergeometric distribution-based enrichment analyses to explore the underlying biological relevance of differentially expressed genes. The Benjamini-Hochberg (BH) derived step-up procedure of false discovery rate (FDR) [42] was used to assess the significance of the enrichments.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) analysis was performed in 96-well plate using a SYBR Premix Ex Taq (Takara, Shiga, Japan) according to the supplier’s instructions. PCR was carried out on an Applied Biosystems 7300 Real Time Instrument (Applied Biosystems, Foster City, CA). Cycling parameters were 95°C for 10 s and then 40 cycles of 95°C for 5 s and 60°C for 31 s. Quantifications were performed in triplicate, and human β-actin was used as an internal control. All the sequences of the primers used in this study were listed in Table 2. To test the primer efficiency, 0.0384, 0.192, 0.96, 4.8, 24, 120 ng cDNA were added as PCR template in triplicate, after amplification, CT (cycle threshold) value versus DNA amount were plotted to calculate the slope, the real time PCR efficiency was calculated based on the equation: Efficiency = 10(−1/slope) [43]. Statistical analysis was performed using Student’s t-test. Error bars in graphs represent SEM.
Protein preparation and western blotting
To prepare total protein extracts, tissue specimens were homogenated in lysis buffer containing 150 mM NaCl, 20 mM Tris–HCl (pH 7.5), 1 mM EDTA, 0.1% SDS, and protease inhibitors (Sigma, St. Louis, MO). For cell proteins, cells were washed with phosphate-buffered saline (PBS) and isolated in RIPA buffer containing 50 mM Tris–HCl (pH 8), 150 mM NaCl, 5 mM MgCl2, 2 mM EDTA, 1 mM NaF, 1% NP-40, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitor cocktail, and phosphatase inhibitor cocktail (Sigma, St Louis, MO). The protein samples were loaded onto 10% SDS-PAGE and electronically transferred into polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). The membranes were blocked with 10% non-fat milk in Tris-buffered saline with Tween 20 (TBS-T) for 2 h and then probed with the following primary antibodies: anti-PAK3 antibody (1:1000 dilutions, Santa Cruz Biotechnology, Santa Cruz, CA), Erk, p-Erk, p38, p-p38, JNK, p-JNK, and c-Jun antibodies (1:1000 dilutions, Cell Signaling, Beverly, MA). Then the membranes were washed with TBS-T, and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h. The signals were visualized with enhanced chemiluminescence (ECL) reagents according to the manufacturer’s protocol (Amersham Pharmacia Biotech). Band intensities were quantified by densitometry analysis using Bio-Rad GS-710 densitometer with QuantityOne 4.5 Software.
Immunohistochemistry
Formalin-fixed and paraffin-embedded sections were deparaffinized in xylene and rehydrated through descending ethanol concentration to distilled water. Endogenous peroxidase was quenched by incubating the slides in 0.3% H2O2 for 30 min at room temperature. After being blocked with 2% normal rabbit serum for 30 min, the slides were incubated overnight in a solution of PAK3 polyclonal antibody (1:200, Santa Cruz Biotechnology, Santa Cruz, CA). Next, the sections were covered with biotinylated secondary antibody and incubated for 30 min at room temperature. After the sections were finally incubated with ABC complex/HRP for 30 min at room temperature, they were left to develop with 3-30-diaminobenzidine (DAB). The images were acquired using an Olympus system.
Cell culture, plasmid, and transfection
NIH 3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen life technologies, Carlsbad, CA) with 10% fetal bovine serum in 5% CO2 at 37°C. Mouse Pak3 was amplified from mouse brain cDNA and subcloned into pcDNA 3.1+ vector. The constructs were transfected into NIH3T3 cells by using Lipofectamine 2000 according to the manufacturer’ instructions (Invitrogen life technologies, Carlsbad, CA) and selected with G418 (Promega, Madison, WI, USA) 48 h after transfection.
