
Abstract
It has been proposed to cautiously supplement with vitamin D to any patient with asymptomatic primary hyperparathyroidism (PHTP) and a plasma 25-hydroxyvitamin D [25(OH)D] concentration <50 nmol/l. Evidence about the safeness of this intervention is limited to two studies. Our aim was to prospectively assess the biochemical effects of one-year 25(OH)D supplementation in this context. Twenty-seven patients were included in this study. Calcifediol was started at a dose of 480–960 IU/24 h (8–16 μg/24 h) and adjusted up to a maximum of 960 IU/24 h (16 μg/24 h). Basal calcium, phosphate, albumin, total alkaline phosphatase (ALP), creatinine, 24 h calcium urinary excretion, intact PTH (iPTH) and 25(OH)D were measured before and during vitamin D supplementation. The mean basal 25(OH)D was 28.7 ± 8.0 nmol/l, and at 12 months was 71.5 ± 32.5 nmol/l (P = 0.00 vs. baseline). After 3, 6 and 12 months iPTH levels were 141.7 ± 108.4 ng/l (P = 0.00 vs. baseline), 131.1 ± 95.7 ng/l (P = 0.03 vs. baseline) and 162.2 ± 139.3 ng/l (P = ns vs. baseline). Mean calcium did not change. Mean urinary calcium excretion increased significantly (basal: 5.7 ± 2.9 mmol/24 h, 12 months: 7.9 ± 4.9 mmol/24 h, P = 0.02). Cautious calcifediol supplementation significantly increased mean 25(OH)D and temporarily reduced mean iPTH. It did not change mean serum calcium, but urinary calcium excretion increased significantly. We suggest that serum calcium and 24 h calciuria be measured at regular intervals in patients with PHTP, while on calcifediol supplementation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plasma 25-hydroxyvitamin D [25(OH)D] level is frequently low in patients with primary hyperparathyroidism (PHTP) [1–4]. The pathophysiological mechanisms responsible for the low serum 25(OH)D level in PHPT are not completely clarified. The data obtained from several studies suggest that vitamin D status might influence the clinical and biological expression of PHTP [1, 2, 4–15] and even recovery after surgical treatment [11, 16, 17], although some authors have not found such an association [18, 19].
In the 2nd Workshop on the Management of Asymptomatic PHPT [20] the panel proposed to cautiously supplement with vitamin D to any asymptomatic patient with a cut-off 25(OH)D concentrations <50 nmol/l. The panel stated that “serum calcium concentration must be monitored frequently, on the chance that in some patients serum calcium levels could rise further”. Four years later one study demonstrated that, at least in some countries, the recommendation was far from being applied [4], which has been largely attributed to concerns that such treatment might worsen the hypercalcemia and/or hypercalciuria. The above-mentioned recommendation is again stated in the recently published 3rd Workshop in Hyperparathyroidism [21].
Few studies have addressed the effects of 25(OH)D normalization in this context [22–25] and, although it seems physiologically reasonable, it has not been shown that it has clinical beneficial effects on patients who are not planning to undergo surgical treatment.
The aim of this study was to prospectively assess the biochemical effects of 25(OH)D supplementation in patients with asymptomatic normo or hypercalcemic PHTP, and concomitant vitamin D deficiency.
Results
Table 1 shows the baseline characteristics of the patients. Eight patients were normocalcemic when the study began, but PHPT had been confirmed, at least twice, before enrolment. Six of them became hypercalcemic during the period of study.
Calcifediol was withdrawn in four patients after 3 months (in two because of serum calcium >2.9 mmol/l, in two due to 24 h calciuria >10 mmol), in one patient after 6 months (due to 24 h calciuria >10 mmol), and in six patients after 12 months (due to 24 h calciuria >10 mmol). In all the cases the data available before calcifediol was withdrawn was included in the analysis performed until that time point.
The mean basal level of 25(OH)D was 28.7 ± 8.0 nmol/l. The mean levels at 3, 6 and 12 months were 62.9 ± 25.0 nmol/l (P = 0.00 vs. baseline), 62.7 ± 26.77 nmol/l (P = 0.00 vs. baseline) and 71.5 ± 32.5 nmol/l (P = 0.00 vs. baseline), respectively. After 12 months, not all patients achieved 25(OH)D levels ≥50 nmol/l (Fig. 1). There was a weak inverse correlation between vitamin D and iPTH at baseline (r = −0.41; P = 0.04) and at 12 months (r = −0.49; P = 0.04), as if there was some relationship between 25(OH)D and iPTH. Basal vitamin D was also inversely correlated with the degree of iPTH reduction at 3 months (r = −0.45, P = 0.04).

