
Abstract
Objective
The purpose of this study was to evaluate the lifetime evolution of the radioclinical features in a large family with van Buchem disease.
Design and patients
The study population included 13 patients, ranging between 6 and 69 years. The evolution of the clinical features has been assessed by retrospective analysis of the clinical records of the patients. The age-related evolution of the cortical hyperostosis and defective modeling at the tubular bones was evaluated by morphometric analysis of hand films in 9 patients, compared with 9 control individuals. Progression of sclerosis of the craniofacial bones was evaluated by analysis of the skull radiographs of eleven van Buchem patients, taken at different age.
Results and conclusions
Radioclinical features, including sclerosis of the cranial and tubular bones and cranial nerve deficit, become more prominent in older patients. Defective modeling of tubular bones, cortical thickness and medullary width progress with age. Radioclinical abnormalities of van Buchem patients become more prominent in older patients, which suggests that the van Buchem gene is very actively involved in bone metabolism throughout life. Morphometric analysis of the plain films supports the hypothesis that the physiological function of the van Buchem gene is to inhibit bone formation and possibly to regulate bone remodeling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Van Buchem disease is a rare autosomal-recessive sclerosing bone dysplasia that was initially described by van Buchem et al. in 1955 [1]. It is also known as hyperostosis corticalis generalisata, and is classified among the craniotubular hyperostoses that are characterized by bone overgrowth of the calvaria, the skull base, the mandible, the ribs, the clavicles and the diaphyses of the long bones.
The purpose of this study was to evaluate the lifetime evolution of the radioclinical features of van Buchem disease, with special emphasis on the age-related variation in hyperostosis of the cranial and tubular bones and on the defective modeling of the tubular bones.
Patients and methods
Thirteen van Buchem patients, from eight sibships, were included in this study. Twelve patients were living in a small Dutch ethnically isolated village, in which the current population has grown from a restricted number of ancestors. All 12 patients share the same ancestors, a couple that married in 1751 (Fig. 1). One additional patient, also originating from The Netherlands, was not connected to this pedigree. This village has been geographically isolated, as it was an island in the Zuyderzee until 1941, the year it merged with the Dutch continent. This makes this village an ethnic isolate thereby explaining the high incidence of this rare disease in that particular area.

Pedigree including 12 van Buchem patients, over ten generations. Blackened symbols represent affected individuals
The current age of the patients ranged between 14 and 69 years, but radiological documents of these patients were available from the age of 6–69 years. Eight patients were male and five were female.
The diagnosis of van Buchem disease was based on phenotypic radioclinical abnormalities. All patients were proven to have the chromosome 17q12–21 deletion that has been assumed to cause the disease by suppressing the expression of the Sost gene [2].
The records of these 13 patients were reviewed retrospectively for the lifetime evolution of both clinical and radiological features. Radiographs of the hand (taken at a focus-screen distance of 1 m) and of the wrist were available for 9 patients, as well as several radiographs or CT scans of the skull (available in 11 patients), of the long bones, and of the axial skeleton were available for a limited number of patients. The CT scans of the petrous bone were available for 4 patients (Table 1). The lifetime evolution of the radioclinical characteristics was evaluated on a qualitative basis for several patients.
The plain films in 9 patients, at different ages, were scored for five parameters (Table 2) and were compared with those of 9 normal control individuals (Table 3):
-
1.
The relative diaphyseal broadening of the metacarpal bones and of the phalanges, in comparison with their distal epiphyses, was evaluated by calculation of both metacarpal and phalangeal dia-epiphyseal ratio. The metacarpal dia-epiphyseal ratio consists of the maximal mid-diaphyseal width of the second metacarpal divided by its maximal distal epiphyseal width, whereas the phalangeal dia-epiphyseal ratio consists of the maximal mid-diaphyseal width of the base phalanx of the second finger divided by the maximal width of its distal epiphysis (Fig. 2).
Fig. 2 
Schematic presentation of the metacarpal and phalangeal dia-epiphyseal ratio. The metacarpal dia-epiphyseal ratio (a/b) consists of the maximal width of the mid-diaphysis of metacarpal 2 divided by the maximal width of the distal epiphysis of metacarpal 2, whereas the phalangeal dia-epiphyseal ratio (c/d) consists of the maximal width of the mid-diaphysis of the base phalanx of the second finger divided by the maximal width of its distal epiphysis
-
2.
The cortical thickness at the radial side of the second metacarpal at the level of the mid-diaphysis.
-
3.
The medullary cavity width at the level of the mid-diaphysis of the second metacarpal.
-
4.
The presence or absence of periosteal apposition (periosteal excrescences).