Kinase activity assay
Cell lysates were extracted from the transfected NIH3T3 cells and incubated with antibody against PAK3, and then pulled-down with Sepharose beads (Santa Cruz Biotechnology, Santa Cruz, CA). The beads were washed and resuspended in 20 μl of the kinase buffer containing 2 mg of myelin basic protein (MBP) (Sigma, St. Louis, MO), and then incubated at 30°C for 20 min in the presence of 10 mCi of [γ-32P]ATP (6,000 Ci/mmol; Amersham Pharmacia Biotech, Piscataway, NJ). The reactions were terminated by boiling in Laemmli’s SDS sample buffer. The γ-32P-labeled MBP was loaded on 12% SDS-PAGE and visualized by autoradiography.
Wound healing assay
The transfected NIH3T3 cells were seeded in 6-well plates the day before wounding. When at confluence, cell monolayers were wounded by manually scraping the cells with a pipette tip. The cell culture was then replaced with fresh medium. Cells were allowed to migrate in complete medium in the absence or presence of JNK inhibitor, SP600125 (40 μM, Sigma, St Louis, MO, USA). The wound size was monitored by microscopy at different times.
Invasion assay
The 24-well BD Biocoat Matrigel invasion chambers with 8 μm polycarbonated filters (Becton–Dickinson, Bedford, MA) were used according to the manufacture’s instructions. In brief, cells were placed on Matrigel coated chambers. Complete DMEM with 10% FBS was added in the lower wells. Cells were incubated at 37°C for 20 h with 5% CO2. Non-invading cells on the upper surface of the filter were wiped with a cotton swab. Invasive cells that penetrated through the filter and migrated to the bottom side of the membrane were fixed and stained with Hematoxylin & Eosin (HE). The migrated cells were counted in 3 fields for each transwell filter under microscopy at ×200. Quantification of the rate of cell invasion was calculated by dividing the number of cells invading through matrigel insert membrane by the number of cells migrating through control insert membrane. Statistical analysis was performed using Student’s t-test.
References
S.B. Baylin, G. Mendelsohn, Ectopic (inappropriate) hormone production by tumors: mechanisms involved and the biological and clinical implications. Endocr. Rev. 1, 45–77 (1980)
A.M. Isidori, A. Lenzi, Ectopic ACTH syndrome. Arq. Bras. Endocrinol. Metabol. 51, 1217–1225 (2007)
W.Q. Wang, L. Ye, Y.F. Bi, H.Y. Zhao, S.Y. Sun, Z.Y. Tang, Y.J. Zhao, W.Q. Fang, Z.Y. Chen, K.M. Chen, X.L. Jin, G. Ning, Six cases of ectopic ACTH syndrome caused by thymic carcinoid. J. Endocrinol. Invest. 29, 293–297 (2006)
Y.F. Bi, L. Ye, Y.H. Chen, G. Ning, S.Y. Sun, L. Jiang, N. Zhu, W.W. Zhou, W.Q. Wang, Characteristics of ectopic adrenocorticotropic hormone syndrome due to thymic carcinoid and analysis of the molecular mechanism. Chin. Med. J. 121, 667–670 (2008)
I. Ilias, D.J. Torpy, K. Pacak, N. Mullen, R.A. Wesley, L.K. Nieman, Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J. Clin. Endocrinol. Metab. 90, 4955–4962 (2005)
A.M. Isidori, G.A. Kaltsas, C. Pozza, V. Frajese, J. Newell-Price, R.H. Reznek, P.J. Jenkins, J.P. Monson, A.B. Grossman, G.M. Besser, The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J. Clin. Endocrinol. Metab. 91, 371–377 (2006)
D.Y. Wang, D.B. Chang, S.H. Kuo, P.C. Yang, Y.C. Lee, H.C. Hsu, K.T. Luh, Carcinoid tumours of the thymus. Thorax 49, 357–360 (1994)
V.T. de Montpreville, P. Macchiarini, E. Dulmet, Thymic neuroendocrine carcinoma (carcinoid): a clinicopathologic study of fourteen cases. J. Thorac. Cardiovasc. Surg. 111, 134–141 (1996)