Absolute values of 25(OH)D in the patients at baseline (0), after 3 months (1) and 12 months (3) of calcifediol supplementation. Values for individual patients are connected by the solid lines. The dotted line represents the level of serum 25(OH)D the panel of the 3rd Workshop on the Management of Asymptomatic PHPT proposed to try to achieve
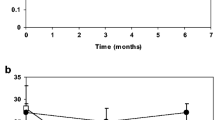
The mean basal iPTH was 187.8 ± 134.8 ng/l. After 3, 6 and 12 months of treatment, mean iPTH levels were 141.7 ± 108.4 ng/l (P = 0.00 vs. baseline), 131.1 ± 95.7 ng/l (P = 0.03 vs. baseline), and 162.2 ± 139.3 ng/l (P = ns vs. baseline), respectively (Fig. 2). There was a significant, but not progressive, decrease in iPTH levels during the first 6 months of treatment, and then iPTH increased, so that at 12 months mean iPTH was not significantly different from baseline. The higher the basal iPTH was, the deeper the reduction in iPTH was observed at 6 months in response to vitamin D (r = 0.78, P = 0.00), but we also observed an inverse correlation between basal iPTH and the degree of iPTH reduction at 12 months (r = −0.71, P = 0.00). We found no correlation between the degree of change in plasma vitamin D and the degree of change in iPTH at any time during the study.

Changes in serum iPTH after 3, 6 and 12 months of calcifediol supplementation. Each data point represents the change from baseline in an individual patient at the indicated time point. The horizontal bars represent group mean and 95% confidence interval. *P < 0.05 vs. baseline. **P < 0.01 vs. baseline
During the one-year treatment with calcifediol, mean serum calcium did not change (basal: 2.70 ± 0.13 mmol/l; 3 months: 2.70 ± 0.13 mmol/l; 6 months: 2.73 ± 0.10 mmol/l; 12 months: 2.73 ± 0.10 mmol/l). In two patients, serum calcium rose over 0.2 mmol/l above upper limit of normal (from 2.7 to 3.1, and from 2.7 to 3.0 mmol/l, respectively) after 3 months, although none of them developed any calcium-related complaints. Vitamin D supplementation was withdrawn in both patients, and serum calcium decreased to basal levels. No other case of calcium >2.9 mmol/l was found afterwards. Serum phosphorus and total ALP did not change significantly during the study.
Mean 24 h urinary calcium excretion increased significantly with vitamin D supplementation (basal: 5.7 ± 2.9 mmol; 3 months: 6.9 ± 3.8 mmol, P = 0.02; 6 months: 7.0 ± 4.7 mmol, P = ns vs. baseline; 12 months: 7.9 ± 4.9 mmol, P = 0.02 vs. baseline) (Fig. 3). Although there was a tendency towards a progressive increase in the urinary calcium excretion throughout the study, this increase was not significant. There was no correlation between 24 h urinary calcium excretion, or changes in 24 h urinary calcium excretion, and either absolute values of (or changes in) vitamin D or iPTH at any point of the study.

Changes in 24 h urinary calcium excretion after 3, 6 and 12 months of calcifediol supplementation. Each data point represents the change from baseline in an individual patient at the indicated time point. The horizontal bars represent group mean and 95% confidence interval. *P < 0.05 vs. baseline. **P < 0.01 vs. baseline
Table 2 shows the biochemical data after 3, 6 and 12 months of vitamin D supplementation.
The accumulated incidence of calciuria >10 mmol/24 h was 7.4% at 3 months, 11.1% at 6 months, and 33.3% at 12 months. In three patients it developed while taking calcifediol 480 IU/d and the dose of vitamin D was withdrawn; in the other six it developed while taking 720 or 960 IU/d, and the dose was reduced. In every case calciuria returned to basal levels. None of these patients developed complaints related to hypercalciuria during the period of study.