A Pearson correlation was performed to assess the correlation between age and the above-mentioned measurements.
To evaluate if the metacarpal and phalangeal dia-epiphyseal ratio, cortical thickness, and medullary cavity width were significantly different in patients compared with controls, a bilateral unpaired-samples t-test was performed.
In order to evaluate the intra-observer variability for each parameter, the measurements were repeated ten times and the averages with according variation coefficient (parameter reflecting the intra-observer variability) are displayed in Tables 2 and 3. No significant intra-observer variability for any parameter was observed.
The inter-observer variability was assessed by comparison (paired t-test) of ten consecutive measurements performed by two independent radiologists. No significant inter-observer variability for metacarpal and phalangeal dia-epiphyseal ratio, nor for medullary cavity width, were observed; however, for cortical thickness, a slight inter-observer variability between the two radiologists was observed.
Results
Clinical findings and lifetime evolution
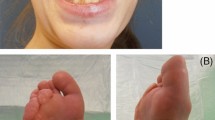
In all patients (n=13), craniofacial abnormalities, including a high forehead, frontal bossing and a widened and thickened chin, were noted (Fig. 3).

Clinical pictures of the same van Buchem patient (patient 8) at the age of A 45 years and B 65 years. The characteristic craniofacial abnormalities consisting of a high forehead, bilateral facial nerve paralysis, and a protruding chin are worsening with age
Facial nerve deficit on at least one side was present in all 13 cases.
Sensorineural hearing loss was present in 6 patients, whereas mixed hearing loss was present in two other cases. Only 1 patient had pure conductive hearing loss. Four patients had hearings thresholds at normal levels.
The lifetime evolution of these characteristics was reviewed retrospectively by evaluation of clinical pictures of the patient’s face (n=6) and by reviewing their medical records.
Both craniofacial abnormalities and cranial nerve deficit seemed to start at puberty and became more prominent throughout life (Fig. 3). The loss of function of the facial nerve followed a pattern of multiple exarcerbations and partial remissions in several patients.
Radiological findings
Eleven patients were examined by skull radiographs or CT scans of the head or of the petrous bone. All patients showed an increased thickness and hyperostosis of the calvaria, the skull base, and of the mandible (Fig. 4). A CT scan of the petrous bone was available for 4 patients. These scans showed narrowing of both the internal acoustic canal and the facial canal (Fig. 5). Sclerosis was symmetrical in all patients, and the pneumatization of the paranasal sinuses was normal.

Lateral radiograph of a postmortem specimen of the skull of a patient with van Buchem disease. Extensive sclerosis of the calvaria, skull base, and the mandible is seen. There is loss of the diploë of the calvaria. (From [1])

Coronal CT scan of the petrous bone of van Buchem patient (patient 6). Due to the extensive sclerosis of the petrous bone, there is narrowing of the internal acoustic canal and facial canal (arrow), leading to encroachment of the facial nerve and vestibulocochlear nerve
These radiological features were obviously more prominent in older patients (see Table 1).
On the plain films of 9 patients, diaphyseal broadening of both metacarpal bones and phalanges was calculated by means of the dia-epiphyseal ratio. Diaphyseal broadening is a parameter for the modeling defect that is observed at the diaphyses of the tubular bones in van Buchem patients. The progressive relative diaphyseal broadening reflects the pure diaphyseal involvement of the long tubular bones, whereas the epiphysis is typically spared (Fig. 6). This metacarpal ratio slowly declined with age in control individuals (Pearson correlation coefficient R2=0.42), reflecting nearly constant remodeling of cortical bone, but progressively increased with age in patients (Pearson correlation coefficient R2=0.42), beginning in the second decade.

