
Abstract
Autoimmune type 1 diabetes is characterized by selective destruction of insulin-secreting beta cells in the pancreas of genetically susceptible individuals. The mechanisms underlying the development of type 1 diabetes are not fully understood. However, a widely accepted point is that type 1 diabetes is caused by a combination of genetic and environmental factors. Although most type 1 diabetes patients do not have a family history, genetic susceptibility does play a vital role in beta cell autoimmunity and destruction. Human leukocyte antigen (HLA) regions are the strongest genetic determinants, which can contribute 40–50 % of the genetic risk to type 1 diabetes. Other genes, including INS also contribute to disease risk. The mechanisms of the susceptible genes in type 1 diabetes may relate to their respective roles in antigen presentation, beta cell autoimmunity, immune tolerance, and autoreactive T cell response. Environmental susceptibility factors also contribute to the risk of developing type 1 diabetes. From an epigenetic standpoint, the pathologic mechanisms involved in the development of type 1 diabetes may include DNA methylation, histone modification, microRNA, and molecular mimicry. These mechanisms may act through regulating of gene expression, thereby affecting the immune system response toward islet beta cells. One of the characteristics of type 1 diabetes is the recognition of islet autoantigens by autoreactive CD4+ and CD8+ T cells and autoantibodies. Autoantibodies against islet autoantigens are involved in autoantigen processing and presentation by HLA molecules. This review will mainly focus on the molecular mechanism by which genetic, epigenetic, and environmental factors contribute to the risk of type 1 diabetes.
Similar content being viewed by others
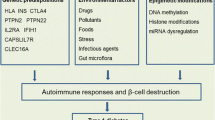
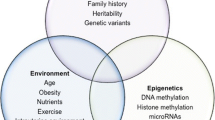
Avoid common mistakes on your manuscript.
Introduction
Diabetes is now known to be an organ-specific autoimmune disease, but the phenotype differs in children versus adults. The two major autoimmune diabetic conditions include type 1 diabetes, which generally but not exclusively affects children, and latent autoimmune diabetes of adults (LADA) [1].
Almost 40 years ago, islet cell antibodies against type 1 diabetes (T1D)-specific antigens were found in the serum of T1D patients, suggesting that the beta cell loss of T1D was autoimmune in nature [2]. T1D is generally thought to be characterized by autoimmune destruction of insulin-producing pancreatic beta cells mediated by an autoantibody to islet cell antigens [3, 4]. The resultant loss of insulin causes an overproduction of glucose and a decreased cellular uptake of glucose, resulting in hyperglycemia. Loss of insulin also leads to an increase in fat breakdown and fatty acid oxidation, which, in turn, causes overproduction of ketones [5]. Overproduction of ketones leads to diabetic ketoacidosis, and lifetime exogenous insulin treatment is required for the treatment of diabetes patients.
T1D can be present at any age. The incidence of T1D in children has been increasing over the past several decades [6]. It is considered one of the most common chronic childhood diseases [7]. Abundant research on T1D has historically originated out of Europe and North America, including countries such as Finland, Norway, Sweden, UK, Canada, and the USA [8, 9]. The incidence and prevalence of T1D vary substantially worldwide [10, 11]. For example, the incidence of T1D is 60 cases in Finland and 40 cases in Sardinia per 100,000 people each year, while the incidence of T1D in China and Venezuela has been reported to be as low as 0.1/100,000 per year [1, 12]. The mechanisms underlying the increased incidence of T1D in selected countries are unknown but have been attributed to environmental influences [4].
LADA was first reported over 27 years ago [13]. LADA patients are defined as glutamic acid decarboxylase antibody (GADA)-positive, initially without insulin treatment for at least 6 months, diagnosed over the age of 30 years according to the criteria of Immunology of Diabetes Society (IDS) [14]. LADA is a slowly progressive form of autoimmune diabetes in adults. The progression of autoimmune beta cell loss is associated with the development of islet cell autoantibodies in a manner similar to T1D, but the clinical features are more consistent with type 2 diabetes (T2D) [15]. LADA patients do not require insulin treatment during the first 6 months after diagnosis [16, 17]. Many other names have been used to describe this condition, including diabetes mellitus type 1.5 [18], non-insulin requiring autoimmune diabetes (NIRAD) [19], slowly progressive T1D (SPT1D) [20], and autoimmune diabetes in adults (ADA) [16].
The relationship between LADA, T1D, and T2D remains controversial [21, 22]. LADA was once considered a slowly progressing subtype of T1D. However, the clinical features more resemble T2D. It is suggested that LADA is different from both classic T1D and T2D [23]. Studies from our group [14] and others [24] have demonstrated that human leukocyte antigen (HLA) protective haplotypes are less frequent in LADA. However, other studies have shown that LADA share similar susceptibility genes to classic T1D [25–28] and T2D [29]. Some researchers believe that diabetes occurs on a continuum. Our results suggested that the susceptible haplotypes of the HLA-DQ gene present a continuous spectrum from T1D, through LADA, to T2D [30]. Autoimmune diabetes is not triggered by a single factor but results from a complex interaction between genetic and environmental factors. The molecular mechanisms involved in susceptibility and the development of autoimmune diabetes, T1D in particular, are complex and redundant immune pathways.
Genetics
TID is caused by both genetic and environmental factors. Genetic susceptibility plays a vital role in the pathogenesis of T1D. It was reported that the risk of diabetes in sibling is 6 %, which is 15 times higher than that in the general Caucasian population [31]. The concordance rate for monozygotic twins (30–40 %) is much higher than that for dizygotic twins (6–8 %) [32, 33]. These observations suggest that the genetics is a significant risk factor. Over 50 susceptibility regions have been identified to associate with T1D (Table 1). The major susceptibility genes to T1D are located in HLA region. The first reports regarding the association between HLA and T1D were published 40 years ago [70], which spawned extensive research from all regions of the world to determine which alleles of HLA are associated with T1D. In addition to HLA, other genes, such as INS, cytotoxic T-lymphocyte-associated protein 4 (CTLA4), and PTPN22, also contribute to the risk of T1D.
HLA Association
HLA is located on human chromosome 6p21.3, spans about 4,000 kb, and contains over 200 genes (Fig. 1). Certain HLA genes are reported to have immune response functions to environmental pathogens and in autoimmune diseases [72, 73]. The genes that encode class I (A, B, and C) and class II (DP, DQ, and DR) molecules are important in self and non-self-immune recognition. Nine thousand five hundred and forty-six polymorphisms of the HLA region have been reported so far (Table 2). The extreme polymorphism of the HLA makes it an invaluable tool for T1D association studies. HLA class I molecules are widely expressed as single chain proteins that can present intracellular antigen to CD8+ T cells.

Representation map of the HLA region on human chromosome 6p21. HLA genes confer ~50 % risk to T1D. HLA genes are arranged in three classes, class I, class III, and class II. The class I (A, B, and C) and class II (DR, DQ, and DP) genes are reported to be associated with T1D. Adapted from Mehers 2008 [71] and Kelly 2003 [5]
HLA class II molecules are heterodimers expressed mainly on professional antigen-presenting cells. They are composed of α and β chains and are responsible for presenting extracellular antigen to CD4+ T cells [2]. The strongest association with the development of T1D is in the HLA class II loci, which can contribute about 40–50 % risk to the T1D susceptibility [2, 75]. The precise mechanisms by which the HLA class II genes confer susceptibility to the loss of islet beta cells are largely unknown, but the binding properties of key peptides derived from proinsulin, insulinoma-associated antigen 2 (IA-2), glutamic acid decarboxylase (GAD), and zinc transporter 8 (ZnT8) to antigen-presenting cells may play a role [68].
Specific combinations of alleles, genotypes, and haplotypes of the class II genes may contribute to the risk of T1D. DRB1 and DQB1 are considered to be associated with T1D in people from almost all regions of the world [76]. It has been shown that both susceptible and protective alleles may be found at the DRB1, DQA1, and DQB1 loci, including DQB1*0602, DQB1*0302, DRB1*0301, DRB1*0401, and DRB1*0405 alleles [77, 78]. Specifically, DRB1*0401, DRB1*0402, and DRB1*0405 have been suggested to confer susceptibility to T1D, while DRB1*0403 and DRB1*0406 confer protection from T1D [79, 80]. However, the susceptible and protective alleles in Asians are different from the Caucasian population. Susceptibility and protective class II alleles in Japanese populations with T1D include DQB1*0301, DQB1*0602, DRB1*1501, and DRB1*1502 [81, 82]. In the Korean population, these alleles include DQB1*0301, DQB1*0503, DQB1*0601, DQB1*0602, DRB1*0803, DRB1*1202, and DRB1*1405 [83, 84]. In the Chinese populations, susceptibility and protective alleles include DQB1*0301, DQB1*0402, DQB1*0501, DQB1*0503, DQB1*0601, DQB1*0602, DRB1*0403, and DRB1*0406 [85, 86]. Independent effects of HLA-A and HLA-B may also increase the risk of T1D independent of HLA class II genes [47].
With regard to genotypes and haplotypes, specific combinations of alleles at the DRB1, DQA1, and DQB1 loci contribute to the risk of developing T1D. DQA1*0501-DQB1*0201 and DQA1*0301-DQB1*0302 encode the HLA-DQ2 and HLA-DQ8 molecules, respectively. HLA-DRB1*03 and HLA-DRB1*04, which encode DR3 and DR4 molecules, are in linkage disequilibrium with DQ2 and DQ8, respectively. These alleles form the DR3-DQ2 and DR4-DQ8 haplotypes, respectively [5]. The highest risk of DR-DQ haplotypes for T1D are DRB1*0301-DQA1*0501-DQB1*0201 and DRB1*0401-DQA1*0301-DQB1*0302 [77, 87]. Studies from different countries have shown that association of class II allele at the DRB1, DQA1, and DQB1 loci may vary among countries and ethnic origins [79]. For example, the DRB1*0301-DQB1*0201 and DRB1*0401-DQB1*0302 haplotypes are consistently associated with T1D in Caucasian individuals, while the DRB1*0405-DQB1*0401 and DRB1*0901-DQB1*0303 haplotypes are associated with Japanese individuals and East Asian populations [76, 81, 88]. Other haplotypes that are associated with T1D, including DRB1*0801-DQA1*040-DQB1*0402, DRB1*0405-DQB1*0401, DRB1*0901-DQA1*0301-DQB1*0303, DRB1*0802-DQB1*0302, and DRB1*0901-DQB1*0303, also vary among different countries [89–91].
In contrast with classic T1D, the association between HLA and LADA is not as well understood. Studies have demonstrated that HLA genetics are related to LADA in Caucasian populations [16, 21, 23]. A study conducted in a large well-characterized LADA cohort found that patterns of HLA-DRB1 and HLA-DQB1 loci in LADA are similar to that of T1D [26]. The authors found that DRB1*0301-DQB1*0201 and DRB1*0401-DQB1*0302 haplotypes are the main susceptibility haplotypes in LADA, while DRB1*1501-DQB1*0602 is a protective haplotype in LADA. The highest risk HLA genotypes for T1D, DR3/DR4 and DQ2/DQ8, were more prevalent in LADA compared with normal controls; however, the extent of the association was not the same when compared with T1D [16].