R.J. Rieker, S. Aulmann, R. Penzel, P.A. Schnabel, H. Blaeker, I. Esposito, A. Morresi-Hauf, H.F. Otto, E. Hecker, H. Dienemann, P. Schirmacher, G. Mechtersheimer, Chromosomal imbalances in sporadic neuroendocrine tumours of the thymus. Cancer Lett. 223, 169–174 (2005)
B.T. Teh, J. Zedenius, S. Kytola, B. Skogseid, J. Trotter, H. Choplin, S. Twigg, F. Farnebo, S. Giraud, D. Cameron, B. Robinson, A. Calender, C. Larsson, P. Salmela, Thymic carcinoids in multiple endocrine neoplasia type 1. Ann. Surg. 228, 99–105 (1998)
P.D. Leotlela, A. Jauch, H. Holtgreve-Grez, R.V. Thakker, Genetics of neuroendocrine and carcinoid tumours. Endocr. Relat. Cancer 10, 437–450 (2003)
L. Ye, X. Li, X. Kong, W. Wang, Y. Bi, L. Hu, B. Cui, G. Ning, Hypomethylation in the promoter region of POMC gene correlates with ectopic overexpression in thymic carcinoids. J. Endocrinol. 185, 337–343 (2005)
Y.F. Bi, R.X. Liu, L. Ye, H. Fang, X. Li, W. Wang, J. Zhang, K.K. Wang, L. Jiang, T. Su, Z.Y. Chen, G. Ning, Gene expression profiles of thymic neuroendocrine tumors (carcinoids) with ectopic ACTH syndrome reveal novel molecular mechanism. Endocr. Relat. Cancer 16, 1273–1282 (2009)
E. Manser, C. Chong, Z.S. Zhao, T. Leung, G. Michael, C. Hall, L. Lim, Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J. Biol. Chem. 270, 25070–25078 (1995)
R. Kumar, A.E. Gururaj, C.J. Barnes, p21-Activated kinases in cancer. Nat. Rev. Cancer 6, 459–471 (2006)
S. Balasenthil, A.A. Sahin, C.J. Barnes, R.A. Wang, R.G. Pestell, R.K. Vadlamudi, R. Kumar, p21-Activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J. Biol. Chem. 279, 1422–1428 (2004)
Y.P. Ching, V.Y. Leong, M.F. Lee, H.T. Xu, D.Y. Jin, I.O. Ng, P21-activated protein kinase is overexpressed in hepatocellular carcinoma and enhances cancer metastasis involving c-Jun NH2-terminal kinase activation and paxillin phosphorylation. Cancer Res. 67, 3601–3608 (2007)
V. Rousseau, O. Goupille, N. Morin, J.V. Barnier, A new constitutively active brain PAK3 isoform displays modified specificities toward Rac and Cdc42 GTPases. J. Biol. Chem. 278, 3912–3920 (2003)
K.M. Allen, J.G. Gleeson, S. Bagrodia, M.W. Partington, J.C. MacMillan, R.A. Cerione, J.C. Mulley, C.A. Walsh, PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 20, 25–30 (1998)
J. Meng, Y. Meng, A. Hanna, C. Janus, Z. Jia, Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J. Neurosci. 25, 6641–6650 (2005)
J. Souopgui, M. Solter, T. Pieler, XPak3 promotes cell cycle withdrawal during primary neurogenesis in Xenopus laevis. EMBO J. 21, 6429–6439 (2002)
K. Kageyama, S. Sakihara, T. Suda, Regulation and role of p21-activated kinase 3 by corticotropin-releasing factor in mouse pituitary. Regul. Pept. 152, 88–94 (2009)
P. Kreis, E. Thevenot, V. Rousseau, B. Boda, D. Muller, J.V. Barnier, The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc42-dependent pathway. J. Biol. Chem. 282, 21497–21506 (2007)
R. Node-Langlois, D. Muller, B. Boda, Sequential implication of the mental retardation proteins ARHGEF6 and PAK3 in spine morphogenesis. J. Cell Sci. 119, 4986–4993 (2006)
R.A. Wang, H. Zhang, S. Balasenthil, D. Medina, R. Kumar, PAK1 hyperactivation is sufficient for mammary gland tumor formation. Oncogene 25, 2931–2936 (2006)
L. Adam, R. Vadlamudi, M. Mandal, J. Chernoff, R. Kumar, Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J. Biol. Chem. 275, 12041–12050 (2000)