Radiograph of the right hand in a van Buchem patient (patient 7) at the age of 52 years. There is thickening of the cortex of the metacarpal bones, as well as of the phalanges. The epiphyses are typically spared. Apart from the cortical hyperostosis, there is a diaphyseal modeling defect. Note also periosteal excrescences at the ulnar side of metacarpal bones (arrows)
A significant difference between control individuals and patients was observed (p<0.001; bilateral unpaired-samples t-test), which reflects the progression of the defective diaphyseal modeling with age in patients (Figs. 7, 8; Tables 2, 3).

Age evolution of the metacarpal dia-epiphyseal ratio in van Buchem patients and its corresponding linear regression curve

Age evolution of the metacarpal dia-epiphyseal ratio in controls and its corresponding linear regression curve
The differences between patients and control individuals in age-related evolution of phalangeal dia-epiphyseal index (Figs. 9, 10), which also represents a parameter for the modeling defect at the phalangeal diaphyses, is even more pronounced (p<0.001; bilateral unpaired-samples t-test).

Age evolution of the phalangeal dia-epiphyseal ratio in van Buchem patients and its corresponding linear regression curve

Age evolution of the phalangeal dia-epiphyseal ratio in controls and its corresponding linear regression curve
In control individuals, no correlation between age and the phalangeal dia-epiphyseal index is observed (Pearson correlation coefficient R2<0.01), whereas a positive correlation with age is seen in patients (Pearson correlation coefficient R2=0.80).
Cortical thickness was 1.5 mm for both patients and controls at the age of 6 years, but increased in patients with age (Fig. 11; Pearson correlation coefficient R2=0.47), whereas no such correlation was observed in the control population (Pearson correlation coefficient R2=0.03; Fig. 12).

Age evolution of the cortical thickness in van Buchem patients and its corresponding linear regression curve

Age evolution of the cortical thickness in controls and its corresponding linear regression curve
The difference between control individuals and patients is significant (p=0.001; bilateral unpaired-samples t-test).
The measurement of cortical thickness seems to be a less reproducible parameter with respect to the other parameters that were measured, since a slight inter-observer variability was observed; however, because the intra-observer variability is still acceptable, there is adequate evidence for a significant age-related increase in cortical thickness in van Buchem patients.
The medullary cavity diameter was found to be growing in patients in function of their age (although the correlation with age is weak; Pearson correlation coefficient R2=0.2), whereas no correlation was found in control individuals (Pearson correlation coefficient R2=0.01; Figs. 13, 14). Again, a significant difference between patients and control individuals was observed (p<0.001; bilateral unpaired-samples t-test).

Age evolution of the width of the medullary cavity in van Buchem patients and its corresponding linear regression curve

Age evolution of the width of the medullary cavity in controls and its corresponding linear regression curve
Periosteal excrescences at the metacarpal bones (Fig. 6) and, to a lesser degree, at the phalanges, were only seen in older patients (Table 2, 3).
Other radiological features (Figs. 15, 16, 17) observed in our population of 13 van Buchem patients are summarized in Table 4.

Standard radiograph of the A chest and the B right clavicle in patient 13 at the age of 42 years. Marked thickening and hyperostosis of the clavicles, especially at the medial side. Note also thickening and hyperostosis of the ribs

Standard radiographs of the right elbow of the same van Buchem patient (patient 3) at the age of A 14 years and B 21 years. There is progressive hyperostosis at the diaphysis, resulting in narrowing of the medullary cavity