Discussion
In our series, supplementation with 480–960 IU/d of calcifediol to patients with mild PHPT and concomitant vitamin D deficiency, significantly increased mean 25(OH)D and temporarily reduced mean iPTH levels. These doses of calcifediol we employed were sufficient to increase mean 25(OH)D levels above 50 mmol/l after 3, 6 and 12 months of treatment, although not every individual patient achieved this level. Vitamin D supplementation did not change mean calcium, but accumulated incidence of serum calcium over 0.2 mmol/l above upper limit of normal was 7.4%. Mean urinary calcium excretion increased significantly and the accumulated incidence of 24 h calciuria >10 mmol was 33.3% at 12 months. Since these findings prompted us to reduce or withdraw calcitriol supplementation in those patients, we are not certain whether those changes would be temporary, permanent, or could even have worsened had the patients continued taking the drug. To our knowledge there are only two other studies about the biochemical effects of long-term vitamin D supplementation in this context, and they report discrepant results.
In both the 2nd [20] and the 3rd Workshop on the Management of Asymptomatic PHPT [21], the panels proposed to cautiously supplement with vitamin D to any asymptomatic patient with a cut-off 25-OHD concentrations <50 nmol/l before other management decision is taken. Only a few studies have addressed the effects of this intervention [22, 24] and evidence to support this is limited to two [23, 25].
Grey et al. [23] reported results similar to ours as far as mean calcium, phosphate, and 25(OH)D are concerned, but they found a permanent decline in iPTH levels while on vitamin D repletion. Tucci [25] did not find a significant decrease in iPTH after 10 weeks of treatment. In our study there was a significant decrease in iPTH during the first 6 months of treatment and then iPTH increased, so that at 12 months mean iPTH was not different from baseline. This was so even though 25(OH)D at 12 months was not significantly different from 25(OH)D levels at 3 and 6 months. We do not know whether this finding was influenced by the limited size of our sample, or whether in fact there was an escape from the effect of the intervention. We found no relationship between the degree of change in vitamin D and the degree of change in iPTH, but there was an inverse correlation between iPTH and vitamin D both at baseline and at 12 months, as if there was some relation between vitamin D and iPTH. Contrary to what Grey et al. reported, we found no changes in total ALP in response to vitamin D, but total ALP might not have been a sensitive approach to detect subtle changes in bone turnover, especially in patients with very limited bone disease, as ours were.
Tucci [25] reports a nonsignificant decrease in urine calcium/creatinine ratios. Grey et al. [23] reported a small, nonsignificant increase in urinary calcium at 6 months that returned to baseline levels at 12 months, and in only two patients did calcium excretion rose to exceed 10 mmol/24 h. In contrast, we found a significant increase in 24 h urinary calcium excretion that persisted throughout the period of study, and the accumulated incidence of calciuria >10 mmol/d was 33.3% at 12 months. We could not find any relationship between changes in 25(OH)D and changes in urinary calcium excretion, but the fact that 24 h calciuria decreased to the basal levels after calcifediol withdrawal, and that it remained stable afterwards, supports the hypothesis that vitamin supplementation was related to its development, rather than to progression of the disease.
We can only hypothesize about the reasons for the discrepancies reported among the three studies. Differences in the populations studied, the therapeutic regimes employed, and the small sample sizes may account, to some extent, for some of the divergences.
In the 3rd Workshop on the Management of Asymptomatic Primary Hyperparathyroidism the panel states that, in the absence of kidney stones, urinary calcium excretion >10 mmol/24 h should no longer be considered as an indication for surgical treatment, and that urinary calcium excretion need not be monitored during follow-up. Nevertheless it acknowledges that some physicians still regard 24 h urinary calcium excretion >10 mmol as an indication for surgery. We suggest that 24 h urinary calcium excretion be monitored at regular intervals while on vitamin D supplementation. Although the significance of the increase in urinary calcium excretion is uncertain, it still has to be shown that vitamin D supplementation in this context has beneficial clinical effects.
We do not think that the fact that some patients were on biphosphonate treatment at enrollment invalidates our results. In fact, alendronate has not been shown to significantly modify plasma calcium levels [26–28] and neither alendronate nor arisedronate has been shown to increase urinary calcium excretion [28, 29]. Biphosphonates do not significantly alter serum calcium or PTH during long-term therapy [30] and all our patients had been on biphosphonates for more than 6 months, when they entered the study.