When comparing LADA with juvenile-onset T1D, the highest risk genotype DQ2/DQ8 was less frequent in LADA, which is more consistent with late onset diabetes [23, 92]. However, when comparing LADA with adult-onset T1D, there were no consistent differences in HLA class II, which suggest that they may share similar HLA genetic backgrounds [25, 93, 94]. Other reports suggest differences in HLA association between LADA and classic T1D, and some studies have also shown that LADA patients have an increased frequency of DRB1*0602, suggesting that DRB1*0602 may play a protective role in delaying the onset of autoimmune diabetes [29, 95]. HLA-DRB1*03 and HLA-DRB1*04 alleles in T1D patients are higher than in LADA [96]. In contrast to the strong association of HLA with autoimmune diabetes in Caucasians, the most susceptible HLA-DQ genes in Chinese LADA were moderate-risk haplotypes, including DQA1*03-DQB1*0303 and DQA1*05-DQB1*0201 in our studies [14, 30]. This suggests that the immunogenotype association of Chinese LADA is more moderate than that in Caucasians.
In conclusion, HLA associations with T1D and LADA are extremely complex, with many alleles and haplotypes affecting diabetes risk [5]. The mechanisms of HLA-DR and HLA-DQ molecules’ association with T1D and LADA may be related to the primary role of HLA in antigen presentation to CD4+ T cells. If HLA cannot present an antigen, the antigen cannot, through classical pathways, become an autoantigen in our body. Different antigens are recognized by the immune system and presented by different HLA molecules. The structural differences between the susceptible and protective HLA molecules may vary with autoantigens and the T cell receptors (TCRs) of autoreactive T cells and thus contribute to disease risk.
Non-HLA Association
Although association studies have shown that the HLA region is the most important genetic factor conferring risk or protectivity toward the development of T1D and related disorders, other T1D susceptibility genes, such as INS, PTPN22, CTLA4, IFIH1, CLEC161, and PTPN2, have also been described (see Table 1).
INS
The first strong non-HLA association with T1D was found at polymorphisms within the INS gene on chromosome 11p15.5: a variable number of tandem repeats (VNTRs) located 596 bp upstream of the translational start site of the INS gene [54, 97]. The VNTRs in the INS gene are found in three forms, class I alleles (20–63 repeats), class II alleles (64–139 repeats), and class III alleles (140–210 repeats) [98]. The class I alleles are associated with susceptibility to T1D, while the class III alleles are associated with protection against T1D. The mechanism by which the VNTRs in the INS gene affect the risk of T1D is unknown. However, it has been shown that the VNTRs can regulate insulin messenger RNA (mRNA) transcription in the pancreas and thymus. Class I alleles are associated with high mRNA expression levels in the pancreas and low levels in the thymus, while class III alleles are associated with lower levels of insulin mRNA in the pancreas but higher levels in the thymus [99, 100].
Insulin and its precursor, preproinsulin, are potential target autoantigens for beta cell destruction (Fig. 2). Low levels of proinsulin in the thymus may affect the positive selection of T cells in the thymus, which will cause migration of CD4+, proinsulin-specific T lymphocytes to the periphery and increase the risk for developing T1D. On the other hand, high levels of proinsulin in the thymus may promote negative selection of insulin-specific autoreactive T lymphocytes, leading to immune tolerance [101] and a decrease risk for the development of T1D.

Development of the autoimmune response. During thymocyte maturation, positive and negative selection takes place in the thymus. This process requires the interaction between the MHC molecules on the APCs, autoantigens (proinsulin as an example here), and the TCR on the surface of thymocyte. Large percentages (98 %) of thymocytes are removed by apoptosis (negative selection). The rest (2 %) thymocytes are positively selected and then migrated into peripheral as mature T cells. The mature T cells will then develop into CD4+ and CD8+ T cells and regulatory T cells. The balance between regulatory and effector T cells plays an important role in the development of both T1D and LADA. Note that susceptibility genes, including HLA, CTLA4, and PTPN22 are involved in this process. Adapted from Ounissi-Benkalha 2008 [2]
CTLA4
The association between T1D and CTLA4, located on chromosome 2q33, was confirmed by several studies [42, 102]. CTLA4 protein is a costimulatory receptor on CD4+ T cell surface, which can bind B7 ligands that activate CD28, an important molecule in T cell costimulation (Fig. 2). CTLA4 plays a role in producing a negative signal to inhibit T cell activation and has a crucial role in the function of CD4+ T regulatory cells [103]. The intracellular part of CTLA4 interacts with the intracellular part of CD3 receptor to initiate phosphorylation of several downstream molecules, leading to activation of T cells after their binding to HLA molecules on antigen presenting cells (APCs) [2].
Changes in expression of CTLA4 can increase T cell self-reactivity and may play a role in autoimmune diabetes [102, 104]. Studies have shown an association between CTLA4 A49G polymorphisms and T1D [105]. The A49G polymorphism in exon 1 of CTLA4 causes substitution of alanine with threonine in the signal sequence, leading to incorrect expression of the mutant protein and the subsequent reduction of CTLA4 cell surface expression. CTLA4 has been implicated in multiple autoimmune diseases [102, 106], including, but not limited to, rheumatoid arthritis (RA), systemic lupus erythematosus, and Addison’s disease [107].
PTPN22
PTPN22, located on chromosome 1p13, encodes lymphoid tyrosine phosphatase (LYP), and mutations of this gene have been associated with T1D [108]. LYP is mainly expressed in T cells and plays a role in inhibiting TCR signaling by dephosphorylation of three kinases in the TCR signaling pathway. LYP also interacts with C-terminal Src tyrosine kinase (Csk) to downregulate T cell activation (Fig. 2) [109]. The non-synonymous single nucleotide polymorphism (SNP) C1858T results in a substitution mutation of arginine for tryptophan (R620W). Functional studies have found that the gain-of-function R620W mutation can increase phosphatase activity [110]. This mutation leads to increased inhibition of TCR signaling, which will reduce CD4+ T cell activation and potentially leads to increased autoimmunity [111]. In addition to T1D, PTPN22 gene has also been reported to be associated with other autoimmune diseases [112], including RA, Grave’s disease, and SLE [113, 114].
Other Susceptible Genes
Interleukin 2 receptor alpha (IL2RA), which encodes the alpha chain of the IL-2 receptor complex locus (CD25) and locates on chromosome 10p15, was identified as a major non-MHC risk gene associated with T1D. CD25 is expressed on regulatory naive T cells, memory T cells, and activated monocytes [115]. CD25 is responsible for binding of IL-2 and plays a role in the proliferation of regulatory T cells. CD25 regulates the activity of effector T cells through regulatory cells, and mutations in CD25 may potentially lead to the development of autoimmunity. Expression of CD25 on the surface of the regulatory T cells is important in regulating T cell proliferation in response to an immunogenic stimulus [2, 71, 116].
Small ubiquitin-like modifier 4 (SUMO4) has also been reported to be a risk factor for T1D [50]. However, inconsistent results have also been reported [117]. The substitution of methionine to valine (M55V) in SUMO4 has been proposed as a causative variant associated with T1D. This substitution causes a significant reduction of sumoylation capacity and higher NF-κB activity as well as elevated secretion of IL12B [50]. Subsequent studies have found that SUMO4 sumoylates IκBα and negatively regulates NF-κB transcriptional activity [118]. The transcription factor NF-κB has a central regulatory role in the immune response [119] and is involved in the development of autoimmune diabetes.
Several studies have found polymorphisms in signal transducers and activators of transcription (STAT) to be associated with T1D [120]. STAT4 is expressed in activated peripheral blood monocytes cells (PBMCs), dendritic cells (DCs), and macrophages at sites of inflammation [121]. STAT4 directly interacts with the IL-12 receptor and plays an important role in the IL-12 signaling pathway [122]. IL-12 is an immunoregulatory cytokine which takes part in the generation of Th1 cells and cytotoxic lymphocytes and leads to the production of proinflammatory cytokines [123].
Although the INS gene is the only non-HLA gene consistently demonstrated to be associated with LADA [27], other associations have been reported [28, 124]. The influence of non-HLA genes on LADA may, in fact, be more significant than in T1D. A study from Germany showed that PTPN22, STAT4, CTLA4, IL2RA, INS, ERBB3, SH2B3, and CLEC16A are all associated with LADA [125]. Other studies suggest that HLA-related genes play more of a role in the onset of T1D in children, whereas non-HLA genes play more of a role in the onset of LADA and T1D in young adults [29, 126]. Different HLA associations could not explain the differences between LADA and classic T1D, and further studies are needed to clarify the role of genetics in LADA.
Environment
Environmental factors may not only impact disease but can also interact with genetic factors to affect the development and progression of human disease. The importance of environmental factors in the pathogenesis of T1D is suggested by (1) the low concordance rate (30–40 %) in monozygotic twins [127, 128], (2) only 10 % of genetically individuals with susceptible HLA genes eventually progress to diabetes [129], (3) a 15-fold difference in the disease incidence among Caucasians living in Europe, and (4) population studies which show that the incidence of diabetes increased after migration to a high-incidence region [130]. Studies have shown that the proportion of patients with high-risk HLA has decreased, while the proportion of patients with low-risk and protective HLA has increased [131, 132]. These data suggest an increased environmental risk. In Europe, the lowest annual rate occurs in Macedonia, amounting to 3.2/100,000 under 15 years, while the highest rate is 60/100,000 in Finland. The 15-fold difference cannot be explained by genetic factors alone [133].
Environmental factors that have been attributed to autoimmune diseases include a wide range of chemicals, pathogens, drugs, toxins, diet, stress, viral infection, organ phosphates, heavy metals, and solvents [134–138]. Other non-environmental factors that can contribute to the pathogenesis of T1D and other autoimmune diseases, and which may be affected by the environment, include weight, puberty, increased linear growth, body mass index, and other parameters of body habitus. Studies have examined the role of chemicals in the environment in the development of T1D, such as N-nitroso compounds, air pollutants, and persistent organic pollutants [139, 140]. Environmental chemicals may affect the development and function of the immune system, leading to autoimmunity and contributing to the development of T1D [141].
Diet is associated with T1D, with particular risk factors being cow’s milk and wheat gluten, while the protective effects of breastfeeding and various nutrients have also been demonstrated [142, 143]. The gut immune system plays an important role in the development of autoimmune diabetes, and the intestinal walls of patients with T1D have been found to be more leaky than those of non-diabetic patients [144].
Psychological stress is associated with T1D-related autoimmunity at early ages. Studies suggest that psychological stress may accelerate the appearance of T1D, contribute to the induction or progression of T1D-associated autoimmunity, and induce beta cell stress or adversely impact the immune system, as a mechanism for the development of the autoimmune state [145].
Height, weight, and BMI have been associated with an increased incidence of T1D [146]. A number of viral infections have been associated with T1D and/or autoantibodies in humans, including enterovirus, rubella, mumps, rotavirus, and cytomegalovirus (CMV). A meta-analysis found a clinically significant association between enterovirus infection and T1D autoimmunity [147]. Other studies also found increased enterovirus RNA in the blood of children with T1D compared to control children, suggesting that enterovirus plays a role in the pathogenesis of T1D, by initiation of the process leading to beta cell damage [148, 149].