C. Greenman, P. Stephens, R. Smith, G.L. Dalgliesh, C. Hunter, G. Bignell, H. Davies, J. Teague, A. Butler, C. Stevens, S. Edkins, S. O’Meara, I. Vastrik, E.E. Schmidt, T. Avis, S. Barthorpe, G. Bhamra, G. Buck, B. Choudhury, J. Clements, J. Cole, E. Dicks, S. Forbes, K. Gray, K. Halliday, R. Harrison, K. Hills, J. Hinton, A. Jenkinson, D. Jones, A. Menzies, T. Mironenko, J. Perry, K. Raine, D. Richardson, R. Shepherd, A. Small, C. Tofts, J. Varian, T. Webb, S. West, S. Widaa, A. Yates, D.P. Cahill, D.N. Louis, P. Goldstraw, A.G. Nicholson, F. Brasseur, L. Looijenga, B.L. Weber, Y.E. Chiew, A. DeFazio, M.F. Greaves, A.R. Green, P. Campbell, E. Birney, D.F. Easton, G. Chenevix-Trench, M.H. Tan, S.K. Khoo, B.T. Teh, S.T. Yuen, S.Y. Leung, R. Wooster, P.A. Futreal, M.R. Stratton, Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007)
Y. Asano, A. Jimenez-Dalmaroni, T.B. Liverpool, M.C. Marchetti, L. Giomi, A. Kiger, T. Duke, B. Baum, Pak3 inhibits local actin filament formation to regulate global cell polarity. HFSP J. 3, 194–203 (2009)
I. Cobos, U. Borello, J.L. Rubenstein, Dlx transcription factors promote migration through repression of axon and dendrite growth. Neuron 54, 873–888 (2007)
M.A. Sells, U.G. Knaus, S. Bagrodia, D.M. Ambrose, G.M. Bokoch, J. Chernoff, Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7, 202–210 (1997)
A. Nayal, D.J. Webb, C.M. Brown, E.M. Schaefer, M. Vicente-Manzanares, A.R. Horwitz, Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J. Cell Biol. 173, 587–589 (2006)
A. Obermeier, S. Ahmed, E. Manser, S.C. Yen, C. Hall, L. Lim, PAK promotes morphological changes by acting upstream of Rac. EMBO J. 17, 4328–4339 (1998)
C.A. Moran, S. Suster, Neuroendocrine carcinomas (carcinoid tumor) of the thymus. A clinicopathologic analysis of 80 cases. Am. J. Clin. Pathol. 114, 100–110 (2000)
C. Huang, K. Jacobson, M.D. Schaller, MAP kinases and cell migration. J. Cell Sci. 117, 4619–4628 (2004)
C. Hofmann, M. Shepelev, J. Chernoff, The genetics of Pak. J. Cell Sci. 117, 4343–4354 (2004)
S. Bagrodia, B. Derijard, R.J. Davis, R.A. Cerione, Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 270, 27995–27998 (1995)
C. Huang, Z. Rajfur, C. Borchers, M.D. Schaller, K. Jacobson, JNK phosphorylates paxillin and regulates cell migration. Nature 424, 219–223 (2003)
H. Mi, N. Guo, A. Kejariwal, P.D. Thomas, PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 35, D247–D252 (2007)
Gene Ontology Consortium, The Gene Ontology project in 2008. Nucleic Acids Res. 36, D440–444 (2008)
M. Kanehisa, S. Goto, S. Kawashima, Y. Okuno, M. Hattori, The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280 (2004)
K.D. Dahlquist, N. Salomonis, K. Vranizan, S.C. Lawlor, B.R. Conklin, GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat. Genet. 31, 19–20 (2002)
Y. Benjamini, Y. Hochberg, Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 (1995)
R. Rasmussen, Quantification on the LightCycler, in Rapid Cycle Real-time PCR, Methods and Applications, ed. by S. Meuer, C. Wittwer, K. Nakagawara (Springer, Heidelberg, 2001), pp. 21–34
Acknowledgments
We greatly appreciate the patients for their cooperation in this study. This work was supported by the grants from the National Natural Science Foundation of China (No. 30725037, 30771018 and 30871203). The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Rui-xin Liu and Wei-qing Wang contributed equally to this paper.
Rights and permissions
About this article
Cite this article
Liu, Rx., Wang, Wq., Ye, L. et al. p21-Activated kinase 3 is overexpressed in thymic neuroendocrine tumors (carcinoids) with ectopic ACTH syndrome and participates in cell migration. Endocr 38, 38–47 (2010). https://doi.org/10.1007/s12020-010-9324-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-010-9324-6