As far as the dose of vitamin D is concerned, we chose doses in the range of preventive or maintenance measures to avoid deficiency, rather than those recommended to treat deficiency [31], following the advice of the 2nd Workshop on Asymptomatic Primary Hyperparathyroidism [20]. We found that dose to be sufficient to increase mean 25(OH)D plasma level to those recommended in PHPT, although this threshold was not achieved in every individual patient. The apparently low requirements of vitamin D in our patients could be due to several factors. First, in the other two studies cholecalciferol (vitamin D3) and ergocalciferol (vitamin D2) were prescribed, while we used 25(OH)D directly. Some studies suggest that ergocalciferol increases serum 25(OH)D less efficiently than cholecalciferol. In addition, vitamin D2 is not accurately measured in all vitamin D assays. Second, 25(OH)D concentrations are associated with body composition variables, especially body fat [32], so that adiposity affects vitamin D requirements in deficient states. Differences in body fat might have influenced the doses required. Third, different vitamin D input conditions seem to have effect on the efficiency of the 25-hydroxylation of vitamin D [33, 34]. Finally, mean 25(OH)D levels achieved during vitamin D supplementation were lower in our patients than those achieved in Grey’s and Tucci’s studies.
In conclusion, supplementation with 480–960 IU/d of calcifediol to patients with mild PHPT significantly increased mean 25(OH)D and temporarily reduced mean iPTH levels. In agreement with previous studies, vitamin D supplementation did not change mean serum calcium, but accumulated incidence of serum calcium over 0.2 mmol/l above upper limit of normal was 7.4%. Contrary to what has been reported, urinary calcium excretion increased significantly in response to vitamin D repletion and the accumulated incidence of calciuria >10 mmol/d was 33.3% at 12 months. We suggest that serum calcium and 24 h calciuria be measured at regular intervals in patients with PHTP, while on calcifediol supplementation.
Patients and methods
Patients
Twenty-seven consecutive patients referred to the Endocrinology Department, and that fulfilled the inclusion criteria, were included. PHPT was defined band hypercalcemia in the presence of inappropriately normal or elevated levels of iPTH. The inclusion criteria were basal serum calcium <2.9 mmol/l, basal 24 h urinary calcium excretion <10 mmol, glomerular filtration rate >60 ml/min (Cockroft–Gault equation), and 25(OH)D concentration <50 nmol/l.
Six patients were on biphosphonates (alendronate or risedronate) for more than 6 months when they entered the study, and no change was made on these treatments during the period of the study. No patient was taking any medication known to interfere with calcium metabolism, and no such a drug was started during the period of the study. The patients were not considered for surgical treatment either because they did not fulfill the 2nd Workshop on the Management of Asymptomatic PHPT criteria for surgical treatment, because anesthetic risk was considered unacceptable, or because they refused the procedure.
Since the intervention constitutes an ordinary recommendation of both the 2nd and the 3rd Workshop on the Management of Asymptomatic PHPT, approval was not requested at the local Ethical Committee, but every patient gave informed consent to the collection, analysis and publication of his personal data.
Vitamin D supplementation
Vitamin D supplementation was implemented with calcifediol [25(OH)D3] solution (1 drop = 4 μg = 240 IU). Calcifediol was started at a dose of 480–960 IU/24 h (2–4 drops/24 h) on the basis of the severity and of the deficit and the other basal results. Vitamin D dose was then adjusted on a 3-month basis. The patients in whom calcium plasma levels rose to ≥2.9 mmol/l (0.2 mmol/l above the upper limit of normal) or who developed 24 h calcium urinary and excretion ≥10 mmol/24 h, calcifediol dose was reduced (if taking 720 or 960 IU/d) or withdrawn (if taking 480 IU/d), and changes in calcium and urinary calcium excretion were monitored. In the patients in whom 25(OH)D levels did not increase >50 nmol/l (and as long as their serum calcium was <2.9 mmol/l and their 24 h urinary and calcium excretion was <10 mmol), the calcifediol dose was increased up to a maximum dose of 960 IU/24 h.