How exactly these factors influence the development of T1D is unclear. However, evidence has shown that the mechanism by which these environmental factors induce T1D may include epigenetic modification (DNA methylation, histone modification, and microRNA), reaction with the self-component to generate novel antigen molecules, and molecular mimicry [150], which is based on the cross-reactivity between environmental antigens and autoantigens. This has been supported by the detection of serum autoantibodies that also recognize pathogenic epitopes. For example, the anti-GAD autoantibody obtained from T1D patients also reacts with CMV.
Epigenetics
Environmental factors can induce epigenetic changes, which regulate gene expression and affect immune cell function. For this reason, epigenetics provides a source of molecular mechanisms that can explain the environmental effects on the development of autoimmune diabetes [150, 151]. Epigenetics focuses on the mechanisms that influence gene expression and cell function without a change in the DNA sequence. There are three main epigenetic modifications, including DNA methylation, histone modification, and microRNA. All of them are associated with transcriptional regulation and determination of the cellular transcriptome, thereby contributing to cell function [152].
DNA Methylation
DNA methylation is a biochemical process involving the addition of a methyl group to the fifth carbon of cytosine DNA nucleotides in CpG dinucleotide islands by using S-adenosylmethionine (SAM). DNA methylation is carried out by specific enzymes called DNA methyltransferases (DNMTs) including DNMT1, DNMT3a, DNMT3b, DNMT3L, and DNMT2. 5-Methylcytosine can be converted to 5-hydroxymethylcytosine and cytosine by the ten-eleven translocation (TET) family of proteins [153]. Generally, DNMTs have two classes, including maintenance DNA methyltransferases and de novo methyltransferases.
DNMT1 is responsible for the maintenance of methylation patterns during DNA replication, while DNMT3a and DNMT3b are responsible for de novo methylation [154]. Methyl-CpG-binding domain (MBD) proteins also regulate methylation together with DNMTs [128]. There are two types of demethylation—passive demethylation and active demethylation. The maintenance methyltransferase DNMT1 has a preference for hemi-methylated DNA. If DNMT1 is inhibited or absent during DNA replication, the newly synthesized DNA will not be methylated, and this will cause passive demethylation. Active demethylation can occur through the enzymatic replacement of 5-methylcytosine (5meC) with cytosine with the help of TET and thymine-DNA-glycosylase (TDG) [155]. The balance between methylation and demethylation is important to the growth and development, as aberrant methylation may cause serious diseases [156, 157], such as cancer, autoimmune diseases, and neurodegenerative diseases.
A genome-wide DNA methylation analysis of diabetic nephropathy in T1D was performed in 2010. Nineteen CpG sites were found to correlate with the development of diabetic nephropathy in T1D [158]. Another genome-wide DNA methylation profile was performed by using CD14+ monocytes from 15 T1D-discordant monozygotic twin pairs. It was discovered that 132 different CpG sites significantly correlated with the diabetic state, including 58 hypermethylated T1D–methylation variable positions (MVPs), such as TNF and TRAF6, and 74 hypomethylated T1D–MVPs, such as GAD65 and HLA-DQB1 [159]. DNA methylation has also been found to decrease TLR9 stimulation of FOXP3 expression, through attenuation of IRF-7 binding activity in T1D [160].
Specific DNA methylation changes in T1D have also been found [161]. It has been proposed that the methylation level in the human INS gene acts as a biomarker in predicting beta cell death [162, 163]. Our group found that the genomic DNA methylation in CD4+ T cells from LADA patients was significantly increased compared to controls. DNMT3b mRNA levels were higher in CD4+ T cells from LADA patients, whereas FOXP3 expression was decreased, and the FOXP3 promoter region was hypermethylated in CD4+ T cells [162]. All these studies suggest the importance of aberrant DNA methylation in the development of autoimmune diabetes [164]. This mechanism may explain the effect of DNA methylation in the expression of autoimmune diabetes-related genes, such as INS and FOXP3.
Histone Deacetylation
Histone proteins are subjected to a wide variety of posttranslational modification, including lysine acetylation, lysine and arginine methylation, serine and threonine phosphorylation, lysine ubiquitination, and sumoylation. These modifications work together to alter the function of the nucleosome. Among histone modifications, acetylation/deacetylation is the most common gene expression regulatory mechanism. This process is catalyzed by histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes respectively [165].
Another modification is lysine methylation within the histone tail. The number of methyl groups in lysine residues has an impact on gene expression. For example, three methyl groups on the lysine 4 residue of histone H3 has been associated with transcriptional activation. However, the triple methylation of residues 9 or 27 has been associated with transcriptional inhibition [166]. Other modifications, such as phosphorylation, ubiquitination, and sumoylation can also modulate gene expression. A chromatin immunoprecipitation (ChIP) linked to microarray (ChIP-on-chip) approach [167] was used to analyze histone methylation patterns in blood lymphocytes and monocytes from patients with T1D. The expression of a series of genes was found to be significantly increased in H3K9me2 in lymphocytes but not in monocytes from patients with T1D. Many of these genes were associated with autoimmune and inflammation-related pathways, such as TGF-beta, NF-κB, p38 MAPK, toll-like receptor, and interleukin 6.
Expression of the T1D susceptibility gene CTLA4 was also increased in their study [168]. Another study analyzed the T1D mellitus (T1DM)-specific gene expression and the relationship with T1DM autoimmunity. It was found that CD4+ T cells of patients with T1DM were downregulated, specifically affecting key immune functions and the cell cycle. At the same time, gene expression of HDAC was decreased [169]. The H3K9Ac status of HLA-DRB1 and HLA-DQB1 has also been shown to be associated with T1D [170]. This study suggests that the promoters and/or enhancers of key susceptible genes may be important determinants in their functional association to T1D susceptibility, suggesting that the interaction between genetics and epigenetics may play a role in the development of T1D.
MicroRNAs
MicroRNAs are short non-coding RNA sequences (22 nucleotides) found in plants and animals. MicroRNAs are transcriptional and posttranscriptional regulators of gene expression [171]. MicroRNA binds to the complementary sequences in the 3′ UTR of multiple target mRNAs, causing the degradation of the target gene. They are crucial regulators of immune function, including development, differentiation, proliferation, and apoptosis [172–174]. Dysregulated microRNA expression patterns have been found in patients with T1D [175, 176]. It has been shown that dysregulated microRNA will cause aberrant immune function and may be involved in the development of T1D.
Increased expression levels of miR-326 in the peripheral blood lymphocytes from patients with T1D have been reported. The predicted targets are involved in immune regulation, indicating that miR-326 may be associated with ongoing islet autoimmunity [177]. Increased expression of miRNA-510 and decreased expressions of both miRNA-342 and miRNA-191 were identified in regulatory T cells of T1D patients [178]. Another study found that miR-21a and miR-93 were downregulated in PBMCs from patients with T1D [179].
More recently, a study compared the expression level of serum microRNAs from new onset T1D children and healthy controls. Twelve increased human microRNAs in T1D patients (miR-152, miR-30a-5p, miR-181a, miR-24, miR-148a, miR-210, miR-27a, miR-29a, miR-26a, miR-27b, miR-25, and miR-200a) were identified. Several of these microRNAs were linked to apoptosis and beta cell function [180]. MicroRNA-21 was reported to prevent T1D by blocking pancreatic beta cell death. It has been proposed that the NF-κB-microRNA-21-PDCD4 axis plays a vital role in T1D and represents a unique therapeutic target for disease treatment (Table 3) [182]. The contribution of microRNA to immune system function and the development of autoimmunity are becoming more and more evident [183]. It is believed that the study of microRNA in T1D will elucidate new mechanisms involved in the development of T1D.
Environmental factors can affect epigenetic mechanisms of gene expression and the development of T1D. These mechanisms can regulate gene expression and thus affect the development and function of immune and islet beta cells. The increased research on epigenetics in recent years will provide a new perspective and a possibility that epigenetic modification can act as a potential diabetes therapeutic target [184].
Mechanisms of Autoimmunity in T1D
Molecular Mimicry
Viral infections are thought to be a major environmental factor influencing the development of T1D. Common viruses identified to be associated with T1D include enteroviruses such as coxsackievirus B [185], rotavirus [186], mumps virus [187], and CMV [188]. It is unknown whether viruses act as an accelerator during the ongoing immune process initiated by other factors [189] or whether they are able to initiate the entire autoimmune process. It is difficult to establish which immunological processes link viral infections to disease initiation and progression. A commonly discussed mechanism by which viruses may take part in the development of T1D is molecular mimicry. Molecular mimicry is based on structural similarity (amino acid sequence or conformational structure) between pathogen and autoantigen [190]. The virus, which may share a similar epitope with certain structures on the beta cells, can mimic autoantigens and thus activate T cells, inducing a cross-reactive autoimmune response [189].
Molecular mimicry has been thought to be a primary mechanism contributing to many autoimmune diseases, including T1D, systemic lupus erythematosus, multiple sclerosis, and Sjogren’s syndrome. The potential cross-reactivity between the non-structural P2-C protein of coxsackievirus and the autoantigen GAD65 was found in T1D [191]. Another study showed that the homologous peptides for cross-reactivity are immunogenic. However, the homology between GAD65 and P2-C was not associated with significant functional consequences [192].
Similarity and cross-reactivity between the VP1-protein of enteroviruses and the beta cell autoantigen tyrosine phosphatase IA-2 have also been reported, and it was postulated that enterovirus infection may also alter immune responses [193]. The similarity between GAD65 and human CMV (hCMV) major DNA-binding protein has also been demonstrated. The hCMV-derived epitope can be naturally processed by dendritic cells and recognized by GAD65 reactive T cells [194].
All these data suggest that molecular mimicry may play a role in the development of T1D. However, functional studies, which involve experimentally inducing T cells with a viral peptide to mimic the islet autoantigen, are still needed to demonstrate the role of molecular mimicry role in the development of T1D. Whether molecular mimicry can only enhance autoimmunity, or can initiate autoimmunity, or both, is unknown. We should also keep in mind that molecular mimicry between virus and autoantigens may not be the sole mechanism for the development of autoimmunity; other mechanisms may also play a major role in initiating the immune response and in the pathogenesis of T1D.
Autoantigens and Autoantibodies
One of the characteristics of T1D is the recognition of beta cell proteins as autoantigens by autoreactive CD4+ and CD8+ T cells and autoantibodies. Several autoantigens have been attributed to T1D. These autoantigens include propreinsulin, GAD65, IA-2 [195], ZnT8, non-specific islet cell autoantigens (ICAs), imogen 38, pancreatic duodenal homeobox factor 1 (PDX1), chromogranin A (CHGA), islet specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), heat shock protein 60 (hsp60) [196], and islet cell antigen 69 (ICA69). It has been shown that many proteins mentioned above are potential targets of the immune system.