Standard radiographs of the left wrist of the same van Buchem patient (patient 3) at the age of A 14 years and B 17 years. There is a progressive modeling defect at the diaphysis with broadening of ulna and radius
Discussion
The prevalence of van Buchem disease is very low, with less than 30 cases reported [1]. The largest study of 15 patients was reported by van Buchem et al. in 1976 [3]. Besides a few isolated cases [5, 6, 7, 8, 9], one other family with four affected patients was described by Dixon et al. in 1982 [4].
Six of our patients have previously been described in detail by van Buchem et al. [3].
In a previous study, the genetic linkage between the disease and markers from chromosome 17q12-q21 was proved [1], which was followed by the identification of the underlying genetic defect [2].
Clinically, the most striking features are craniofacial abnormalities and cranial nerve deficit. All 13 patients displayed a high forehead, with frontal bossing and a widened and thickened chin (Fig. 3). Cranial nerve deficit, especially facial nerve deficit, was present in all cases, whereas sensorineural hearing loss was less common. Other manifestations of cranial nerve deficit of the second, fifth, and tenth cranial nerves, like visual problems, due to optic atrophy, and neuralgic pain have been described in the literature but were not found in this series [11]. In our population, the clinical symptoms started at puberty, although in a previous case report [7], facial paralysis at the age of 2 months has been described as the first clinical sign of van Buchem disease. These clinical features became more prominent with older age (Fig. 3).
The clinical symptoms of van Buchem disease are very similar to those of sclerosteosis. This autosomal-recessive disorder also consists of a craniotubular hyperostosis, resulting in cranial nerve compression and even in more severe intracranial pressure [12]. Severe headaches may occur as a result of raised intracranial pressure, and may even be complicated by sudden death [13].
The onset of the skeletal deformities in sclerosteosis has been compared to van Buchem disease, starting at the age of 5 years, and progressing steadily thereafter [13]. Additional features of sclerosteosis, that are not recognized in van Buchem patients, are excessive height, even gigantism [14], and hand abnormalities, including syndactyly of the digits, radial deviation of the terminal phalanges, and dysplastic or absent nails [15].
Van Buchem disease is classified as a form of craniotubular hyperostosis [10].
The cranial bones display sclerosis of the calvarium, of the skull base, and of the mandible (Fig. 4). The diploë is lost and sclerosis of the skull bones results in stenosis of the cranial neuroforamina, which causes encroachment of the cranial nerves (Fig. 5).
The epiphyses of the long bones are typically spared, whereas the cortical bone of the diaphysis is thickened (Fig. 6). In previous publications, it has been described that sclerosis of the tubular bones, of the ribs, and of the clavicles predominates on the endosteal side of the diaphyseal cortex, thereby resulting in narrowing of the medullary canal [3, 8, 9].
This finding may be true for the calvaria, where we have the impression that the diploë is completely lost; however, our results show that medullary cavity width in the metacarpal tubular bones progresses with age (Fig. 13), despite the increase in cortical thickness (Fig. 11). The cortical sclerosis on the tubular bones of the hands is well reflected in the elevated value of the cortical sclerosis in van Buchem patients, compared with normal controls (Figs. 11, 12; Tables 2, 3).
On the other hand, some periosteal spikes may be present on the altered bones, resulting in a very rough bone surface of the diaphyses (Fig. 6). This is particularly true in older patients (Table 2).
Although, classically, van Buchem disease is referred to as an endosteal hyperostosis, in which overproduction of cortical bone is referred to as a defective endosteal resorption of cortical bone rather than as an increase in periosteal apposition, this is not confirmed by the presence of relative widening with age of the medullary cavity, nor by the occurrence of periosteal spikes at older age.
From the radiological point of view, this finding seems to support the hypothesis that the thickening of cortical bone is due to an increased osteoblast function, resulting in overproduction of bone at the periosteal side of the cortical bone, rather than defective bone resorption at the endosteal side of cortical bone, due to an osteoclastic dysfunction. This may indirectly favor the hypothesis that the physiological function of the van Buchem gene lies in inhibiting the osteoblast, rather than in stimulating osteoclasts. This is in line with the preliminary functional data related to the Sost gene.