Laboratory analysis
After an overnight fast, basal calcium, phosphate, albumin, alkaline phosphatase, creatinine, 24 h calcium urinary excretion, iPTH, and 25(OH)D were measured. After vitamin D supplementation was started, the same studies were performed on a 3-month basis.
Serum calcium, phosphate, albumin, alkaline phosphatase, creatinine, and 24 h calcium urinary excretion were measured using a multichannel autoanalyzer. iPTH was measured by band chemiluminescence immunoassay (Centaur Analyzer®, Siemens Healthcare Diagnostics, Tarrytown, NAND, USA). 25(OH)D was measured by band a 2-site chemiluminescence immunoassay (DiaSorin LIAISON® 25-OH Vitamin D, DiaSorin, Stillwater, MN, USA).
Statistical analysis
The SPSS software 16.0 (Chicago, IL, USA) was used to produce statistical analysis. Changes in the quantitative variables along the period of study were assessed using Wilcoxon’s signed-rank test. Relationships between biochemical variables were assessed using Rho Spearman’s correlation coefficient. Data are expressed as mean ± SD. P values ≤ 0.05 were considered to be significant.
References
S.J. Silverberg, E. Shane, D.W. Dempster, J.P. Bilezikian, Am. J. Med. 107, 561–567 (1999)
B. Moosgaard, P. Vestergaard, L. Heickendorff, F. Melsen, P. Christiansen, L. Mosekilde, Clin. Endocrinol. (Oxf) 63, 506–513 (2005)
A.D. Bussey, J.M. Bruder, Endocr. Pract. 11, 37–42 (2005)
P. Boudou, F. Ibrahim, C. Cormier, E. Sarfati, J.C. Souberbielle, J. Endocrinol. Invest. 29, 511–515 (2006)
D.S. Rao, M. Honasoge, G.W. Divine, E.R. Phillips, M.W. Lee, M.R. Ansari, G.B. Talpos, A.M. Parfitt, J. Clin. Endocrinol. Metab. 85, 1054–1058 (2000)
D.S. Rao, G. Agarwal, G.B. Talpos, E.R. Phillips, F. Bandeira, S.K. Mishra, A. Mithal, J. Bone Miner. Res. 17(Suppl 2), N75–N80 (2002)
E. Nordenström, J. Westerdahl, B. Lindergård, P. Lindblom, A. Bergenfelz, World J. Surg. 26, 1463–1467 (2002)
F. Bandeira, G. Caldas, E. Freese, L. Griz, M. Faria, C. Bandeira, Endocr. Pract. 8, 266–270 (2002)
H. Raef, S. Ingemansson, S. Sobhi, A. Sultan, M. Ahmedm, M. Chaudhry, J. Endocrinol. Invest. 27, 807–812 (2002)
B. Moosgaard, P. Vestergaard, L. Heickendorff, F. Melsen, P. Christiansen, L. Mosekilde, Eur. J. Endocrinol. 155, 237–244 (2006)
N. Ozbey, Y. Erbil, E. Ademoğlu, S. Ozarmağan, U. Barbaros, A. Bozbora, World J. Surg. 30, 321–326 (2006)
T.D. Beyer, E.L. Chen, N. Nulibol, R.A. Prinz, C.C. Solorzano, J. Surg. Res. 143, 145–150 (2007)
Y. Inoue, H. Kaji, I. Hisa, T. Tobimatsu, J. Naito, M.F. Iu, T. Sugimoto, K. Chihara, Endocr. J. 55, 57–65 (2008)
B. Moosgaard, S.E. Christensen, P. Vestergaard, L. Heickendorff, P. Christiansen, L. Mosekilde, Clin. Endocrinol. (Oxf) 68, 707–715 (2008)
F. Bandeira, L.H. Griz, C. Bandeira, J. Pinho, C.S. Lucena, C. Alencar, A.C. Thé, E.T. Diniz, J. Clin. Densitom. 12, 195–199 (2009)
Z.A. Stewart, A. Blackford, H. Somervell, K. Friedman, E. Garrett-Mayer, A.P. Dackiw, M.A. Zeiger, Surgery 138, 1018–1025 (2005)