Several methods have been used to characterize autoantigens in T1D [197]. The first method involves the detection of islet cell autoantibodies, such as GAD65, ICA69, IA-2, and carboxypeptidase H (CPH). The second method is based on the detection of islet autoreactive T cells. Imogen 38 and IGRP were identified as beta cell autoantigens by detection of pancreatic islet autoreactive CD4+ T cells. The third method employed a technique used in cell biology, which is based on selective expression of β cell proteins defined by complementary DNA subtraction libraries or microarrays. Previously reported autoantigens, imogen 38, IGRP, IA-2, IA-2β, and ZnT8, were confirmed or identified by this method. The fourth method is through a process known as inverse translation or adoptive transfer. GAD65 and insulin were confirmed by adoptive transfer of autoreactive T cells. Proteomic strategy was used as a fifth method for identifying islet autoantigens [198].
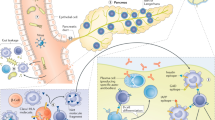
It has been proposed that β cell loss is caused by lymphocytic infiltration of the islet by dendritic cells, macrophages, and T lymphocytes [2]. Autoreactive T lymphocyte cells specific for beta cell autoantigens, such as insulin, GAD, IA-2, and ZnT8 have been identified [199, 200]. Studies have shown that CD4+ helper and CD8+ cytotoxic T lymphocytes play an important role in the pathogenesis of T1D [201, 202] (Fig. 2). The main factors initiating autoreactive responses are not clear; however, it is well accepted that specific autoantigens are processed by APCs. APCs include DCs, macrophages, and B cells in the pancreatic islets. The autoantigens are then presented to naive T cells by diabetes-associated HLA molecules to contribute to priming and expansion of pathogenic T cells and generation of autoreactive CD4+ T cells. These activated CD4+ T cells will then produce cytokines and subsequently activate beta-cell-specific cytotoxic CD8+ T cells. The activated T cells will be recruited to islets and stimulate macrophages and other T cells, contributing to the destruction of islet beta cells [196, 203].
Fundamental molecular mechanisms still remain unclear [204]. For example, what is the role of the autoantigen-specific CD4+ T cell response [196]?. Which autoantigen is primary in initiating T1D? A series of studies suggest that proinsulin or insulin is the primary autoantigen. The specific expression of insulin in islet beta cells makes it a good candidate. Other autoantigens are expressed elsewhere. We should also keep in mind that the true primary autoantigen in T1D has not yet been definitively identified. The importance of identifying T1D-associated autoantigens will help us understand the molecular mechanisms by which beta cells are destroyed by the immune system during the development of T1D. Identification of autoantigens is also important for the development of autoantigen-specific tolerance induction immunotherapy and for establishing diagnostic and predictive markers of T1D.
The autoimmune nature of T1D is supported by the appearance of autoreactive T cells and autoantibodies. Most patients with T1D develop humoral and cellular immune responses to islet autoantigens such as GAD65 and insulin [205, 206]. The presence of antibodies to islet autoantigens can occur many years before clinical diagnosis. Autoantibodies against these islet autoantigens are present in the serum of 90 % of patients with T1D [207–209]. It is not clear if these autoantibodies play a pathogenic role in the development of T1D or if they are merely an epiphenomenon.
The first autoantibodies reported were islet cell autoantibodies [210]. ICAs are detected by reacting serum with sections of human pancreas and then staining for these autoantibodies. Anti-insulin antibodies were found in patients with T1D without exogenous insulin. Other studies indicate that anti-insulin antibodies are present several years before clinical diagnosis. Anti-GAD antibodies were first reported in patients with stiff man syndrome and were subsequently reported in patients with T1D [211]. ZnT8 was identified as an autoantigen from microarray data; our results and others show that ZnT8 autoantibodies is a predictive and diagnostic marker associated with T1D [212].
Autoantibodies may play an important role in autoantigen processing and presentation by HLA molecules. Several experiments have shown that the T cell response to autoantigen is enhanced or shifted in the presence of autoantibodies. This suggests that disease-associated GAD65 antibody can modulate the GAD65 presentation to the T cells and it may be a potential mechanism for the breakdown of islet beta cell tolerance [213]. Autoantibodies are widely used in disease prediction and diagnosis. It is accepted that the number of positive antibodies in patients is more important in predicting disease than the particular autoantibody. One experiment analyzed 45 new onset patients, 882 first degree relatives, and 217 controls, and found that 98 % had one or two antibodies and 76 % had two or three autoantibodies when they are diagnosed [214]. Our results suggest that combination testing of IAA with GADA and IA-2A may improve the LADA diagnosis rate [215].
T1D is caused by autoimmune destruction of beta cells in genetically susceptible individuals. Several studies have shown that HLA alleles are associated with autoantibodies [216]. This suggests that HLA molecules may participate in regulating the generation of autoantibodies against a specific autoantigen. The relationship between HLA and autoantibodies still needs to be explored further in order to better understand the inter-relationship between these two important pathogenic mechanisms.
Discussion
Accumulating data suggests that autoimmune T1D is the result of interaction between genetic susceptibility and environmental factors. Both genetic and environmental factors are vital for the development of autoimmune T1D. The findings in this field will accelerate our understanding of T1D. The improved understanding will help elucidate new methods of predicting the risk of developing T1D, as well as novel treatment methods and methods to prevent the onset or progression of the disease process. Currently, prediction of T1D is possible by the detection of autoantibodies in relatives of T1D patients [217]. However, negative autoantibody results do not rule out the possibility of developing T1D. Environmental factors are also important factors in the pathogenesis of T1D. Therefore, the modification of environmental exposures in the global population or in populations with high genetic susceptibility, while a massive undertaking, may be a strategy for the prevention of sporadic and familial T1D.
It has been suggested that the HLA genes are, by far, the strongest genetic determinants to T1D. HLA was identified by its role in transplant rejection. HLA has been suggested to be involved in over 100 diseases, including many autoimmune diseases, such as RA, multiple sclerosis, and T1D; infectious diseases, such as AIDS; and other diseases, such as narcolepsy [74]. The main genes associated with susceptibility of T1D are the HLA class II genes, HLA-DRB1, HLA-DQA1, and HLA-DQB1.
Alleles at the class I locus A and locus B have also been shown to play a role in T1D susceptibility. Previous research has suggested that T1D is associated with class I A*24 alleles [218]. B*3906 alleles appear to be the alleles most commonly associated with T1D [219]. The class III region does not have classical HLA loci but includes several immunologically relevant genes, such as tumor necrosis factor-α (TNFA) gene and complement C4-encoding genes C4A and C4Bc SNPs in the -238 and -308 positions in the promoter region of the TNFA gene have been reported to be associated with T1D with conflicting results. It is recommended that all studies regarding genetic susceptibility genes in T1D be accompanied by consideration of HLA genetic susceptibility in the interpretation of the data [74].
About 50 additional non-HLA loci have been found to contribute to the development of T1D. Certain susceptible genes were found based on the candidate gene strategy, which takes into account the immunological or islet-related function of the candidate genes. Many susceptible genes were identified by genome-wide association studies (GWAS). It is estimated that there are about 5–10 million frequent variants in the human genome, with most of them being SNPs. However, only a few dozen SNPs are expected to be involved in susceptibility to T1D. The identification of these SNPs had been a significant challenge until the development of high-throughput SNP genotyping arrays. Given the huge number of SNPs used in GWAS, a very large number of T1D cases and health controls are needed to get a genome-wide statistical significance [2].
GWAS is based on common variants (high-population frequency and low contribution to disease) in the human genome [220, 221]. Several large GWAS have been performed, with the greatest amount of data coming from the T1D genetics consortium (T1DGC) [34, 222]. The most significant GWAS associations were found to be in the HLA region and the insulin gene, which were previously identified by linkage analysis and candidate gene strategy. A repository of GWAS genes associated with T1D can be found at www.t1dbase.org. Most of these genes are involved in immunological and metabolic function [68]. With the advancement of high-throughput next-generation sequencing technologies, rare variant SNPs (low-population frequency and high contribution to disease) can be rapidly identified. These results will represent a significant achievement in the development of methodology that will contribute to our understanding of the genetic susceptibility to T1D.
Although GWAS have identified many common variants which are associated with T1D, the reason for familial clustering of T1D is largely unknown. A new methodology to help explain this takes advantage of copy number variation (CNV). CNVs are a form of structural variation, leading to the cell having a different number of copies of one or more sections of the DNA. CNVs may affect the expression of surrounding genes. Several studies have reported an association between CNV and autoimmune diseases in humans, such as systemic lupus, psoriasis, Crohn’s disease, RA, and T1D [223]. Recently, a genome-wide CNV analysis was performed in 20 unrelated adults with T1D and 20 control subjects. Nine CNVs were identified to be either enriched or depleted in patients with T1D or who were at high risk for T1D [224]. These CNV regions may contain genetic variants which can contribute to disease onset and be used to predict the risk of developing T1D. We believe that knowledge of CNVs involved in T1D could also improve our understanding of the mechanisms of autoimmune diabetes.
The mechanisms of susceptibility genes in T1D may be related to their role in presenting antigens and in autoreactive T cell responses. However, the key autoantigens and T cell populations which are vital in the initiation and amplification of β cell loss are still unclear. This is a significant unmet need in our quest for answers to the questions regarding pathogenesis of T1D. Other susceptibility genes that should be considered in the future include the BACH2 gene, which is specifically expressed in β cells.
MicroRNAs play an important role in regulating the expression of target genes. These short inhibitory RNA sequences have the capability to influence many biological processes, including the maintenance of immune homeostasis and immune cell differentiation and maturation. This regulatory role of microRNA is essential for the maintenance of physiological systems. MicroRNAs have been associated with aberrant expression in many autoimmune diseases, including multiple sclerosis, RA, and systemic lupus erythematosus [225–227]. These findings suggest that microRNAs play critical roles in the pathogenesis of autoimmune diseases. Aberrant expressions of microRNAs were identified in autoimmune diabetes (Table 3), and these findings may provide a new perspective on molecular mechanism of autoimmune diseases and highlight the development of microRNA-based disease interventions.
Posttranslational modifications (PTMs) have also been proposed to be important in the development of T1D. We know that proteins can be modified after they are translated and result in new antigens, and the antigens which undergo modifications are recognized by T cells or antibodies as new antigens [228]. PTMs have been shown to be involved in several autoimmune diseases, including RA, multiple sclerosis, and celiac disease [229, 230]. Evidence has also shown that PTMs may also be important in T1D. Cells undergoing stress are more prone to PTM changes. The β cell is susceptible to ER and oxidative stress, and it is a good candidate for PTMs. The β-cell-specific autoantigen, insulin, is present in high concentration in β cells, and is thus a target for PTMs [231]. A recent paper demonstrated that β cell proteins which undergo PTMs may be involved in β cell destruction in T1D [232]. However, this conclusion has been challenged in another paper, suggesting that there is no evidence of a causal effect of PTMs of β cell proteins in T1D [233]. Further experimental studies are required to clarify whether and how PTMs are involved in the pathogenesis of T1D.