In addition to increased bone density, a modeling defect with lack of constriction of the diaphyses of long bones occurs. This is well reflected by the increase of the metacarpal and phalangeal dia-epiphyseal index in patients, compared with control individuals (Figs. 7, 8, 9, 10; Tables 2, 3). This implies that another important physiological function of the gene might be to regulate bone remodeling.
Other bony elements (of the axial skeleton) are less affected. There may be sclerosis of the posterior elements of the spine and some sclerosis of the pelvis.
The radiological abnormalities of the craniofacial bones and the tubular bones usually become obvious at the end of the first decade of life and at the beginning of the second decade, and are progressive throughout life. The progression of the cortical sclerosis and defective modeling at the tubular bones of the hands with age is well illustrated both by the evolution of the metacarpal and phalangeal dia-epiphyseal index and the cortical thickness.
It is remarkable to notice that fractures are not described in our study population, despite knowledge of severe trauma antecedents in several patients.
The observation that both clinical and radiological features of van Buchem disease progress with age suggests that the van Buchem gene is not a developmental gene but rather a gene that is very actively involved in bone metabolism, also at older age; therefore, the Sost gene and its pathway might lead to the development of new tools in fracture treatment and therapy for osteoporosis [1].
The limitations of this study are the semiquantitative analysis of the imaging files. Indeed, not all the patients were examined by CT scan of the petrous bone and we were unable to perform standard radiographs of the whole skeleton at fixed intervals of the patients, because this study was performed retrospectively, and because of the different ages of the patients; however, for reasons of radioprotection, it would be unethical to perform serial skeletal surveys and CT scans of the petrous bones, as this would not alter patient’s prognosis.
Conclusion
Van Buchem disease is a rare autosomal-recessive disease that is radiologically characterized by hyperostosis of the cranial and the tubular bones, whereas craniofacial abnormalities and cranial nerve deficit are the most striking clinical features. The disease progresses with age, and the radioclinical features become more prominent in older patients, suggesting that the van Buchem gene is very actively involved in bone metabolism.
The presence of periosteal spikes and the age-related increase in medullary cavity width may support the hypothesis that the cortical sclerosis may be related to relative overproduction of bone at the periosteal side of bone, rather than to a defective endosteal bone resorption.
References
Van Hul W, Balemans W, Van Hul E et al. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am J Hum Genet 1998; 62:391–399.
Balemans W, Patel N, Ebeling M et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 2002; 39:91–93.
Van Buchem FSP, Prick JJG, Jaspar HH. Hyperostosis corticalis generalisata familiaris (van Buchem’s disease). Excerpta Medica, Amsterdam, 1976.
Dixon JM, Cull RE, Gamble P. Two cases of van Buchem’s disease. J Neurol Neurosurg Psychiatry 1982; 45:913–918.
Lopez AG, Pinero ML, Varo FM. Hiperostosis cortical generalizada (Van Buchem). Rev Clin Esp 1985; 177:293–294.
Miguez AM, Esteban BM, Ramallo VG et al. Silla turca vacia parcial an la enfermedad de van Buchem. Med Clin (Barc) 1986; 87:719–721.
Fryns JP, Vandenberghe H. Facial paralysis at the age of 2 months as a first clinical sign of van Buchem disease (endosteal hyperostosis). Eur J Pediatr 1988; 147:99–100.
Cook JV, Phelps PD, Chandy J. Van Buchem’s disease with classical radiological features and appearances on cranial computed tomography. Br J Radiol 1989; 62:74–77.
Bettini R, Sessa V, Mingardi R, Molinari A, Anzani P, Vezzetti V. Endosteal hyperostosis with recessive transmission (Van Buchem’s disease): a case report. Recenti Prog Med 1991; 82:24–28.
Beighton P. Inherited disorders of the skeleton. Churchill Livingstone, Avon, 1988.
Van Buchem FS. Hyperostosis corticalis generalisata: eight new cases. Acta Med Scand 1971; 189:257–267.
Beighton P, Davidson J, Durr L, Hamersma H. Sclerosteosis: an autosomal recessive disorder. Clin Genet 1977; 11:1–7.
Nager GT, Stein SA, Dorst JP et al. Sclerosteosis involving the temporal bone: clinical and radiologic aspects. Am J Otolaryngol 1983; 4:1–17.
Beighton P, Barnard A, Hamersma H, van der Wouden A. The syndromic status of sclerosteosis and van Buchem disease. Clin Genet 1984; 25:175–181.
Balemans W, Van Den Ende J, Freire Paes-Alves A et al. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am J Hum Genet 1999; 64:1661–1669.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vanhoenacker, F.M., Balemans, W., Tan, G.J. et al. Van Buchem disease: lifetime evolution of radioclinical features. Skeletal Radiol 32, 708–718 (2003). https://doi.org/10.1007/s00256-003-0675-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00256-003-0675-4