E. Kandil, A.P. Tufaro, K.A. Carson, F. Lin, H. Somervell, T. Farrag, A. Dackiw, M. Zeiger, R.P. Tufano, Arch. Otolaryngol. Head Neck Surg. 134, 1071–1075 (2008)
H. Yamashita, S. Noguchi, S. Uchino, S. Watanabe, E. Koike, T. Murakami, T. Fujihira, Y. Koga, T. Masatsugu, H. Yamashita, World J. Surg. 26, 937–941 (2002)
V. Carnevale, G. Manfredi, E. Romagnoli, S. De Geronimo, F. Paglia, J. Pepe, A. Scillitani, E. D’Erasmo, S. Minisola, Clin. Endocrinol. (Oxf) 60, 81–86 (2004)
J.P. Bilezikian, J.T. Potts Jr, Gel-H Fuleihan, M. Kleerekoper, R. Neer, M. Peacock, J. Rastad, S.J. Silverberg, R. Udelsman, S.A. Wells, J. Clin. Endocrinol. Metab. 87, 5353–5361 (2002)
R. Eastell, A. Arnold, M.L. Brandi, E.M. Brown, P. D’Amour, D.A. Hanley, D.S. Rao, M.R. Rubin, D. Goltzman, S.J. Silverberg, S.J. Marx, M. Peacock, L. Mosekilde, R. Bouillon, E.M. Lewiecki, J. Clin. Endocrinol. Metab. 2, 340–350 (2009)
V. Kantorovich, M.A. Gacad, L.L. Seeger, J.S. Adams, J. Clin. Endocrinol. Metab. 85, 3541–3543 (2000)
A. Grey, J. Lucas, A. Horne, G. Gamble, J.S. Davidson, I.R. Reid, J. Clin. Endocrinol. Metab. 90, 2122–2126 (2005)
E.G. Grubbs, S. Rafeeq, C. Jiménez, L. Feng, J.E. Lee, D.B. Evans, N.D. Perrier, Surgery 144, 852–858 (2008)
J. Tucci, Eur. J. Endocrinol. (2009) [Epub ahead of print]
C.R. Parker, P.J. Blackwell, K.J. Fairbairn, D.J. Hosking, J. Clin. Endocrinol. Metab. 87, 4482–4489 (2002)
C.C. Chow, W.B. Chan, J.K. Li, N.N. Chan, M.H. Chan, G.T. Ko, K.W. Lo, C.S. Cokram, J. Clin. Endocrinol. Metab. 88, 581–587 (2003)
A.A. Khan, J.P. Bilezikian, A.W. Kung, M.M. Ahmed, S.J. Dubois, A.Y. Ho, D. Schussheim, M.R. Rubin, A.M. Shaikh, S.J. Silverberg, T.I. Standish, Z. Syed, Z.A. Syed, J. Clin. Endocrinol. Metab. 89, 3319–3325 (2004)
M. Sarioglu, C. Tuzun, Z. Unlu, C. Tokiz, F. Taneli, B.S. Uyanik, Rheumatol. Int. 26, 195–200 (2006)
A. Khan, A. Grey, D. Shoback, J. Clin. Endocrinol. Metab. 94, 373–381 (2009)
J.M. Bruder, T.A. Guise, G.R. Mundy, Mineral metabolism, in Endocrinology and Metabolism, 4th edn., ed. by P. Felig, L.A. Frohman, L.A. Frohman (McGraw-Hill, New York, 2001), pp. 1079–1177
N. Vilarrasa, J. Maravall, A. Estepa, R. Sánchez, C. Masdevall, M.A. Navarro, P. Alía, J. Soler, J.M. Gómez, J. Endocrinol. Invest. 30, 653–658 (2007)
R.P. Heaney, L.A. Armas, J.R. Shary, N.H. Bell, N. Binkley, B.W. Hollis, Am. J. Clin. Nutr. 87, 1738–1742 (2008)
T. Pekkarinen, V.V. Välimäki, S. Aarum, U. Turpeinen, E. Hämäläinen, E. Löyttyniemi, M.J. Välimäki, Clin. Endocrinol. (Oxf). (2009) May 25 [Epub ahead of print]
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Isidro, M.L., Ruano, B. Biochemical effects of calcifediol supplementation in mild, asymptomatic, hyperparathyroidism with concomitant vitamin D deficiency. Endocr 36, 305–310 (2009). https://doi.org/10.1007/s12020-009-9211-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-009-9211-1