One of the characteristics of T1D is the recognition of islet autoantigens by autoreactive CD4+ and CD8+ T cells and autoantibodies. CD4+ T cells and CD8+ cytotoxic T lymphocytes play an important role in the pathogenesis of T1D. It is generally believed that in T1D, specific autoantigens are processed and presented by APCs to naive T cells, leading to activation of CD4+ T cells [234]. This results in the production of inflammatory cytokines leading to the activation of beta-cell-specific CD8+ T cells. These activated T cells are then recruited to islets and stimulate macrophages and other T cells, resulting in the damage to islet beta cells [235].
The primary autoantigen involved is a controversial issue, and it has not been definitively identified. Identification of this autoantigen will help us understand the molecular mechanisms on how beta cells are destroyed by the immune system. It will also help in the development of new strategies for autoantigen-specific tolerance induction immunotherapy and for the diagnosis and prognosis of T1D. Autoantibodies against islet autoantigens may play an important role in autoantigen processing and presentation by HLA molecules.
The gut microbiota refers to the microbe population living in our intestine. One third of gut microbiota is common to the majority of humans, while two thirds are specific to the individual. Both the gut and pancreas are involved in the intestinal immune system, so it is expected that there may be an association between autoimmune diabetes and the gut [236]. The gut microbiota has been implicated in a variety of autoimmune diseases, including RA, T1D, and systemic lupus erythematosus [237, 238].
Significant differences in gut microbiota were found between children with T1D and healthy controls by PCR-DGGE and real-time quantitative PCR methods. In a recent study, the number of Clostridium, Bacteroides, and Veillonella was increased, while the number of Lactobacillus and Bifidobacterium was decreased in children with T1D. The quantity of bacteria which is critical to maintenance of gut integrity was decreased in the children with T1D [239].
Gut microbiota can affect the function of innate and adaptive immune systems. Studies have shown that the balance between Th17 and Treg cells is dependent on the composition of gut microbiota [240, 241]. Altered gut microbiota can cause increased gut permeability and decreased butyrate and mucus production, imbalance of T cells, and eventually may lead to β cell destruction [242]. More research is still needed to elucidate the role of gut microbiota in the development of T1D. The research of microbiota will provide us with new methods for the prevention and treatment of T1D by targeting the gut immune system.
It is important for us to understand that the interaction between genetic and environmental factors is important not only in the initiation of β cell autoimmunity, but may also be involved in the disease process of T1D. Another consideration to keep in mind is that there may be interactions among environment factors [129].
Conclusions
It is known that the development of autoimmune T1D is a complex process. The molecular mechanisms of autoimmune responses, beta cell autoimmunity, immune tolerance, and the causes of autoimmune diseases are numerous and complicated [243]. The interaction between genetic factors and environmental factors are crucial in the development of autoimmune T1D. Advances in genetics, epigenetics, autoreactive T cells, and new autoantigen discovery are important research goals that will drive new methods of diagnosis and treatment of autoimmune diseases such as T1D.
References
Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G, Group ES (2009) Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet 373:2027–2033
Ounissi-Benkalha H, Polychronakos C (2008) The molecular genetics of type 1 diabetes: new genes and emerging mechanisms. Trends Mol Med 14:268–275
Todd JA (2010) Etiology of type 1 diabetes. Immunity 32:457–467
Atkinson MA, Eisenbarth GS, Michels AW (2014) Type 1 diabetes. Lancet 383:69–82
Kelly MA, Rayner ML, Mijovic CH, Barnett AH (2003) Molecular aspects of type 1 diabetes. Mol Pathol: MP 56:1–10
Dabelea D (2009) The accelerating epidemic of childhood diabetes. Lancet 373:1999–2000
Gale EA (2005) Type 1 diabetes in the young: the harvest of sorrow goes on. Diabetologia 48:1435–1438
Podar T, Solntsev A, Reunanen A, Urbonaite B, Zalinkevicius R, Karvonen M et al (2000) Mortality in patients with childhood-onset type 1 diabetes in Finland, Estonia, and Lithuania: follow-up of nationwide cohorts. Diabetes Care 23:290–294
Karvonen M, Viik-Kajander M, Moltchanova E, Libman I, LaPorte R, Tuomilehto J (2000) Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care 23:1516–1526
Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ (2010) Epidemiology of type 1 diabetes. Endocrinol Metab Clin N Am 39:481–497
Levy-Marchal C, Patterson CC, Green A, Europe EASG, Diabetes (2001) Geographical variation of presentation at diagnosis of type I diabetes in children: the EURODIAB study. European and Dibetes. Diabetologia 44(Suppl 3):B75–B80
Yang Z, Wang K, Li T, Sun W, Li Y, Chang YF et al (1998) Childhood diabetes in China. Enormous variation by place and ethnic group. Diabetes Care 21:525–529
Groop LC, Bottazzo GF, Doniach D (1986) Islet cell antibodies identify latent type I diabetes in patients aged 35–75 years at diagnosis. Diabetes 35:237–241
Zhou Z, Xiang Y, Ji L, Jia W, Ning G, Huang G et al (2013) Frequency, immunogenetics, and clinical characteristics of latent autoimmune diabetes in China (LADA China study): a nationwide, multicenter, clinic-based cross-sectional study. Diabetes 62:543–550
Brahmkshatriya PP, Mehta AA, Saboo BD, Goyal RK (2012) Characteristics and prevalence of latent autoimmune diabetes in adults (LADA). ISRN Pharmacol 2012:580202
Fourlanos S, Dotta F, Greenbaum CJ, Palmer JP, Rolandsson O, Colman PG et al (2005) Latent autoimmune diabetes in adults (LADA) should be less latent. Diabetologia 48:2206–2212
Redondo MJ (2013) LADA: time for a new definition. Diabetes 62:339–340
Purushothaman R, Ramchandani N, Kazachkova I, Ten S (2007) Prevalence and clinical features of type 1.5 diabetes mellitus in children. J Pediatr Endocrinol Metab: JPEM 20:981–987
Lampasona V, Petrone A, Tiberti C, Capizzi M, Spoletini M, di Pietro S et al (2010) Zinc transporter 8 antibodies complement GAD and IA-2 antibodies in the identification and characterization of adult-onset autoimmune diabetes: non insulin requiring autoimmune diabetes (NIRAD) 4. Diabetes Care 33:104–108
Munakata Y, Yamada T, Takahashi K, Tsukita S, Takahashi K, Sawada S et al (2012) A case of slowly progressive type 1 diabetes with insulin independence maintained for 10years with alpha-glucosidase inhibitor monotherapy. Intern Med 51:3391–3394
Gale EA (2005) Latent autoimmune diabetes in adults: a guide for the perplexed. Diabetologia 48:2195–2199
Leslie RD, Williams R, Pozzilli P (2006) Clinical review: Type 1 diabetes and latent autoimmune diabetes in adults: one end of the rainbow. J Clin Endocrinol Metab 91:1654–1659
Tuomi T, Carlsson A, Li H, Isomaa B, Miettinen A, Nilsson A et al (1999) Clinical and genetic characteristics of type 2 diabetes with and without GAD antibodies. Diabetes 48:150–157
Stenstrom G, Berger B, Borg H, Fernlund P, Dorman JS, Sundkvist G (2002) HLA-DQ genotypes in classic type 1 diabetes and in latent autoimmune diabetes of the adult. Am J Epidemiol 156:787–796
Hosszufalusi N, Vatay A, Rajczy K, Prohaszka Z, Pozsonyi E, Horvath L et al (2003) Similar genetic features and different islet cell autoantibody pattern of latent autoimmune diabetes in adults (LADA) compared with adult-onset type 1 diabetes with rapid progression. Diabetes Care 26:452–457
Desai M, Zeggini E, Horton VA, Owen KR, Hattersley AT, Levy JC et al (2007) An association analysis of the HLA gene region in latent autoimmune diabetes in adults. Diabetologia 50:68–73
Desai M, Zeggini E, Horton VA, Owen KR, Hattersley AT, Levy JC et al (2006) The variable number of tandem repeats upstream of the insulin gene is a susceptibility locus for latent autoimmune diabetes in adults. Diabetes 55:1890–1894
Petrone A, Suraci C, Capizzi M, Giaccari A, Bosi E, Tiberti C et al (2008) The protein tyrosine phosphatase nonreceptor 22 (PTPN22) is associated with high GAD antibody titer in latent autoimmune diabetes in adults: non insulin requiring autoimmune diabetes (NIRAD) study 3. Diabetes Care 31:534–538
Cervin C, Lyssenko V, Bakhtadze E, Lindholm E, Nilsson P, Tuomi T et al (2008) Genetic similarities between latent autoimmune diabetes in adults, type 1 diabetes, and type 2 diabetes. Diabetes 57:1433–1437
Lin J, Zhou ZG, Wang JP, Zhang C, Huang G (2008) From Type 1, through LADA, to type 2 diabetes: a continuous spectrum? Ann N Y Acad Sci 1150:99–102
Risch N (1987) Assessing the role of HLA-linked and unlinked determinants of disease. Am J Hum Genet 40:1–14
Olmos P, A’Hern R, Heaton DA, Millward BA, Risley D, Pyke DA et al (1988) The significance of the concordance rate for type 1 (insulin-dependent) diabetes in identical twins. Diabetologia 31:747–750
Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS et al (2001) Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 44:354–362
Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA et al (2009) Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41:703–707
Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH et al (2008) Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med 359:2767–2777
Zoledziewska M, Perra C, Orru V, Moi L, Frongia P, Congia M et al (2008) Further evidence of a primary, causal association of the PTPN22 620W variant with type 1 diabetes. Diabetes 57:229–234
Bradfield JP, Qu HQ, Wang K, Zhang H, Sleiman PM, Kim CE et al (2011) A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet 7:e1002293
Cooper JD, Howson JM, Smyth D, Walker NM, Stevens H, Yang JH et al (2012) Confirmation of novel type 1 diabetes risk loci in families. Diabetologia 55:996–1000
Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V et al (2007) Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 39:857–864
Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ et al (2006) A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 38:617–619
Gestermann N, Mekinian A, Comets E, Loiseau P, Puechal X, Hachulla E et al (2010) STAT4 is a confirmed genetic risk factor for Sjogren’s syndrome and could be involved in type 1 interferon pathway signaling. Genes Immun 11:432–438
Nistico L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E et al (1996) The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet 5:1075–1080
Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE et al (2008) Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet 40:1399–1401
Yang JH, Downes K, Howson JM, Nutland S, Stevens HE, Walker NM et al (2011) Evidence of association with type 1 diabetes in the SLC11A1 gene region. BMC Med Genet 12:59
Baschal EE, Sarkar SA, Boyle TA, Siebert JC, Jasinski JM, Grabek KR et al (2011) Replication and further characterization of a Type 1 diabetes-associated locus at the telomeric end of the major histocompatibility complex. J Diabetes 3:238–247
Cheung YH, Watkinson J, Anastassiou D (2011) Conditional meta-analysis stratifying on detailed HLA genotypes identifies a novel type 1 diabetes locus around TCF19 in the MHC. Hum Genet 129:161–176
Howson JM, Walker NM, Clayton D, Todd JA, Type 1 Diabetes Genetics C (2009) Confirmation of HLA class II independent type 1 diabetes associations in the major histocompatibility complex including HLA-B and HLA-A. Diabetes Obes Metab 11(Suppl 1):31–45
Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE et al (2007) Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 450:887–892
Fung EY, Smyth DJ, Howson JM, Cooper JD, Walker NM, Stevens H et al (2009) Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun 10:188–191
Guo D, Li M, Zhang Y, Yang P, Eckenrode S, Hopkins D et al (2004) A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat Genet 36:837–841
Swafford AD, Howson JM, Davison LJ, Wallace C, Smyth DJ, Schuilenburg H et al (2011) An allele of IKZF1 (Ikaros) conferring susceptibility to childhood acute lymphoblastic leukemia protects against type 1 diabetes. Diabetes 60:1041–1044
Reddy MV, Wang H, Liu S, Bode B, Reed JC, Steed RD et al (2011) Association between type 1 diabetes and GWAS SNPs in the southeast US Caucasian population. Genes Immun 12:208–212
Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R et al (2007) Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet 39:1074–1082
Bell GI, Horita S, Karam JH (1984) A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 33:176–183
Barratt BJ, Payne F, Lowe CE, Hermann R, Healy BC, Harold D et al (2004) Remapping the insulin gene/IDDM2 locus in type 1 diabetes. Diabetes 53:1884–1889
Hakonarson H, Qu HQ, Bradfield JP, Marchand L, Kim CE, Glessner JT et al (2008) A novel susceptibility locus for type 1 diabetes on Chr12q13 identified by a genome-wide association study. Diabetes 57:1143–1146
Keene KL, Quinlan AR, Hou X, Hall IM, Mychaleckyj JC, Onengut-Gumuscu S et al (2012) Evidence for two independent associations with type 1 diabetes at the 12q13 locus. Genes Immun 13:66–70
Espino-Paisan L, de la Calle H, Fernandez-Arquero M, Figueredo MA, de la Concha EG, Urcelay E et al (2011) Polymorphisms in chromosome region 12q13 and their influence on age at onset of type 1 diabetes. Diabetologia 54:2033–2037
Lavrikova EY, Nikitin AG, Kuraeva TL, Peterkova VA, Tsitlidze NM, Chistiakov DA et al (2011) The carriage of the type 1 diabetes-associated R262W variant of human LNK correlates with increased proliferation of peripheral blood monocytes in diabetic patients. Pediatr Diabetes 12:127–132
Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H et al (2010) A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature 467:460–464
Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG (2010) The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet 42:68–71
Qu HQ, Grant SF, Bradfield JP, Kim C, Frackelton E, Hakonarson H et al (2009) Association of RASGRP1 with type 1 diabetes is revealed by combined follow-up of two genome-wide studies. J Med Genet 46:553–554
Wang K, Baldassano R, Zhang H, Qu HQ, Imielinski M, Kugathasan S et al (2010) Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum Mol Genet 19:2059–2067
Smyth DJ, Cooper JD, Howson JM, Clarke P, Downes K, Mistry T et al (2011) FUT2 nonsecretor status links type 1 diabetes susceptibility and resistance to infection. Diabetes 60:3081–3084
Concannon P, Onengut-Gumuscu S, Todd JA, Smyth DJ, Pociot F, Bergholdt R et al (2008) A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes 57:2858–2861
Turunen JA, Wessman M, Forsblom C, Kilpikari R, Parkkonen M, Pontynen N et al (2006) Association analysis of the AIRE and insulin genes in Finnish type 1 diabetic patients. Immunogenetics 58:331–338
Cooper JD, Walker NM, Smyth DJ, Downes K, Healy BC, Todd JA et al (2009) Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium genome-wide association study in type I diabetes families. Genes Immun 10(Suppl 1):S85–S94
Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G et al (2010) Genetics of type 1 diabetes: what’s next? Diabetes 59:1561–1571
Morahan G (2012) Insights into type 1 diabetes provided by genetic analyses. Curr Opin Endocrinol Diabetes Obes 19:263–270
Singal DP, Blajchman MA (1973) Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 22:429–432
Mehers KL, Gillespie KM (2008) The genetic basis for type 1 diabetes. Br Med Bull 88:115–129
Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK et al (2004) Gene map of the extended human MHC. Nat Rev Genet 5:889–899
Complete sequence and gene map of a human major histocompatibility complex. The MHC sequencing consortium. Nature. 1999;401:921-3
Noble JA, Erlich HA (2012) Genetics of type 1 diabetes. Cold Spring Harb Perspect Med 2:a007732
van Belle TL, Coppieters KT, von Herrath MG (2011) Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91:79–118
Thomson G, Valdes AM, Noble JA, Kockum I, Grote MN, Najman J et al (2007) Relative predispositional effects of HLA class II DRB1-DQB1 haplotypes and genotypes on type 1 diabetes: a meta-analysis. Tissue Antigens 70:110–127
Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P et al (2008) HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 57:1084–1092
Mimbacas A, Perez-Bravo F, Santos JL, Pisciottano C, Grignola R, Javiel G et al (2004) The association between HLA DQ genetic polymorphism and type 1 diabetes in a case-parent study conducted in an admixed population. Eur J Epidemiol 19:931–934
She JX (1996) Susceptibility to type I diabetes: HLA-DQ and DR revisited. Immunol Today 17:323–329
Undlien DE, Friede T, Rammensee HG, Joner G, Dahl-Jorgensen K, Sovik O et al (1997) HLA-encoded genetic predisposition in IDDM: DR4 subtypes may be associated with different degrees of protection. Diabetes 46:143–149
Kawabata Y, Ikegami H, Kawaguchi Y, Fujisawa T, Shintani M, Ono M et al (2002) Asian-specific HLA haplotypes reveal heterogeneity of the contribution of HLA-DR and -DQ haplotypes to susceptibility to type 1 diabetes. Diabetes 51:545–551
Katahira M, Ishiguro T, Segawa S, Kuzuya-Nagao K, Hara I, Nishisaki T (2008) Reevaluation of human leukocyte antigen DR-DQ haplotype and genotype in type 1 diabetes in the Japanese population. Horm Res 69:284–289
Park YS, Wang CY, Ko KW, Yang SW, Park M, Yang MC et al (1998) Combinations of HLA DR and DQ molecules determine the susceptibility to insulin-dependent diabetes mellitus in Koreans. Hum Immunol 59:794–801
Park Y, She JX, Wang CY, Lee H, Babu S, Erlich HA et al (2000) Common susceptibility and transmission pattern of human leukocyte antigen DRB1-DQB1 haplotypes to Korean and Caucasian patients with type 1 diabetes. J Clin Endocrinol Metab 85:4538–4542
Zhang XM, Wang HY, Luo YY, Ji LN (2009) HLA-DQ, DR allele polymorphism of type 1 diabetes in the Chinese population: a meta-analysis. Chin Med J 122:980–986
Huang HS, Peng JT, She JY, Zhang LP, Chao CC, Liu KH et al (1995) HLA-encoded susceptibility to insulin-dependent diabetes mellitus is determined by DR and DQ genes as well as their linkage disequilibria in a Chinese population. Hum Immunol 44:210–219
Noble JA, Valdes AM, Cook M, Klitz W, Thomson G, Erlich HA (1996) The role of HLA class II genes in insulin-dependent diabetes mellitus: molecular analysis of 180 Caucasian, multiplex families. Am J Hum Genet 59:1134–1148
Ikegami H, Kawabata Y, Noso S, Fujisawa T, Ogihara T (2007) Genetics of type 1 diabetes in Asian and Caucasian populations. Diabetes Res Clin Pract 77(Suppl 1):S116–S121
Undlien DE, Kockum I, Ronningen KS, Lowe R, Saanjeevi CB, Graham J et al (1999) HLA associations in type 1 diabetes among patients not carrying high-risk DR3-DQ2 or DR4-DQ8 haplotypes. Tissue Antigens 54:543–551
Kawasaki E, Noble J, Erlich H, Mulgrew CL, Fain PR, Eisenbarth GS (1998) Transmission of DQ haplotypes to patients with type 1 diabetes. Diabetes 47:1971–1973
Awata T, Kuzuya T, Matsuda A, Iwamoto Y, Kanazawa Y (1992) Genetic analysis of HLA class II alleles and susceptibility to type 1 (insulin-dependent) diabetes mellitus in Japanese subjects. Diabetologia 35:419–424
Karjalainen J, Salmela P, Ilonen J, Surcel HM, Knip M (1989) A comparison of childhood and adult type I diabetes mellitus. N Engl J Med 320:881–886
Cerna M, Novota P, Kolostova K, Cejkova P, Zdarsky E, Novakova D et al (2003) HLA in Czech adult patients with autoimmune diabetes mellitus: comparison with Czech children with type 1 diabetes and patients with type 2 diabetes. Eur J immunogenet 30:401–407
Fukui M, Kitagawa Y, Nakamura N, Yoshikawa T (2003) Clinical and genetic heterogeneity of latent autoimmune diabetes in adults. Diabetes Care 26:2223, author reply 4
Andersen MK, Lundgren V, Turunen JA, Forsblom C, Isomaa B, Groop PH et al (2010) Latent autoimmune diabetes in adults differs genetically from classical type 1 diabetes diagnosed after the age of 35years. Diabetes Care 33:2062–2064
Weber P, Meluzinova H, Kubesova H, Ambrosova P, Polcarova V, Cejkova P et al (2010) Type 1 diabetes and LADA–occurrence of HLA-DRB1 *03 and DRB1 *04 alleles in two age different groups of diabetics. Adv Gerontol Usp Gerontol Ross Akad Nauk Gerontol Obshch 23:243–248
Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE et al (1995) Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet 9:284–292
Durinovic-Bello I, Wu RP, Gersuk VH, Sanda S, Shilling HG, Nepom GT (2010) Insulin gene VNTR genotype associates with frequency and phenotype of the autoimmune response to proinsulin. Genes Immun 11:188–193
Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG et al (1997) Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet 15:289–292
Pugliese A, Zeller M, Fernandez A Jr, Zalcberg LJ, Bartlett RJ, Ricordi C et al (1997) The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet 15:293–297
Vafiadis P, Ounissi-Benkalha H, Palumbo M, Grabs R, Rousseau M, Goodyer CG et al (2001) Class III alleles of the variable number of tandem repeat insulin polymorphism associated with silencing of thymic insulin predispose to type 1 diabetes. J Clin Endocrinol Metab 86:3705–3710
Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G et al (2003) Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423:506–511
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z et al (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science 322:271–275
Atabani SF, Thio CL, Divanovic S, Trompette A, Belkaid Y, Thomas DL et al (2005) Association of CTLA4 polymorphism with regulatory T cell frequency. Eur J Immunol 35:2157–2162
Korolija M, Renar IP, Hadzija M, Medvidovic EP, Pavkovic P, Jokic M et al (2009) Association of PTPN22 C1858T and CTLA-4 A49G polymorphisms with type 1 diabetes in Croatians. Diabetes Res Clin Pract 86:e54–e57
Cutolo M, Nadler SG (2013) Advances in CTLA-4-Ig-mediated modulation of inflammatory cell and immune response activation in rheumatoid arthritis. Autoimmun Rev 12:758–767
Romo-Tena J, Gomez-Martin D, Alcocer-Varela J (2013) CTLA-4 and autoimmunity: new insights into the dual regulator of tolerance. Autoimmun Rev 12:1171–1176
Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM et al (2004) Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 53:3020–3023
Gregersen PK, Behrens TW (2006) Genetics of autoimmune diseases—disorders of immune homeostasis. Nat Rev Genet 7:917–928
Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P et al (2005) Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet 37:1317–1319
Gregersen PK (2005) Gaining insight into PTPN22 and autoimmunity. Nat Genet 37:1300–1302
Chung SA, Criswell LA (2007) PTPN22: its role in SLE and autoimmunity. Autoimmunity 40:582–590
Gianchecchi E, Palombi M, Fierabracci A (2013) The putative role of the C1858T polymorphism of protein tyrosine phosphatase PTPN22 gene in autoimmunity. Autoimmun Rev 12:717–725
Zheng J, Petersen F, Yu X (2014) The role of PTPN22 in autoimmunity: learning from mice. Autoimmun Rev 13:266–271
Corthay A (2009) How do regulatory T cells work? Scand J Immunol 70:326–336
Askenasy N (2013) Enhanced killing activity of regulatory T cells ameliorates inflammation and autoimmunity. Autoimmun Rev 12:972–975
Smyth DJ, Howson JM, Lowe CE, Walker NM, Lam AC, Nutland S et al (2005) Assessing the validity of the association between the SUMO4 M55V variant and risk of type 1 diabetes. Nat Genet 37:110–111, author reply 2-3
Guo D, Han J, Adam BL, Colburn NH, Wang MH, Dong Z et al (2005) Proteomic analysis of SUMO4 substrates in HEK293 cells under serum starvation-induced stress. Biochem Biophys Res Commun 337:1308–1318
Caamano J, Hunter CA (2002) NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev 15:414–429
Lee HS, Park H, Yang S, Kim D, Park Y (2008) STAT4 polymorphism is associated with early-onset type 1 diabetes, but not with late-onset type 1 diabetes. Ann N Y Acad Sci 1150:93–98
Frucht DM, Aringer M, Galon J, Danning C, Brown M, Fan S et al (2000) Stat4 is expressed in activated peripheral blood monocytes, dendritic cells, and macrophages at sites of Th1-mediated inflammation. J Immunol 164:4659–4664
Yang Z, Chen M, Ellett JD, Fialkow LB, Carter JD, McDuffie M et al (2004) Autoimmune diabetes is blocked in Stat4-deficient mice. J Autoimmun 22:191–200
Trinchieri G (1994) Interleukin-12: a cytokine produced by antigen-presenting cells with immunoregulatory functions in the generation of T-helper cells type 1 and cytotoxic lymphocytes. Blood 84:4008–4027
Pettersen E, Skorpen F, Kvaloy K, Midthjell K, Grill V (2010) Genetic heterogeneity in latent autoimmune diabetes is linked to various degrees of autoimmune activity: results from the Nord-Trondelag Health Study. Diabetes 59:302–310
Howson JM, Rosinger S, Smyth DJ, Boehm BO, Group A-ES, Todd JA (2011) Genetic analysis of adult-onset autoimmune diabetes. Diabetes 60:2645–2653
Okruszko A, Szepietowska B, Wawrusiewicz-Kurylonek N, Gorska M, Kretowski A, Szelachowska M (2012) HLA-DR, HLA-DQB1 and PTPN22 gene polymorphism: association with age at onset for autoimmune diabetes. Arch Med Sci AMS 8:874–878
Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J et al (1992) Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 35:1060–1067
Dang MN, Buzzetti R, Pozzilli P (2013) Epigenetics in autoimmune diseases with focus on type 1 diabetes. Diabetes Metab Res Rev 29:8–18
Knip M, Veijola R, Virtanen SM, Hyoty H, Vaarala O, Akerblom HK (2005) Environmental triggers and determinants of type 1 diabetes. Diabetes 54(Suppl 2):S125–S136
Akerblom HK, Knip M (1998) Putative environmental factors in Type 1 diabetes. Diabetes Metab Rev 14:31–67
Hermann R, Knip M, Veijola R, Simell O, Laine AP, Akerblom HK et al (2003) Temporal changes in the frequencies of HLA genotypes in patients with type 1 diabetes—indication of an increased environmental pressure? Diabetologia 46:420–425
Gillespie KM, Bain SC, Barnett AH, Bingley PJ, Christie MR, Gill GV et al (2004) The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet 364:1699–1700
EURODIAB ACE Study Group (2000) Variation and trends in incidence of childhood diabetes in Europe. Lancet 355:873–876
Karagkouni A, Alevizos M, Theoharides TC (2013) Effect of stress on brain inflammation and multiple sclerosis. Autoimmun Rev 12:947–953
Selmi C (2013) Autoimmunity in 2012. Clin Rev Allergy Immunol 45:290–301
Selmi C, Crotti C, Meroni PL (2013) Less travelled roads in clinical immunology and allergy: drug reactions and the environmental influence. Clin Rev Allergy Immunol 45:1–5
Brooks WH (2012) Autoimmune diseases and polyamines. Clin Rev Allergy Immunol 42:58–70
Rook GA (2012) Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol 42:5–15
Longnecker MP, Daniels JL (2001) Environmental contaminants as etiologic factors for diabetes. Environ Health Perspect 109(Suppl 6):871–876
Virtanen SM, Knip M (2003) Nutritional risk predictors of beta cell autoimmunity and type 1 diabetes at a young age. Am J Clin Nutr 78:1053–1067
Howard SG, Lee DH (2012) What is the role of human contamination by environmental chemicals in the development of type 1 diabetes? J Epidemiol Community Health 66:479–481
Atkinson M, Gale EA (2003) Infant diets and type 1 diabetes: too early, too late, or just too complicated? JAMA J Am Med Assoc 290:1771–1772
Knip M, Virtanen SM, Akerblom HK (2010) Infant feeding and the risk of type 1 diabetes. Am J Clin Nutr 91:1506S–1513S
Vaarala O (2002) The gut immune system and type 1 diabetes. Ann N Y Acad Sci 958:39–46
Sepa A, Ludvigsson J (2006) Psychological stress and the risk of diabetes-related autoimmunity: a review article. Neuroimmunomodulation 13:301–308
Harder T, Roepke K, Diller N, Stechling Y, Dudenhausen JW, Plagemann A (2009) Birth weight, early weight gain, and subsequent risk of type 1 diabetes: systematic review and meta-analysis. Am J Epidemiol 169:1428–1436
Yeung WC, Rawlinson WD, Craig ME (2011) Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ 342:d35
Oikarinen S, Martiskainen M, Tauriainen S, Huhtala H, Ilonen J, Veijola R et al (2011) Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes 60:276–279
Stene LC, Oikarinen S, Hyoty H, Barriga KJ, Norris JM, Klingensmith G et al (2010) Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes 59:3174–3180
Javierre BM, Hernando H, Ballestar E (2011) Environmental triggers and epigenetic deregulation in autoimmune disease. Discov Med 12:535–545
Lu Q (2013) The critical importance of epigenetics in autoimmunity. J Autoimmun 41:1–5
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28:1057–1068
He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q et al (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333:1303–1307
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Wu SC, Zhang Y (2010) Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 11:607–620
Sananbenesi F, Fischer A (2009) The epigenetic bottleneck of neurodegenerative and psychiatric diseases. Biol Chem 390:1145–1153
Robertson KD (2005) DNA methylation and human disease. Nat Rev Genet 6:597–610
Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA (2010) Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genet 3:33
Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D et al (2011) Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet 7:e1002300
Wang Z, Zheng Y, Hou C, Yang L, Li X, Lin J et al (2013) DNA methylation impairs TLR9 induced Foxp3 expression by attenuating IRF-7 binding activity in fulminant type 1 diabetes. J Autoimmun 41:50–59
Fradin D, Le Fur S, Mille C, Naoui N, Groves C, Zelenika D et al (2012) Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS One 7:e36278
Akirav EM, Lebastchi J, Galvan EM, Henegariu O, Akirav M, Ablamunits V et al (2011) Detection of beta cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci U S A 108:19018–19023
Fisher MM, Perez Chumbiauca CN, Mather KJ, Mirmira RG, Tersey SA (2013) Detection of islet beta-cell death in vivo by multiplex PCR analysis of differentially methylated DNA. Endocrinology 154:3476–3481
Stefan M, Zhang W, Concepcion E, Yi Z, Tomer Y (2013) DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J Autoimmun. doi:10.1016/j.jaut.2013.10.001
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML et al (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102:10604–10609
Menegatti E, Berardi D, Messina M, Ferrante I, Giachino O, Spagnolo B et al (2013) Lab-on-a-chip: emerging analytical platforms for immune-mediated diseases. Autoimmun Rev 12:814–820
Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R (2008) Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes 57:3189–3198
Orban T, Kis J, Szereday L, Engelmann P, Farkas K, Jalahej H et al (2007) Reduced CD4+ T-cell-specific gene expression in human type 1 diabetes mellitus. J Autoimmun 28:177–187
Miao F, Chen Z, Zhang L, Liu Z, Wu X, Yuan YC et al (2012) Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J Biol Chem 287:16335–16345
Chen K, Rajewsky N (2007) The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8:93–103
Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9:102–114
Singh RP, Massachi I, Manickavel S, Singh S, Rao NP, Hasan S et al (2013) The role of miRNA in inflammation and autoimmunity. Autoimmun Rev 12:1160–1165
Saito Y, Saito H, Liang G, Friedman JM (2013) Epigenetic alterations and microRNA misexpression in cancer and autoimmune diseases: a critical review. Clin Revi Allergy Immunol. [E-pub ahead of print]
Iborra M, Bernuzzi F, Invernizzi P, Danese S (2012) MicroRNAs in autoimmunity and inflammatory bowel disease: crucial regulators in immune response. Autoimmun Rev 11:305–314
Fernandez-Valverde SL, Taft RJ, Mattick JS (2011) MicroRNAs in beta-cell biology, insulin resistance, diabetes and its complications. Diabetes 60:1825–1831
Sebastiani G, Grieco FA, Spagnuolo I, Galleri L, Cataldo D, Dotta F (2011) Increased expression of microRNA miR-326 in type 1 diabetic patients with ongoing islet autoimmunity. Diabetes Metab Res Rev 27:862–866
Hezova R, Slaby O, Faltejskova P, Mikulkova Z, Buresova I, Raja KR et al (2010) microRNA-342, microRNA-191 and microRNA-510 are differentially expressed in T regulatory cells of type 1 diabetic patients. Cell Immunol 260:70–74
Salas-Perez F, Codner E, Valencia E, Pizarro C, Carrasco E, Perez-Bravo F (2013) MicroRNAs miR-21a and miR-93 are down regulated in peripheral blood mononuclear cells (PBMCs) from patients with type 1 diabetes. Immunobiology 218:733–737
Nielsen LB, Wang C, Sorensen K, Bang-Berthelsen CH, Hansen L, Andersen ML et al (2012) Circulating levels of microRNA from children with newly diagnosed type 1 diabetes and healthy controls: evidence that miR-25 associates to residual beta-cell function and glycaemic control during disease progression. Exp Diabetes Res 2012:896362
Sebastiani G, Spagnuolo I, Patti A, Grieco FA, Cataldo D, Ferretti E et al (2012) MicroRNA expression fingerprint in serum of type 1 diabetic patients. Diabetologia 55:S48
Ruan Q, Wang T, Kameswaran V, Wei Q, Johnson DS, Matschinsky F et al (2011) The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic beta cell death. Proc Natl Acad Sci U S A 108:12030–12035
Jimenez SA, Piera-Velazquez S (2013) Potential role of human-specific genes, human-specific microRNAs and human-specific non-coding regulatory RNAs in the pathogenesis of systemic sclerosis and Sjogren’s syndrome. Autoimmun Rev 12:1046–1051
De Santis M, Selmi C (2012) The therapeutic potential of epigenetics in autoimmune diseases. Clin Rev Allergy Immunol 42:92–101
Hyoty H, Taylor KW (2002) The role of viruses in human diabetes. Diabetologia 45:1353–1361
Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE et al (2000) Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 49:1319–1324
Ramondetti F, Sacco S, Comelli M, Bruno G, Falorni A, Iannilli A et al (2012) Type 1 diabetes and measles, mumps and rubella childhood infections within the Italian Insulin-dependent Diabetes Registry. Diabet Med J Br Diabet Assoc 29:761–766
Aarnisalo J, Veijola R, Vainionpaa R, Simell O, Knip M, Ilonen J (2008) Cytomegalovirus infection in early infancy: risk of induction and progression of autoimmunity associated with type 1 diabetes. Diabetologia 51:769–772
Coppieters KT, Wiberg A, von Herrath MG (2012) Viral infections and molecular mimicry in type 1 diabetes. APMIS 120:941–949
Blank M, Barzilai O, Shoenfeld Y (2007) Molecular mimicry and auto-immunity. Clin Rev Allergy Imunol 32:111–118
Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK (1994) Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J Clin Invest 94:2125–2129
Cusick MF, Libbey JE, Fujinami RS (2012) Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Imunol 42:102–111
Harkonen T, Lankinen H, Davydova B, Hovi T, Roivainen M (2002) Enterovirus infection can induce immune responses that cross-react with beta-cell autoantigen tyrosine phosphatase IA-2/IAR. J Med Virol 66:340–350
Hiemstra HS, Schloot NC, van Veelen PA, Willemen SJ, Franken KL, van Rood JJ et al (2001) Cytomegalovirus in autoimmunity: T cell crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase. Proc Natl Acad Sci U S A 98:3988–3991
Singh B, Delovitch TL (2000) Immune mechanisms that regulate susceptibility to autoimmune type I diabetes. Clin Rev Allergy Imunol 19:247–264
Han S, Donelan W, Wang H, Reeves W, Yang LJ (2013) Novel autoantigens in type 1 diabetes. Am J Transl Res 5:379–392
Roep BO, Peakman M (2012) Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med 2:a007781
Delong T, Baker RL, He J, Haskins K (2013) Novel autoantigens for diabetogenic CD4 T cells in autoimmune diabetes. Immunol Res 55:167–172
Brooks-Worrell B, Warsen A, Palmer JP (2009) Improved T cell assay for identification of type 1 diabetes patients. J Immunol Methods 344:79–83
Knip M, Siljander H (2008) Autoimmune mechanisms in type 1 diabetes. Autoimmun Rev 7:550–557
Wildner G, Kaufmann U (2013) What causes relapses of autoimmune diseases? The etiological role of autoreactive T cells. Autoimmun Rev 12:1070–1075
Sarikonda G, Pettus J, Phatak S, Sachithanantham S, Miller JF, Wesley JD et al (2013) CD8 T-cell reactivity to islet antigens is unique to type 1 while CD4 T-cell reactivity exists in both type 1 and type 2 diabetes. J Autoimmun. doi:10.1016/j.jaut.2013.12.003
Stadinski B, Kappler J, Eisenbarth GS (2010) Molecular targeting of islet autoantigens. Immunity 32:446–456
Askenasy EM, Askenasy N (2013) Is autoimmune diabetes caused by aberrant immune activity or defective suppression of physiological self-reactivity? Autoimmun Rev 12:633–637
Schloot NC, Willemen SJ, Duinkerken G, Drijfhout JW, de Vries RR, Roep BO (2001) Molecular mimicry in type 1 diabetes mellitus revisited: T-cell clones to GAD65 peptides with sequence homology to Coxsackie or proinsulin peptides do not crossreact with homologous counterpart. Hum Immunol 62:299–309
Boettler T, Pagni PP, Jaffe R, Cheng Y, Zerhouni P, von Herrath M (2013) The clinical and immunological significance of GAD-specific autoantibody and T-cell responses in type 1 diabetes. J Autoimmun 44:40–48
Leslie RD, Atkinson MA, Notkins AL (1999) Autoantigens IA-2 and GAD in type I (insulin-dependent) diabetes. Diabetologia 42:3–14
Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK et al (1983) Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 222:1337–1339
Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P et al (2007) The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A 104:17040–17045
Bottazzo GF, Florin-Christensen A, Doniach D (1974) Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 2:1279–1283
Taplin CE, Barker JM (2008) Autoantibodies in type 1 diabetes. Autoimmunity 41:11–18
Yang L, Luo S, Huang G, Peng J, Li X, Yan X et al (2010) The diagnostic value of zinc transporter 8 autoantibody (ZnT8A) for type 1 diabetes in Chinese. Diabetes Metab Res Rev 26:579–584
Pihoker C, Gilliam LK, Hampe CS, Lernmark A (2005) Autoantibodies in diabetes. Diabetes 54(Suppl 2):S52–S61
Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Chase HP et al (1996) Number of autoantibodies (against insulin, GAD or ICA512/IA2) rather than particular autoantibody specificities determines risk of type I diabetes. J Autoimmun 9:379–383
Huang G, Wang X, Li Z, Li H, Li X, Zhou Z (2012) Insulin autoantibody could help to screen latent autoimmune diabetes in adults in phenotypic type 2 diabetes mellitus in Chinese. Acta Diabetol 49:327–331
Graham J, Hagopian WA, Kockum I, Li LS, Sanjeevi CB, Lowe RM et al (2002) Genetic effects on age-dependent onset and islet cell autoantibody markers in type 1 diabetes. Diabetes 51:1346–1355
Miao D, Yu L, Eisenbarth GS (2007) Role of autoantibodies in type 1 diabetes. Front Biosci J Virtual Libr 12:1889–1898
Noble JA, Valdes AM, Bugawan TL, Apple RJ, Thomson G, Erlich HA (2002) The HLA class I A locus affects susceptibility to type 1 diabetes. Hum Immunol 63:657–664
Noble JA, Valdes AM, Varney MD, Carlson JA, Moonsamy P, Fear AL et al (2010) HLA class I and genetic susceptibility to type 1 diabetes: results from the Type 1 Diabetes Genetics Consortium. Diabetes 59:2972–2979
Coit P, Jeffries M, Altorok N, Dozmorov MG, Koelsch KA, Wren JD et al (2013) Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J Autoimmun 43:78–84
Cui Y, Sheng Y, Zhang X (2013) Genetic susceptibility to SLE: recent progress from GWAS. J Autoimmun 41:25–33
Qu HQ, Bradfield JP, Li Q, Kim C, Frackelton E, Grant SF et al (2010) In silico replication of the genome-wide association results of the Type 1 Diabetes Genetics Consortium. Hum Mol Genet 19:2534–2538
Schaschl H, Aitman TJ, Vyse TJ (2009) Copy number variation in the human genome and its implication in autoimmunity. Clin Exp Immunol 156:12–16
Grayson BL, Smith ME, Thomas JW, Wang L, Dexheimer P, Jeffrey J et al (2010) Genome-wide analysis of copy number variation in type 1 diabetes. PLoS One 5:e15393
Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD (2008) MicroRNAs: new regulators of immune cell development and function. Nat Immunol 9:839–845
Pauley KM, Cha S, Chan EK (2009) MicroRNA in autoimmunity and autoimmune diseases. J Autoimmun 32:189–194
Dai R, Ahmed SA (2011) MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl Res J Lab Clin Med 157:163–179
Dahan R, Gebe JA, Preisinger A, James EA, Tendler M, Nepom GT et al (2013) Antigen-specific immunomodulation for type 1 diabetes by novel recombinant antibodies directed against diabetes-associates auto-reactive T cell epitope. J Autoimmun 47:83–93
Dunne JL, Overbergh L, Purcell AW, Mathieu C (2012) Posttranslational modifications of proteins in type 1 diabetes: the next step in finding the cure? Diabetes 61:1907–1914
van Lummel M, Zaldumbide A, Roep BO (2013) Changing faces, unmasking the beta-cell: post-translational modification of antigens in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 20:299–306
Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP et al (2005) The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med 202:1191–1197
Storling J, Overgaard AJ, Brorsson CA, Piva F, Bang-Berthelsen CH, Haase C et al (2013) Do post-translational beta cell protein modifications trigger type 1 diabetes? Diabetologia 56:2347–2354
Lernmark A (2013) Is there evidence for post-translational modification of beta cell autoantigens in the aetiology and pathogenesis of type 1 diabetes? Diabetologia [E-pub ahead of print].
Edwards LJ, Evavold BD (2013) Destabilization of peptide:MHC interaction induces IL-2 resistant anergy in diabetogenic T cells. J Autoimmun 44:82–90
Gravano DM, Hoyer KK (2013) Promotion and prevention of autoimmune disease by CD8+ T cells. J Autoimmun 45:68–79
Vaarala O (2012) Is the origin of type 1 diabetes in the gut? Immunol Cell Biol 90:271–276
Kosiewicz MM, Zirnheld AL, Alard P (2011) Gut microbiota, immunity, and disease: a complex relationship. Front Microbiol 2:180
Pillai S (2013) Rethinking mechanisms of autoimmune pathogenesis. J Autoimmun 45:97–103
Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F et al (2013) Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med 11:46
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139:485–498
Romano-Keeler J, Weitkamp JH, Moore DJ (2012) Regulatory properties of the intestinal microbiome effecting the development and treatment of diabetes. Curr Opin Endocrinol Diabetes Obes 19:73–80
Vaarala O (2013) Human intestinal microbiota and type 1 diabetes. Curr Diabetes Rep 13:601–607
Brooks WH (2012) Mechanisms and pathophysiology of autoimmune disease. Clin Rev Allergy Immunol 42:1–4
Acknowledgments
We would like to thank Dr. Eric Gershwin and Qianjin Lu for their advice and proofreading of the manuscript. This work was supported by the National Natural Science Foundation of China (Grant No. 81170725), the European Foundation for the Study of Diabetes (Grant No. EFSD/CDS/Lilly-2010), Hunan Provincial Natural Science Foundation of China (11JJ7005), Program for Changjiang Scholars and Innovative Research Team in University (IRT1195) and the National Key Technology R&D program (2012BAI02B04).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xie, Z., Chang, C. & Zhou, Z. Molecular Mechanisms in Autoimmune Type 1 Diabetes: a Critical Review. Clinic Rev Allerg Immunol 47, 174–192 (2014). https://doi.org/10.1007/s12016-014-8422-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-014-8422-2